Accelerating drug discovery via organs-on-chips
Chung Yu
Chan
a,
Po-Hsun
Huang
a,
Feng
Guo
a,
Xiaoyun
Ding
a,
Vivek
Kapur
b,
John D.
Mai
c,
Po Ki
Yuen
*d and
Tony Jun
Huang
*a
aDepartment of Engineering Science and Mechanics, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: junhuang@psu.edu; Fax: +1 814 865 9974; Tel: +1 814 863 4209
bDepartment of Veterinary and Biomedical Sciences, The Pennsylvania State University, University Park, PA 16802, USA
cDepartment of Mechanical and Biomedical Engineering, City University of Hong Kong, Kowloon, Hong Kong SAR, PR China
dScience & Technology, Corning Incorporated, Corning, New York 14831-0001, USA. E-mail: yuenp@corning.com; Fax: +1 607 974 5957; Tel: +1 607 974 9680
First published on 5th November 2013
Abstract
Considerable advances have been made in the development of micro-physiological systems that seek to faithfully replicate the complexity and functionality of animal and human physiology in research laboratories. Sometimes referred to as “organs-on-chips”, these systems provide key insights into physiological or pathological processes associated with health maintenance and disease control, and serve as powerful platforms for new drug development and toxicity screening. In this Focus article, we review the state-of-the-art designs and examples for developing multiple “organs-on-chips”, and discuss the potential of this emerging technology to enhance our understanding of human physiology, and to transform and accelerate the drug discovery and preclinical testing process. This Focus article highlights some of the recent technological advances in this field, along with the challenges that must be addressed for these technologies to fully realize their potential.
1. Introduction
Cells are a basic unit of life, and studies of laboratory-grown cells in a monoculture have contributed immeasurably to a better understanding of basic biological and pathological processes associated with life. It is well established that in many living systems, such as human beings, cells are organized into heterotypic functional units – tissues and organs – whose collective responses and functions cannot be emulated by a culture of single cells.1,2 Studies suggest that when multiple cell types are allowed to interact with each other under co-culture conditions, their response to different soluble factors and chemical compounds bear a greater resemblance to what occurs in vivo.3 Hence, there is growing interest in the bioengineering of in vitro systems that are comprised of assemblies of different cells for understanding cellular mechanisms with a greater fidelity, as well as for the replication of the organ functions that more closely resemble those in the human body.4–6 However, conventional in vitro cell culture methods are insufficient in physiological relevance and are not predictive of in vivo behavior in animal models and humans.7,8 Current studies suggest that microfluidic-based approaches have the potential to create an interactive cell microenvironment that mimics cell and organ level organizational structures in vivo because of its user-defined design, relevant length scale, and sophisticated control of a dynamic environment.9–16 Recently, the notion of “organs-on-chips” has been extensively developed and aims to reconstruct the physiological functions at the cellular or organ level and obtain human pharmacokinetic (PK) and pharmacodynamic (PD) response without the use of an animal model.17–20 With growing interest in developing technologies to enable an “organ-on-a-chip”, several micro-devices have been realized that attempt to reconstruct the functional units of various vital organ systems. For example, the recent mimicking of the alveolar–capillary interface, the functional unit of the human lung, has enabled the studies of lung physiology and injury.21,22 Importantly, Hsu et al. demonstrated the full range of physiological mass transport control similar to that found in blood vessels.23 Similar studies have included the reconstruction of hepatic cords, the functional unit of the human liver,24 and many other examples that have revealed the functional, structural, and pathological features of various organs.5,20,25–29Not surprisingly, advances in the design of “organs-on-chips” have already demonstrated their potential for applications in drug discovery and toxicity screening. Usually, in order to assess drug efficacy, an in vitro cell culture model is used to monitor the effect of the drug on the target cells, as well as normal cells in the body.17,30 Though an in vitro cell culture model gives a rapid prediction for the effective concentration of the drug, the data obtained is often too limited and unable to accurately predict the side effects of different drug dosages and interactions on the entire human body or the target organ system. Therefore, animal models are often employed to obtain more comprehensive and systemic responses of the drug or compound. However, given the substantial differences between animal and human physiologies, animal models (particularly the often used rodent models) are increasingly recognized as an imperfect representation of the human system. The consequences of this are considerable. They have led to low success rates in terms of drug efficacy in Phase II and III human clinical trials and have added significantly to the cost and time to develop new therapeutic compounds.17,18,30 Hence, novel approaches to facilitating drug discovery by developing models that are able to more faithfully represent human physiology remains an area of intense research interest.
In this Focus article, we summarize the major techniques involved in developing organs-on-chips along with their application in drug discovery and screening. We discuss recent progress in four areas: (1) integrated micro-devices for cell culture; (2) three-dimensional (3D) cell patterning and culture; (3) multi-layered microfluidic structures; and (4) perfusion-based micro-devices. Within each area, specific examples are provided to illustrate the rationale and characteristics of the individual techniques. In addition, an overview of ongoing efforts in this field and perspectives on future directions, opportunities, and challenges are presented.
2. Integrated micro-devices for cell culture
During the preclinical phase, in vitro cell-based assays have been widely used as the standard method for high-throughput evaluation of new drug candidates due to its simple and low-cost nature in comparison with an in vivo approach.17 However, conventional microplate or well-based cell culture platforms usually neglect the higher-order multicellular interactions.7 In order to fill the gap between conventional in vitro cell cultures and the animal models, many efforts have been made to precisely control the in vitro biological system with micro-scale fabrication techniques.31,32 Taking advantage of advances in semiconductor processing, one can apply microfabrication techniques to cell culture, which provides massively-scalable compartmentalization and demonstrates its potential in the drug screening process.33 In this section, integrated cell culture micro-devices will be introduced, including integrated micro-well arrays and micro cell culture analogs.2.1. Micro-well arrays
In order to address a major drawback due to the lack of multiple organ interactions in the conventional in vitro cell culture system, one direct and simple method is the concept of producing “wells within a well”. This consists of a cell culture plate with larger wells, within each of which are multiple smaller wells.31,34,35 The cells from multiple organs can be deposited and cultured in each individual smaller well. Then a drug-containing medium is added to the larger well, flooding all the smaller wells. Khetani et al. demonstrated such a multi-well, miniaturized system for human liver cell culture and liver toxicity testing.34 Using a soft lithography process, polydimethylsiloxane (PDMS) stencils were first fabricated. Each PDMS stencil consisted of 300 μm-thick membranes with through-holes at the bottom of each well in a 24-well format (Fig. 1(a)). Then the PDMS stencil was sealed against a polystyrene plate and collagen-I was adsorbed into the exposed polystyrene. Next, the PDMS stencil was removed and a 24-well PDMS ‘blank’ was applied. Selective hepatocyte adhesion to the collagenous domains yielded ‘micropatterned’ clusters, which were subsequently surrounded by mouse 3T3-J2 fibroblasts. The diameter of the through-holes in the PDMS stencils determined the size of the collagenous domains and thereby the ratio of the homotypic to the heterotypic interactions in these microscale cultures. With this optimized microscale architecture, the human liver cells maintained their phenotypic functions for several weeks. They also demonstrated utility through assessments of their gene expression profiles, phase I/II metabolism, canalicular transport, secretion of liver-specific products, and susceptibility to hepatotoxins. This approach addresses the need to improve in vitro testing due to the high rate of pre-launch and post-market attrition of pharmaceuticals because of liver toxicity. Ma and colleagues further improved this multi-well array with a multilayer fabrication approach for simultaneous characterization of drug metabolites and a cytotoxicity assay.36 The middle quartz substrate contained embedded separation microchannels and a perforated three-micro-well array with sol–gel bioreactors of human liver microsomes. Assembling this multilayer micro-array device enabled the study of drug metabolism relative to the organ functional units, monitoring of metabolite generation, and assessment of metabolism induced cytotoxicity in the cultured cells.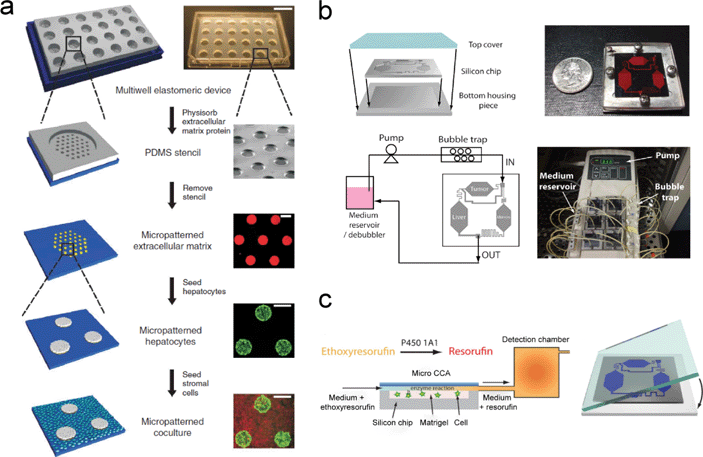 | ||
| Fig. 1 Representative integrated micro-devices for cell culture that have enhanced the drug discovery process. (a) Schematic diagrams along with photomicrographs depicting the fabrication procedures for a micro-patterned culture of liver hepatocytes in a multi-well plate format. (b) Graphical representations and pictures demonstrating the assembly and operational setup of an integrated micro cell culture device (μCCA). (c) Further study utilizing the μCCA in the fluorescent detection of resorufin by P450 1A1 enzymatic conversion. Images from ref. 34, 38 and 40 are reproduced with permissions from Nature Publishing Group, the Royal Society of Chemistry, and John Wiley and Sons, respectively. | ||
2.2. Integrated micro cell culture system
In order to mimic drug PK and PD profiles in humans, a micro cell culture analog (μCCA) approach was developed by the Shuler group.37 In their μCCA device, separated chambers were created to culture cells representing the liver, tumor, and marrow, with microchannels interconnecting the different chambers to simulate the blood flow. The microenvironment provided by the μCCA is more physically similar to an in vivo environment than a conventional monolayer culture. The artificial tumor and liver tissues were created by encapsulating colon cancer cells (HCT-116) and hepatoma cells (HepG2/C3A) in a matrigel and culturing in the respective tumor and the liver chambers in a μCCA. The marrow was built up by culturing alginate-encapsulated myeloblasts (Kasumi-1) in the marrow chamber (Fig. 1(b)). This μCCA device was used to test the cytotoxicity of anticancer drugs while reproducing multi-organ interactions. The cytotoxic effect of Tegafur, an oral prodrug of 5-fluorouracil (5-FU), was demonstrated using this μCCA. The μCCA could reproduce the metabolism of Tegafur into 5-FU in the liver and the consequent death of colon cancer cells by the 5-FU, while a conventional 96-well microtiter plate was unable to do so.38,39 By employing a similar μCCA device, the Shuler group further investigated the enzymatic activity of cytochrome P450 in liver cells with an in situ fluorescence optical detection system (Fig. 1(c)).40Integrated micro-devices allow for the compartmentalization of different tissues, which is critical in studying the systemic response of the specific drug. With either the micropatterned cell clusters or the interconnected chambers, the interaction of multiple organs can be mimicked.39,41–45 We believe that this design is highly compatible with other techniques described in this Focus article for efficient interaction among different tissue types, as well as enabling more accurate predictions of drug responses.
3. Three-dimensional cell patterning and culturing
Besides the need for compartmentalization, it is also challenging to reproduce the full functionality of tissues in conventional two dimensional (2D) cell cultures.46,47 Although these 2D models are advantageous in terms of simplicity and reproducibility, they do not closely resemble or simulate the 3D in vivo-like microenvironments or tissue scaffold.48,49 Efforts have been made to develop cell culture systems which immerse and suspend cells in a more physiologically relevant environment. These interactive environments can promote cell–cell and cell–matrix interactions.50,51 To build such systems, 3D cell patterning and culturing becomes critical to providing a biomimetic environment. Researchers typically use external physical forces, including optical, electromagnetic, and fluidics force, as well as biocompatible scaffolds such as hydrogels, to organize cells within a microstructure to better mimic the natural organ dimensions.3.1. Hydrogel as a biocompatible scaffold
Many materials have been exploited to provide the 3D scaffolds for the cells, with hydrogel as one of the common choices in tissue engineering.50,52 Hydrogels can be natural or synthetic polymers which are biocompatible and contain high proportions of water.49,53 Sung et al. employed laser ablation to fabricate a 3D structure from a plastic mold.54 PDMS and sodium alginate were then used as second and third inverse-replica molds to form a final 3D hydrogel structure (Fig. 2(a)). The advantage of this novel and efficient method was that it allowed the fabrication of a 3D structure with a high aspect ratio and curvature. The mimicking of human intestinal villi was demonstrated by culturing Caco-2 cells, a colon carcinoma cell line. After three weeks of cell culture, a monolayer of Caco-2 cells was formed on the 3D hydrogel structure using the PDMS mold to replicate villi structures. The realization of this 3D gastrointestinal (GI) tract model could be useful to study the permeability of drugs across the villi of the small intestine.54,55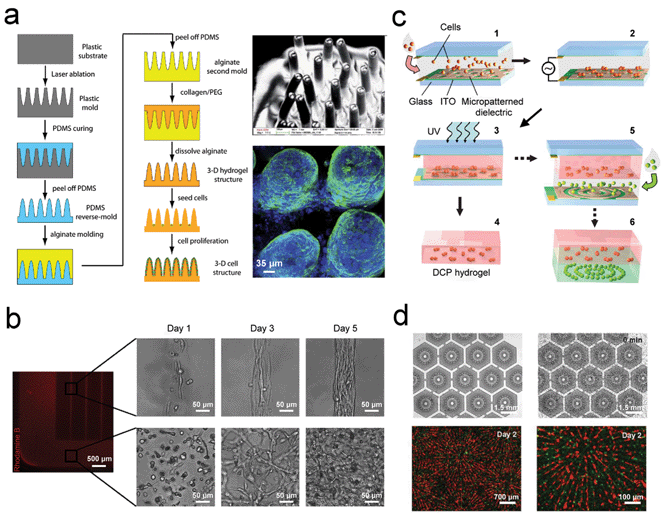 | ||
| Fig. 2 Three-dimensional (3D) cell patterning and culture for improving drug screening applications. (a) Schematic diagrams showing the fabrication process of 3D hydrogel scaffolds by laser ablation (left panel). Scanning electron microscope (SEM) and confocal images of the villi structures are also shown (right panel). (b) Fluorescent-labelled GelMA hydrogel structures illustrating the patterned and unpatterned regions, and the corresponding phase contrast images of 3T3-fibroblasts present in the patterned (upper panel) and unpatterned regions (lower panel). (c) Illustration of the fabrication process for dielectrophoretic cell patterning (DCP). (d) DCP method used to mimic the liver lobule tissue. The electrode arrays used for cell patterning are shown on the top panels while the fluorescent-labelled HepG2 cells (red) and HUVECs (green) co-culture is demonstrated in the bottom panel. Images reproduced from ref. 54 and 61 with permissions from the Royal Society of Chemistry and ref. 56 and 60 from Elsevier and Nature Publishing Group, respectively | ||
A further example of the versatility of hydrogels was revealed by Aubin et al.56 In their design, cells were encapsulated in microengineered 3D gelatin methacrylate (GelMA) hydrogels and were then allowed to self-organize into a functional tissue by their intrinsic capability (Fig. 2(b)). The significance of this study was that by controlling the 3D geometry of the hydrogel, cells were assembled into aligned tissue structures without any additional internal or external stimuli. Annabi et al. also utilized a hydrogel to coat microfluidic channels to culture cardiomyocytes.57 Though the hydrogel was not mainly used as a scaffold in this case, the cell-compatible hydrogel layer was favorable for the seeding of the primary cardiomyocytes which responded positively to the tropoelastin culture substrate. Thus, hydrogels have been shown to be useful in tissue engineering and for organs-on-chips applications.
3.2. Dielectrophoretic cell patterning
The dielectrophoretic (DEP) force is exerted on a particle when it is subject to a non-uniform electric field. This technique has shown great capabilities in the dynamic manipulation of biological objects such as cells. Researchers have presented various 3D cell patterning results by adapting this technique.58,59 Albrecht et al. demonstrated 3D cell patterning in a photopolymerizable polyethylene glycol (PEG) hydrogel using the DEP force.60 A suspension of cells in a non-cross-linked prepolymer solution was sandwiched between two conductive indium tin oxide (ITO)-coated glass slides to form the DEP cell patterning (DCP) chamber. First, one type of cell sample was injected into the chamber and patterned via the DEP force. The cells were then embedded in the hydrogel after a UV light exposure to form a DCP hydrogel layer. Then, a second type of cells was patterned and embedded using the same process to form another DCP hydrogel layer. The two DCP hydrogel layers could also be stacked together to form a 3D multi-cell pattern. In addition, the cells could be linearly clustered in the single DCP hydrogel layer in the vertical direction. For example, a 3D pattern of distinct, fluorescently-labeled fibroblast cells was formed in a cluster array in a layer above and in concentric rings in the layer below (Fig. 2(c)). This technology demonstrated large-scale arrays of various 3D cell patterns for modulating cell–cell interactions, the essential first step in developing a 3D co-culture system for drug screening.Similarly, Ho et al. developed a multi-type cell patterning technique using the DEP force to mimic the basic morphology of liver tissues and to investigate the liver function.61 A stellate-electrode array was designed as a model for cell patterning to mimic the lobules of the liver (Fig. 2(d)). Two steps were involved in forming the heterogeneous lobule-mimetic cell patterns. First, hepatic cells were captured and patterned onto the first set of DEP electrode arrays to form the radial string pattern of cells. Second, endothelial cells were loaded and aligned in between the patterned hepatic cells via applying a DEP force using the second array of DEP electrodes. The final heterogeneous integration of hepatic and endothelial cells was found to mimic the hexagonal lobules of liver tissue. The on-chip heterogeneous integration of hepatic cells (HepG2 cells, red fluorescence) and endothelial cells (HUVECs, green fluorescence) via this lobule-mimetic DEP cell patterning was shown in Fig. 2(d) in various time points and image magnifications. The enzyme activity of CYP450-1A1 was shown to achieve an 80% enhancement in liver function using this biomimetic liver tissue, as compared to a non-patterned co-culture of HepG2 and HUVECs.
3.3. Other cell patterning methods
Besides the use of hydrogels and DEP for cell patterning in a 3D structure, Bratt-Leal et al. demonstrated that magnetic particles can be incorporated within the extracellular space or in stem cells to control multi-cellular aggregates.62 In addition, Yang et al. reported cell patterning by laser diffraction-induced DEP in an optoelectronic device using organic photoconductive substrates.59 This modified version of DEP has the capability of tuning the cell pattern via the generation of specific diffraction patterns by mask designs that cannot be done by traditional physical DEP electrodes. Xie et al. described a novel technique using opto-thermally generated, acoustically-activated surface bubbles to pattern and move cells across a channel.63 Finally, PEG hydrogel has also been widely used as an inert biomaterial to trap and disperse cells due to its excellent properties such as tunable porosity, bioactivity, degradation, and gelation trigger.53Generation of tunable 3D geometries for a cell culture is crucial in replicating the tissue structure in the human body, especially for the restoration of organ-level functionality, which is required for improving drug discovery. While there is no perfect 3D cell patterning technique, we believe that choosing one of the specific techniques for the reconstruction of particular tissues or organ functions would be appropriate and favorable for future drug screening experiments.
4. Layered microfluidic structures
Apart from using hydrogels and cell patterning methods to generate a 3D cell culture, layered structures have also been developed to mimic complicated 3D structures. Such structures typically consist of a membrane with a monolayer of cells sandwiched between side channels. These structures are particularly useful in mimicking biological barriers such as the blood–brain barrier, as well as the respiratory and gastrointestinal tracts in the human body.21,55,64,65 To date, many drug candidates have to pass through these barriers to exert effects on their respective targets, rendering this design extremely useful as a drug screening platform. In addition, muscular thin films (MTFs) have been fabricated to replicate cardiac tissue. Examples of multi-layered structures and MTFs for testing of responses to various drugs will also be given in the following discussion.4.1. Membrane-based multi-layer microfluidic devices
Generally, 2D cell culture models have been widely used in biological studies. However, the lack of a tissue-like microarchitecture significantly degrades the accuracy and reliability of results, creating a need for cell cultures with a 3D structural environment. To better mimic an in vivo microstructure, many membrane-based multi-layer platforms have been reported. Representative studies will be highlighted in the following sections.Kidney tubular cells, when in vivo, are exposed to a fluidic shear stress induced by a luminal flow and provided with nutrients by interstitial flow. In order to create a 3D in vivo cell culture model for the analysis of the renal tubule cells, Jang et al. developed a multi-layer microfluidic device comprising of two PDMS microfluidic channel layers and a polyester porous membrane with pore size of 400 nm to understand the effects of fluidic shear stress on the primary inner medullary collecting duct (IMCD) cells from rats (Fig. 3(a)).66 The porous membrane, which was coated with an extracellular matrix protein, divided the device into two flow channels, a luminal flow channel and an interstitial flow channel. By reproducing the in vivo-microenvironment of kidney tubular cells, phenomena such as enhanced cell polarization, cytoskeletal rearrangement, and reinforced cell junctions for IMCD cells were observed in the presence of 1 dyn cm−2 of fluidic shear stress. To validate the capability of a porous membrane as a support scaffold, water uptake by stimulating aquaporin 2 (AQP2) trafficking and Na+ uptake by activating Na+–K+-pumps were studied and found to be changed after dosing the IMCD cells with vasopressin and aldosterone, respectively. The demonstration of AQP2 trafficking and activation of Na+–K+-pumps not only verified the regulation of water and Na+ uptake by hormonal stimulations, but more importantly, suggested that this device can be a 3D in vivo kidney-like platform for drug screening.
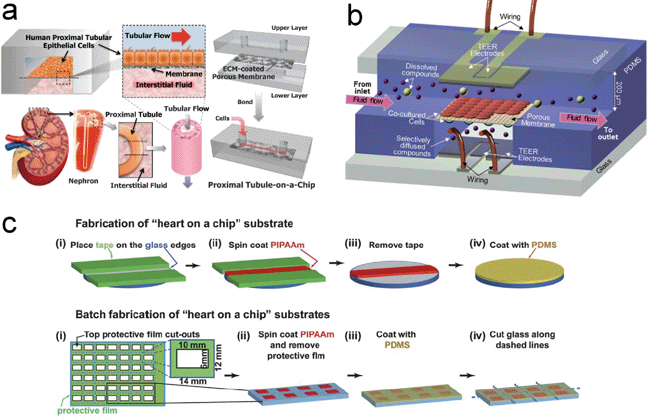 | ||
| Fig. 3 Layered structural devices using organs-on-chips technology for enhancing drug discovery. (a) Schematic design for simulating human kidney function using a proximal tubule-on-a-chip comprising of an apical channel separated from a bottom channel by an ECM-coated porous membrane. (b) Structure of the integrated microfluidic blood brain barrier (μBBB) consisting of two channels for astrocytes and endothelial cells culture with electrodes for trans-endothelial electrical resistance (TEER) measurement. (c) Graphical illustration of the fabrication process flow for muscular thin film (MTF) heart-on-a-chip. Myocytes were cultured on the film for drug dose–response and structural studies. Images reproduced from ref. 66, 69 and 71 with permissions from the Royal Society of Chemistry. | ||
Extending the same device with primary (human) renal proximal tubular epithelial cells, Jang et al. further simulated in vivo kidney-like functions and assessed the effects of fluidic shear stress on drug transport and nephrotoxicity.67 The physiological transport activities of proximal tubular epithelial cells in vivo, including albumin uptake and glucose transport, were observed in this proximal tubule-on-a-chip device under a flow condition, reflecting the successful restoration of proximal tubule cell functions in vitro. Drug toxicity testing was carried out by the injection of 100 μM of cisplatin, a known nephrotoxin, which can cause damage in proximal tubule cells when accumulated. In the presence of 100 μM cisplatin, increased cell injury and apoptosis under both static and fluidic conditions were confirmed by verifying the release of lactate dehydrogenase (LDH), by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, and by Annexin V staining. Of particular importance was the observation that the cells damaged by the accumulation of cisplatin could recover faster in this proximal tubule-on-a-chip device than in the traditional Transwell® culture system. This is similar to the observation that most patients with kidney failure due to cisplatin toxicity will eventually recover. The successful replication of specific kidney functions and the demonstration of cisplatin-induced toxicity suggest that this proximal tubule-on-a-chip could be a useful and reliable platform for assessing drug-induced toxicity in a human kidney.
Aside from the effort to reconstruct a renal tubule system, Ma et al. demonstrated a co-culture model of endothelial cells and astrocytes using an ultra-thin silicon nitride membrane to increase the direct contact between these two cells in order to better understand the properties of the blood–brain barrier (BBB).68 Compared to commercially available membranes for cell cultures, a silicon nitride membrane is advantageous in terms of membrane thickness (one order of magnitude thinner), pore size, pore arrangement, and porosity. A similar system using membrane-based microfluidic devices to mimic the BBB was also developed as a potential candidate in studying the delivery of a preclinical drug (Fig. 3(b)).69 In both studies, the astrocytes and endothelial cells were cultured on opposite sides of the membrane. Trans-endothelial electrical resistance (TEER) measurement, a typical way to assess the barrier properties, indicated that co-culturing these two types of cells on opposite sides of the membrane had a higher resistance than culturing either astrocytes or endothelial cells alone and co-culturing the mixed cell suspensions. This comparison suggested that the separation of these two cell types or, more specifically, the cell bodies, is needed to better reproduce the BBB. Moreover, glial fibrillary acidic protein (GFAP) expression was both observed and the uptake of DiI-Ac-LDL by the endothelial cells was also described in Ma's work. By demonstrating the co-culture of these two cell types in a BBB model and observing the DiI-Ac-LDL uptake of the endothelial cells, this could potentially become an attractive platform to study the properties of the BBB.
4.2. Deformable thin-film-based microfluidic devices
Different types of cells respond differently to mechanical and chemical stimuli. For example, the contraction of cardiomyocytes in vivo is a collective response when exposed to a stimulus. This response varies with different numbers of cardiomyocytes. Although a single cardiomyocyte and isotropic cardiac tissues have been extensively studied for their electrophysiological properties, they are inappropriate candidates for pharmacological study, since they fail to reproduce more physiologically relevant conditions. Recently, a 2D bio-hybrid construct consisting of an anisotropic muscle tissue cultured on a deformable thin film, termed a muscular thin film (MTF), has been developed to observe the contractility of muscle tissue due to various film geometries and tissue architectures. This technique enables a fast and simple characterization of the contractility by simply correlating the curvature of thin film to the generated stresses.70Grosberg et al. developed a MTF-based “heart-on-a-chip” and demonstrated the successful measurement of contractility of neonatal rat ventricular myocytes.71 This heart-on-a-chip was assembled from several separated thin films and was batch fabricated. This enabled the observation and data collection from multiple tissues in the same experiment (Fig. 3(c)). By correlating the curvature of each thin film to the stress generated, the contractility of rat myocytes was characterized. In addition, two different tissue architectures, an isotropic and an anisotropic cell alignment, were compared in terms of contractility and cytoskeleton organization. A chronotropic effect in response to the dosage of epinephrine was observed. Also, different frequencies of contraction were obtained with different concentrations of epinephrine. These experimental results with rat myocytes have proven the usefulness of this device for in vitro contractility characterization and drug-screening of cardiomyocytes.
Using a similar concept, Agarwal et al. semi-automated the operation of a MTF-based heart-on-a-chip, as well as increased the drug screening throughput, to study the in vitro positive inotropic effect of neonatal rat ventricular myocytes in response to different dosages of isoproterenol.72 The semi-automatic operation was enabled by integrating a MTF chip, a temperature control unit, a transparent window for observing thin film deformation, fluidic components, and an electrode into a single microdevice. There were over 35 separated thin films within the single MTF chip, which enabled a higher throughput for drug screening. Experiments were carried out by flushing the MTF chip with ten-fold increments of isoproterenol dosages. The results showed that as the isoproterenol dosage increased, the percentage of twitch stress increased from the baseline. The isoproterenol dose results proved that the modified heart-on-a-chip can become a platform for in vitro pharmacological studies with a high throughput.
While only two types of multi-layer microfluidic devices are highlighted in this article, many researchers have also made great advances toward developing various multi-layer microfluidic devices for mimicking specific organs.48,65,73–75 These multi-layer microfluidic devices have shown their unique advantages to better mimic or reproduce the complicated, yet well-defined 3D structure of specific organs. In short, the development of multi-layer microfluidic devices enables the reconstruction of 3D in vivo-like structures, and more importantly, organ-specific functions and drug-induced responses could be replicated in vitro.
5. Perfusion-based micro-devices
In the interstitial spaces within the human body, cells are constantly bathed in a sea of tissue fluid. Tissue fluid provides an avenue for the cells to access nutrients, soluble factors, as well as the removal of metabolic waste.76 In this regard, the goal of a perfusion-based cell culture technique is to replicate the physiological flow of tissue fluid inside the human body. Instead of flowing the liquid directly to the cells in a channel, two side channels, a nutrient source, and a metabolic waste sink are set up which allow the perfusion of the nutrients and drugs to the cells. Adverse shear stresses on the cells can be minimized in this way since mechanical stimulation can affect the physiology of the cell. These perfusion-based micro-devices facilitate the interpretation of the results by lowering the interfering effects of mechanical stimulation.77 Therefore, researchers employ the above strategy to achieve a more in vivo-like cell microenvironment which would also assist in monitoring drug delivery.5.1. Micro-pillar perfusion cell culture
Many studies have been performed to realize perfusion-based cell cultures and one of the most common approaches is the incorporation of arrays of micro-pillars inside a microfluidic channel.76,78,79 These micro-pillars help retain and enrich the confined cells while at the same time allowing the liquid to flow through the spaces between the micro-pillars. With this approach, 3D cell clusters can be formed and a perfusion cell culture can be achieved. Once the flow in the source and sink channels is steady, a constant chemical gradient can be established which mimics the normal situation in the interstitial spaces.Toh et al. developed a novel 3D microfluidic channel-based cell culture system (3D-μFCCS) and demonstrated the successful restoration of the 3D phenotypes of carcinoma cell lines, primary hepatocytes, and primary progenitor cells (Fig. 4(a)).76 In their design, 3D cell–matrix interactions were achieved by a polyelectrolyte laminar flow and a complex coacervation process so as to maintain the 3D cell structure after cell seeding. In order to test the viability and functionality of the cultured cells, various immunostainings were performed after a continuous perfusion of medium for 72 hours. Differentiated staining of calcein AM and propidium iodide (PI) showed high cell viability inside these devices while the stainings for F-actin and E-cadherin illustrated the maintenance of 3D in vivo-like cyto-architecture and cell–cell interaction in the 3D cell cluster, respectively. Functional analysis of the primary hepatocytes and bone marrow mesenchymal stem cells further demonstrated the feasibility of the 3D-μFCCS for culturing more fragile cell types. Since tumor cells behave differently in 3D, and primary hepatocytes are involved in drug metabolism and toxicity, the results for the carcinoma cell and for the primary hepatocytes in their studies will be of particular interest for use in drug discovery.
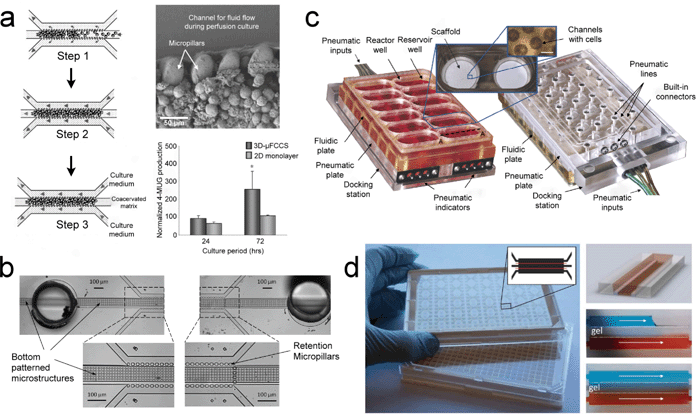 | ||
| Fig. 4 Perfusion-type cell culture devices for the advancement of pharmacological studies. (a) Micro-pillar device for enrichment of cells and subsequent perfusion (left panel). SEM image of the cells in the channel and the functional assessment of UDP-glucuronyltransferase (UGT) activity in primary rat hepatocytes is illustrated (right panel). (b) Analogous setup of the micro-pillar device with additional bottom patterned microstructures to support the growth of primary human hepatocytes without the need for biological or synthetic matrices or coagulants. (c) Detailed setup of the perfused multi-well array for long term culture of primary rat hepatocytes that could be used for liver toxicity and metabolism studies. (d) A 384-well format microfluidic titer plate for possible adoption for high-content screening in an organ-on-a-chip (left panel). The concentration gradient generated by the perfusion of food dye is demonstrated (right panel). Images reproduced from ref. 76, 79, 80 and 82 with permissions from the Royal Society of Chemistry. | ||
Using a comparable approach, a modified device of micro-pillars for a perfusion cell culture of primary human hepatocytes was developed. Goral et al. introduced a bottom patterned microstructure to virtually support a suspension of primary hepatocytes (Fig. 4(b)).79 One of the main differences between the previous study and this one was that a biological or synthetic extracellular matrix was not required in this design. This eliminated the need for an extra experimental step. Structural and functional aspects of primary human hepatocytes, including the membrane polarity, hepatocyte transport function, as well as the bile canalicular network, were extensively studied after seven days of culture. Immunofluorescence of hepatocyte cell surface proteins, connexion 32, and multi-drug resistance-associated protein 2 (MRP2) clearly revealed the formation of gap junctions and the restoration of cell polarity in this perfusion cell culture device. MRP2 function was verified using 5-(and-6)-carboxy-2′,7′-dichloro-fluorescein (CDF), a metabolized product of 5-(and-6)-carboxy-2′,7′-dichloro-fluorescein diacetate (CDFDA), into bile canalicular structures. The MRP2 transporter is responsible for the efflux of drug metabolites, which is related to the Phase III measurement of drug metabolism in the liver. The demonstration of MRP2 function suggests that this device could potentially be a strong platform for evaluating the toxicity of new drugs in the liver.
5.2. Microplate format for perfusion cell culture
A conventional in vitro cell culture platform utilizes a microplate format for ease of use and high-throughput screening. During the development of cell culture platforms, the capabilities of perfusion have also been extended to be compatible with a microplate. There are many advantages of realizing a perfusion microplate, which include the high compatibility with existing automated imaging technologies, more physiological relevance, and elimination of the need for intermittent media exchanges.80–82 Many studies have been carried out in perfusion microplates and some examples will be highlighted in the following sections.In a study of 3D liver tissue engineering, Domansky et al. developed an integrated bioreactor array in a perfused multi-well plate format (Fig. 4(c)).80 Each compartment consisted of a reactor well and a reservoir well, which were fluidically separated from other compartments. Individual reactor wells were coated with ECM to support the formation of tissue while a pneumatic valve was fabricated between the reactor and the reservoir to provide the control needed for the circulation of cell culture medium. Before carrying out cell culture experiments, a model of oxygen consumption and transport was developed with the help of numerical simulations and a ruthenium-based oxygen-sensing layer. Also, a range of acceptable operating parameters was established for different flow rates. Finally, co-cultures of rat hepatocytes and liver sinusoidal endothelial cells (LSEC) were used to demonstrate the performance of this system. Immunostaining of positive functional markers for both cell types showed the feasibility of this design for long-term cell culture, which could help advance drug discovery in microplates.
Another example was the modification of a 96-well microplate for continuous fluidic perfusion without an external pumping system.81 The functional unit of this design contained three wells (i.e., a source well, a cell culture well, and a waste well) that were interconnected by a cellulose membrane. The fluid movement was driven by a higher hydrostatic pressure in the source well (a higher fluid volume) relative to the other wells through the wicking of the membrane. By controlling the volume of the fluids and the dimensions and the pore size of the membrane, different flow rates were achieved, which followed Darcy's law. Besides the demonstration of the viability of C3A cells, two interesting examples related to the soluble factor and drug metabolism were also studied. LADMAC cells were first cultured in the source well while EOC 20 cells were cultured in the cell culture well. Results showed that EOC 20 cells, which required the conditioned medium of the LADMAC cells, were equally viable to the EOC 20 cells that were cultured in a conventional 96-well microplate with daily exchanges of conditioned medium. Further results involved the metabolism of Tegafur, a chemotherapeutic prodrug, by primary human hepatocytes in the source well. The metabolite, 5-FU, was used to kill the HCT 116 colon cancer cells in the cell culture well. The results of this simple, yet useful perfusion microplate design have showed great promise for carrying out multi-organ, metabolism-dependent, toxicity assays.
In addition to the previously described perfusion microplate platforms, Trietsch et al. introduced an elegant phase-guided-stratified 3D cell culture system in a microfluidic titer plate (Fig. 4(d)).82 This platform has the aforementioned advantages of a perfusion microplate. Briefly, the microfluidic channels were fabricated using a dry film resist (DFR)-based process with a phase-guided printed mask. The channels were patterned on laminated glass and then capped with a glass substrate in a hot press. Finally, 384-well plates were adapted to accommodate the microfluidic channels. Similar to other perfusion cell cultures in microfluidic channels, this design sustained a transverse chemical gradient when the two side channels were continuously perfused. A 3D cell culture was also possible with the help of ECM. Cell co-culture and migration studies were observed using this platform. To demonstrate its potential for drug screening, the influence of rifampicin on HepG2 cells was studied. Rifampicin is well-known to cause hepatotoxicity in the liver and its presence would exert an adverse effect on the HepG2 cells. As expected, the results clearly indicated the dose-dependent death of HepG2 cells which, together with the multiple channels on the plate, should further promote the application of this platform for high-throughput drug screening.
Progress has already been made in the use of perfusion cell cultures for applications such as cell co-cultures, restoration of the 3D phenotypical functions and drug metabolism studies. Whereas we discussed only two main types of perfusion cell cultures in this article, we recognized that many other efforts have also been made in this field.83–87 In summary, the promotion of 3D in vivo-like cell environments in long-term perfusion cell cultures should bring benefits and play a substantial role in the use of organs-on-chips for drug discovery.
6. Conclusions and perspectives
Extensive advances have been made in recent years in the development of in vitro micro-physiological systems through the application of innovative bioengineering approaches. We envision that continued innovations and progress in this field will enable the development of novel drug development and screening platforms that are superior to existing in vitro cell culture and animal model systems in mimicking in vivo organ functionality. In the following sections, we present the opportunities (and challenges) for focused innovation to fully realize the potential of organs-on-chips technologies.6.1. Exploring novel techniques
Rational design of the devices is one of the determining factors in the success of organs-on-chips. van der Meer et al. have provided an in-depth discussion of trade-offs between the ease of use and the complexity in a device design. This will ultimately influence the robustness and throughput of the device.8 To facilitate the drug discovery process, we believe that the future direction should focus on exploring new techniques that can be integrated into devices to better support the growth and differentiation of human induced pluripotent stem cells (iPSCs). Recent breakthroughs in our understanding of iPSCs will have an enormous impact on life science studies because of their ability to differentiate into three embryonic germ layers without the controversial use of embryos.88–90 While iPSCs present the unprecedented opportunity to accelerate progress into improving organs-on-chips, such systems still require standardized differentiation protocols and highly trained personnel. Furthermore, the spatiotemporal environment required to successfully culture iPSCs has to be closely regulated for proper and reproducible differentiation.91,92 To overcome this obstacle, techniques that precisely control the spatiotemporal aspect of the biochemical environment in microfluidics have been developed (Fig. 5(a) and (b)).93–97 Some of these devices have demonstrated the successful differentiation of iPSCs in microfluidic channels.98–100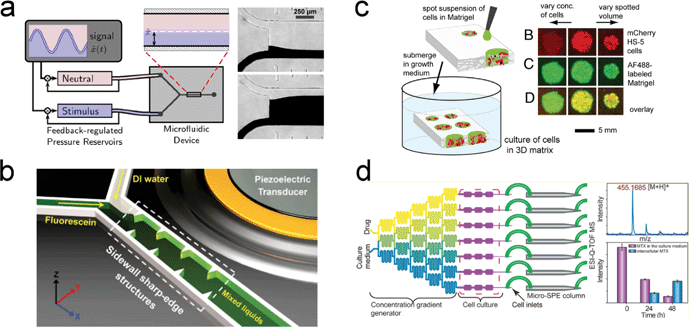 | ||
| Fig. 5 State-of-the-art technologies that can potentially be integrated into current organs-on-chips devices for improving drug discovery results. (a) Diffusion-based and (b) acoustofluidic devices for precise spatiotemporal control of biochemical environments. (c) Paper-based microfluidic structure as an alternative material for future organs-on-chips. (d) Integrated microfluidic device coupled with an electrospray ionization quadrupole time-of-flight mass spectrometer (ESI-Q-TOF MS) to achieve multi-parametric measurements. Images reproduced from ref. 95 with permission from the Public Library of Science; ref. 97 with permissions from the Royal Society of Chemistry; ref. 119 with permissions from the National Academy of Sciences, USA; and ref. 123 with permission from the American Chemical Society. | ||
Another consideration for using iPSCs in organs-on-chips is the 3D organization required for proper differentiation. As discussed in Section 3, many techniques have emerged to pattern cells in a 3D manner. Though iPSCs can be self-assembled into embryoid bodies, it is still desirable to pattern them in advance, for proper mimicking of specific organ functions. Since iPSCs are relatively fragile and susceptible to damage, techniques for their controlled patterning should be relatively non-invasive. In this regard, recent advances in surface acoustic wave (SAW), DCP, and magnetic-based platforms could be applied to iPSCs and produce improved organs-on-chips for drug screening.59,62,101–105 The search for new techniques should carry on until the final goal of realizing organs-on-chips for preclinical trials is achieved.
6.2. Exploring alternative materials
Other than the significances of mimicking the in vivo microenvironment, materials employed in organs-on-chips devices also play an equally important role of reconstructing the organ functions to resemble those in the human body. Currently, PDMS has been widely employed in microfluidic cell culture devices, not just because of its ease of fabrication and its relatively short fabrication time, but also its biocompatibility, gas permeability, and excellent optical transparency. These properties enable cells to be cultured for a long period of time and easily imaged to study their morphology and functions. For example, various organs-on-chips that replicate the functions of the liver, artery, breast and heart were demonstrated using PDMS.26,71,106,107 However, PDMS has its shortcomings. One of these disadvantages is that it absorbs biomolecules non-specifically, thus compromising the accuracy of cytotoxicity studies.108,109 As a result, a flurry of research was initiated to study PDMS surface modifications with particular emphasis on microfluidic applications. These efforts have been aimed at creating longer-lasting surface modifications on PDMS to increase its wettability and to inhibit or reduce its non-specific adsorption of hydrophobic species onto the surfaces with desired functional groups. These functional groups are useful for microfluidic applications such as molecular separation, biomolecular detection via immunoassays, cell culture, and emulsion formation.108,110,111 Therefore, with proper surface modifications, PDMS may have the potential to be the material of choice for organs-on-chips for drug discovery and screening applications.Based on traditional microfabrication methods, silicon substrates are another popular material for organs-on-chips devices. From the early demonstration of liver-function-on-a chip to multi-organ chips to assess metabolism-dependent cytotoxicity of anti-cancer drugs, silicon also plays a critical role in the development of organs-on-chips applications.38,112,113 As the use of organs-on-chips for drug discovery and screening applications grows, the demand has also increased for methods of fabricating prototype devices rapidly with biocompatible materials and novel functional attributes. Porous structures are one attractive feature that can be incorporated into organs-on-chips. As discussed previously, by sandwiching a porous membrane between pieces of PDMS, lung, kidney and BBB functionality was demonstrated on a chip.21,66,69 Although the fabrication, kinetics, and morphology of porous thermoplastic structures have been investigated in the past, there are limited activities ongoing that have produced integrated devices where patterned, interconnected, microporous structures perform both gas diffusion and fluid perfusion functions.114,115 Also, patterned, interconnected, microporous structures enable devices to be fabricated in 2D instead of 3D formats, which can potentially improve device designs and fabrication flexibility.116,117
With the rapid expansion of microfluidic devices for point-of-care and biological applications, other materials (such as polymer and paper) are gaining popularity for use in microfluidic devices due to their relatively low fabrication time and cost. Polystyrene is a ubiquitous material used for tissue culture plasticware and has been the most commonly used thermoplastic in clinical laboratories. It has been employed for decades and used to validate research into cell behavior and functions.118 Berthier et al. provided a critical evaluation of the strengths and limitations of PDMS and polystyrene in relation to the advancement and future impact on microfluidic cell-based studies.109 From their evaluation, they have provided guidelines and suggestions between microfabrication technologies and biological applications. Critical evaluations like the one by Berthier et al. can be invaluable in helping researchers to choose the best material for their organs-on-chips devices. On the other hand, paper-based microfluidic devices are steadily gaining ground for cell culture applications (Fig. 5(c)).119–121 Although paper-based microfluidic devices are very simple to fabricate and could be very inexpensive, it still takes time to evolve to be effectively applied to organs-on-chips applications. With the need to add new on-chip functions and reduce device cost, the material requirement for organs-on-chips applications should present exciting opportunities for new research areas and open doors for new researchers from other fields to contribute innovations to the practical realization of organs-on-chips.
6.3. Multi-parametric approach
Most often it is vital to acquire multiple parameters for a single study. The results from different parameters could complement and support one another so that a more meaningful and accurate conclusion can be drawn. Similarly, biological systems are extremely intricate, highly variable but interconnected. Thus, the validation of any observation or phenomena should be performed with a multi-parametric approach. This notion can also be applied to the organs-on-chips model, especially when the efficacy of a drug has to be accurately predicted. Whereas the previously described state-of-the-art organs-on-chips have already demonstrated their capability to reconstruct human micro-tissues with respect to the function of vital organs, and have shown promise in advancing disease modeling and drug discovery, their output or readout is mostly based on microscopic images such as immunofluorescence or cell viability tests. Certainly, microscopic imaging has numerous advantages, including high molecular specificity; the substantial information that can be collected about cell behaviors and live-cell imaging makes it a popular and powerful tool in life science studies. In the study of drug cytotoxicity and metabolism in these micro-devices, however, a readout that heavily depends on imaging is inadequate to give a comprehensive understanding of the effects of a drug. The study by Khetani et al. discussed earlier in section 2.1. was an example where the toxicity was quantified with multiple assays using measurement modalities other than imaging, yet it still required an independent apparatus for detection, monitoring, and analysis.34 Therefore, it will be beneficial to incorporate other measurement systems to complement the results, such as the integration of mass spectrometric detection on-chip.122–124 One example was an integrated microfluidic device consisting of a cell culture chamber, a cytotoxicity assay chamber, and micro solid-phase extraction columns that were coupled to an electrospray ionization quadrupole time-of-flight mass spectrometer (ESI-Q-TOF MS) (Fig. 5(d)). Both acetaminophen and methotrexate were tested in this system, which characterized the drug metabolite and cytotoxicity concurrently.Besides the detection and analysis of cell metabolism, microfluidic devices coupled to a mass spectrometer can also be implemented for proteomic studies.125 Though the information collected from proteomic studies is not as explicit as that obtained from metabolomics in drug discovery and toxicity, these measurements can be used in further understanding how the signaling pathway of the cell is related to the drug metabolism. With the success of the integration of microfluidic devices to the mass spectrometer described above, we envision that organs-on-chips will achieve breakthroughs in obtaining the extremely important PK–PD values for drugs.
6.4. Towards personalized drug screening
In the previous sections, we have discussed the rationale behind the techniques that have been employed to develop organs-on-chips. From these examples, we believe that certain techniques should be more efficient than others in reconstructing particular organ structures for drug screening. In addition, we believe that scaling effects should also be taken into consideration when designing organs-on-chips.126,127 It is expected that as techniques for generating and handling iPSCs mature, the prospect of using iPSCs for drug discovery significantly improve. Specifically, researchers are focusing on the capability to collect adult cells from individuals and then convert them into iPSCs. From that point, it should be possible to create personalized organs-on-chips, leading to rapid individualized drug screening.128 The benefits of personalized drug screening will be phenomenal. Not only can it yield a system that is specifically tailored to an individual, but it can also more accurately evaluate drug efficacy as well as acute and chronic drug toxicity. With the development of these organs-on-chips technologies, we aspire towards a major breakthrough in personalized drug screening.Acknowledgements
We thank Dr Peng Li and Ms Lauren M. Burt for their kind help and discussion. This research was supported by the National Institutes of Health (NIH) Director's New Innovator Award (1DP2OD007209-01), the National Science Foundation and the Penn State Center for Nanoscale Science (MRSEC).References
- T. Pawson, Nature, 1995, 373, 573–580 CrossRef CAS PubMed.
- N. M. Kumar and N. B. Gilula, Cell, 1996, 84, 381–388 CrossRef CAS.
- F. Guo, J. B. French, P. Li, H. Zhao, C. Y. Chan, J. R. Fick, S. J. Benkovic and T. J. Huang, Lab Chip, 2013, 13, 3152–3162 RSC.
- E. Primiceri, M. S. Chiriaco, R. Rinaldi and G. Maruccio, Lab Chip, 2013, 13, 3789–3802 RSC.
- E. W. K. Young, Integr. Biol., 2013, 5, 1096–1109 RSC.
- R. Khamsi, Nature, 2005, 435, 12–13 CrossRef CAS PubMed.
- D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- A. D. van der Meer and A. van den Berg, Integr. Biol., 2012, 4, 461–470 RSC.
- H. Wang, K. Liu, K. J. Chen, Y. Lu, S. Wang, W. Y. Lin, F. Guo, K. Kamei, Y. C. Chen, M. Ohashi, M. Wang, M. A. Garcia, X. Z. Zhao, C. K. Shen and H. R. Tseng, ACS Nano, 2010, 4, 6235–6243 CrossRef CAS PubMed.
- S. Yang, F. Guo, B. Kiraly, X. Mao, M. Lu, K. W. Leong and T. J. Huang, Lab Chip, 2012, 12, 2097–2102 RSC.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- M. Baker, Nature, 2011, 471, 661–665 CrossRef CAS PubMed.
- Y. Zhao, D. Chen, H. Yue, J. B. French, J. Rufo, S. J. Benkovic and T. J. Huang, Lab Chip, 2013, 13, 2183–2198 RSC.
- P. Neuzil, S. Giselbrecht, K. Lange, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef PubMed.
- Y. H. Zhao, Z. S. Stratton, F. Guo, M. I. Lapsley, C. Y. Chan, S. C. S. Lin and T. J. Huang, Lab Chip, 2013, 13, 17–24 RSC.
- X. L. Mao and T. J. Huang, Lab Chip, 2012, 12, 1412–1416 RSC.
- M. B. Esch, T. L. King and M. L. Shuler, Annu. Rev. Biomed. Eng., 2011, 13, 55–72 CrossRef CAS PubMed.
- J. H. Sung and M. L. Shuler, Bioprocess Biosyst. Eng., 2010, 33, 5–19 CrossRef CAS PubMed.
- P. M. van Midwoud, E. Verpoorte and G. M. M. Groothuis, Integr. Biol., 2011, 3, 509–521 RSC.
- E. M. Materne, A. G. Tonevitsky and U. Marx, Lab Chip, 2013, 13, 3481–3495 RSC.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- D. Huh, H. Fujioka, Y. C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS PubMed.
- Y. H. Hsu, M. L. Moya, P. Abiri, C. C. W. Hughes, S. C. George and A. P. Lee, Lab Chip, 2013, 13, 81–89 RSC.
- P. J. Lee, P. J. Hung and L. P. Lee, Biotechnol. Bioeng., 2007, 97, 1340–1346 CrossRef CAS PubMed.
- N. J. Douville, P. Zamankhan, Y. C. Tung, R. Li, B. L. Vaughan, C. F. Tai, J. White, P. J. Christensen, J. B. Grotberg and S. Takayama, Lab Chip, 2011, 11, 609–619 RSC.
- Y. Nakao, H. Kimura, Y. Sakai and T. Fujii, Biomicrofluidics, 2011, 5, 22212 CrossRef PubMed.
- Y. Huang, J. C. Williams and S. M. Johnson, Lab Chip, 2012, 12, 2103–2117 RSC.
- A. van de Stolpe and J. den Toonder, Lab Chip, 2013, 13, 3449–3470 RSC.
- J. H. Sung, M. B. Esch, J. M. Prot, C. J. Long, A. Smith, J. J. Hickman and M. L. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC.
- C. P. Adams and V. V. Brantner, Health Affairs, 2006, 25, 420–428 CrossRef PubMed.
- A. P. Li, C. Bode and Y. Sakai, Chem.-Biol. Interact., 2004, 150, 129–136 CrossRef CAS PubMed.
- M. R. Dusseiller, D. Schlaepfer, M. Koch, R. Kroschewski and M. Textor, Biomaterials, 2005, 26, 5917–5925 CrossRef CAS PubMed.
- C. Zhang, Z. Q. Zhao, N. A. A. Rahim, D. van Noort and H. Yu, Lab Chip, 2009, 9, 3185–3192 RSC.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS PubMed.
- E. Novik, T. J. Maguire, P. Y. Chao, K. C. Cheng and M. L. Yarmush, Biochem. Pharmacol., 2010, 79, 1036–1044 CrossRef CAS PubMed.
- B. Ma, G. H. Zhang, J. H. Qin and B. C. Lin, Lab Chip, 2009, 9, 232–238 RSC.
- K. Viravaidya and M. L. Shuler, Biotechnol. Prog., 2004, 20, 590–597 CrossRef CAS PubMed.
- J. H. Sung and M. L. Shuler, Lab Chip, 2009, 9, 1385–1394 RSC.
- J. H. Sung, C. Kam and M. L. Shuler, Lab Chip, 2010, 10, 446–455 RSC.
- J. H. Sung, J. R. Choi, D. Kim and M. L. Shuler, Biotechnol. Bioeng., 2009, 104, 516–525 CrossRef CAS PubMed.
- G. J. Mahler, M. B. Esch, R. P. Glahn and M. L. Shuler, Biotechnol. Bioeng., 2009, 104, 193–205 CrossRef CAS PubMed.
- P. Chao, T. Maguire, E. Novik, K. C. Cheng and M. L. Yarmush, Biochem. Pharmacol., 2009, 78, 625–632 CrossRef CAS PubMed.
- D. A. Tatosian and M. L. Shuler, Biotechnol. Bioeng., 2009, 103, 187–198 CrossRef CAS PubMed.
- K. Schimek, M. Busek, S. Brincker, B. Groth, S. Hoffmann, R. Lauster, G. Lindner, A. Lorenz, U. Menzel, F. Sonntag, H. Walles, U. Marx and R. Horland, Lab Chip, 2013, 13, 3588–3598 RSC.
- I. Wagner, E. M. Materne, S. Brincker, U. Sussbier, C. Fradrich, M. Busek, F. Sonntag, D. A. Sakharov, E. V. Trushkin, A. G. Tonevitsky, R. Lauster and U. Marx, Lab Chip, 2013, 13, 3538–3547 RSC.
- J. W. Haycock, Methods Mol. Biol., 2011, 695, 1–15 CAS.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- C. M. Puleo, W. M. Ambrose, T. Takezawa, J. Elisseeff and T. H. Wang, Lab Chip, 2009, 9, 3221–3227 RSC.
- A. P. Golden and J. Tien, Lab Chip, 2007, 7, 720–725 RSC.
- C. J. Bettinger, J. T. Borenstein and R. S. Langer, Mater. Res. Soc. Symp. Proc., 2005, 845, 25–30 CAS.
- N. W. Choi, M. Cabodi, B. Held, J. P. Gleghorn, L. J. Bonassar and A. D. Stroock, Nat. Mater., 2007, 6, 908–915 CrossRef CAS PubMed.
- G. Eng, B. W. Lee, H. Parsa, C. D. Chin, J. Schneider, G. Linkov, S. K. Sia and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4551–4556 CrossRef CAS PubMed.
- A. Sala, P. Hanseler, A. Ranga, M. P. Lutolf, J. Voros, M. Ehrbar and F. E. Weber, Integr. Biol., 2011, 3, 1102–1111 RSC.
- J. H. Sung, J. J. Yu, D. Luo, M. L. Shuler and J. C. March, Lab Chip, 2011, 11, 389–392 RSC.
- J. J. Yu, S. M. Peng, D. Luo and J. C. March, Biotechnol. Bioeng., 2012, 109, 2173–2178 CrossRef CAS PubMed.
- H. Aubin, J. W. Nichol, C. B. Hutson, H. Bae, A. L. Sieminski, D. M. Cropek, P. Akhyari and A. Khademhosseini, Biomaterials, 2010, 31, 6941–6951 CrossRef CAS PubMed.
- N. Annabi, S. Selimovic, J. P. Acevedo Cox, J. Ribas, M. Afshar Bakooshli, D. Heintze, A. S. Weiss, D. Cropek and A. Khademhosseini, Lab Chip, 2013, 13, 3569–3577 RSC.
- K. Park, H. J. Suk, D. Akin and R. Bashir, Lab Chip, 2009, 9, 2224–2229 RSC.
- S. M. Yang, S. Y. Tseng, H. P. Chen, L. Hsu and C. H. Liu, Lab Chip, 2013, 13, 3893–3902 RSC.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS PubMed.
- C. T. Ho, R. Z. Lin, R. J. Chen, C. K. Chin, S. E. Gong, H. Y. Chang, H. L. Peng, L. Hsu, T. R. Yew, S. F. Chang and C. H. Liu, Lab Chip, 2013, 13, 3578–3587 RSC.
- A. M. Bratt-Leal, K. L. Kepple, R. L. Carpenedo, M. T. Cooke and T. C. McDevitt, Integr. Biol., 2011, 3, 1224–1232 RSC.
- Y. Xie, C. Zhao, Y. Zhao, S. Li, J. Rufo, S. Yang, F. Guo and T. J. Huang, Lab Chip, 2013, 13, 1772–1779 RSC.
- R. Cecchelli, V. Berezowski, S. Lundquist, M. Culot, M. Renftel, M. P. Dehouck and L. Fenart, Nat. Rev. Drug Discovery, 2007, 6, 650–661 CrossRef CAS PubMed.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- K. J. Jang and K. Y. Suh, Lab Chip, 2010, 10, 36–42 RSC.
- K. J. Jang, A. P. Mehr, G. A. Hamilton, L. A. McPartlin, S. Chung, K. Y. Suh and D. E. Ingber, Integr. Biol., 2013, 5, 1119–1129 RSC.
- S. H. Ma, L. A. Lepak, R. J. Hussain, W. Shain and M. L. Shuler, Lab Chip, 2005, 5, 74–85 RSC.
- R. Booth and H. Kim, Lab Chip, 2012, 12, 1784–1792 RSC.
- K. Balachandran, P. W. Alford, J. Wylie-Sears, J. A. Goss, A. Grosberg, J. Bischoff, E. Aikawa, R. A. Levine and K. K. Parker, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19943–19948 CrossRef CAS PubMed.
- A. Grosberg, P. W. Alford, M. L. McCain and K. K. Parker, Lab Chip, 2011, 11, 4165–4173 RSC.
- A. Agarwal, J. A. Goss, A. Cho, M. L. McCain and K. K. Parker, Lab Chip, 2013, 13, 3599–3608 RSC.
- A. K. H. Achyuta, A. J. Conway, R. B. Crouse, E. C. Bannister, R. N. Lee, C. P. Katnik, A. A. Behensky, J. Cuevas and S. S. Sundaram, Lab Chip, 2013, 13, 542–553 RSC.
- E. S. Lippmann, A. Al-Ahmad, S. P. Palecek and E. V. Shusta, Fluids Barriers CNS, 2013, 10, 2 CrossRef PubMed.
- B. Prabhakarpandian, M. C. Shen, J. B. Nichols, I. R. Mills, M. Sidoryk-Wegrzynowicz, M. Aschner and K. Pant, Lab Chip, 2013, 13, 1093–1101 RSC.
- Y. C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. van Noort, D. W. Hutmacher and H. R. Yu, Lab Chip, 2007, 7, 302–309 RSC.
- H. Kimura, T. Yamamoto, H. Sakai, Y. Sakai and T. Fujii, Lab Chip, 2008, 8, 741–746 RSC.
- Y. C. Toh, T. C. Lim, D. Tai, G. F. Xiao, D. van Noort and H. R. Yu, Lab Chip, 2009, 9, 2026–2035 RSC.
- V. N. Goral, Y. C. Hsieh, O. N. Petzold, J. S. Clark, P. K. Yuen and R. A. Faris, Lab Chip, 2010, 10, 3380–3386 RSC.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. M. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- V. N. Goral, C. F. Zhou, F. Lai and P. K. Yuen, Lab Chip, 2013, 13, 1039–1043 RSC.
- S. J. Trietsch, G. D. Israels, J. Joore, T. Hankemeier and P. Vulto, Lab Chip, 2013, 13, 3548–3554 RSC.
- L. Ma, J. Barker, C. C. Zhou, W. Li, J. Zhang, B. Y. Lin, G. Foltz, J. Kublbeck and P. Honkakoski, Biomaterials, 2012, 33, 4353–4361 CrossRef CAS PubMed.
- M. Y. Rotenberg, E. Ruvinov, A. Armoza and S. Cohen, Lab Chip, 2012, 12, 2696–2703 RSC.
- J. H. Yeon, H. R. Ryu, M. Chung, Q. P. Hu and N. L. Jeon, Lab Chip, 2012, 12, 2815–2822 RSC.
- S. Giulitti, E. Magrofuoco, L. Prevedello and N. Elvassore, Lab Chip, 2013, 13, 4430–4441 RSC.
- Y. Wang, Y. C. Toh, Q. S. Li, B. Nugraha, B. X. Zheng, T. B. Lu, Y. Gao, M. L. N. Mary and H. R. Yu, Integr. Biol., 2013, 5, 390–401 RSC.
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS PubMed.
- J. B. Kim, V. Sebastiano, G. Wu, M. J. Arauzo-Bravo, P. Sasse, L. Gentile, K. Ko, D. Ruau, M. Ehrich, D. van den Boom, J. Meyer, K. Hubner, C. Bernemann, C. Ortmeier, M. Zenke, B. K. Fleischmann, H. Zaehres and H. R. Scholer, Cell, 2009, 136, 411–419 CrossRef CAS PubMed.
- T. Takebe, K. Sekine, M. Enomura, H. Koike, M. Kimura, T. Ogaeri, R. R. Zhang, Y. Ueno, Y. W. Zheng, N. Koike, S. Aoyama, Y. Adachi and H. Taniguchi, Nature, 2013, 499, 481–484 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- S. K. W. Dertinger, D. T. Chiu, N. L. Jeon and G. M. Whitesides, Anal. Chem., 2001, 73, 1240–1246 CrossRef CAS.
- D. Irimia, D. A. Geba and M. Toner, Anal. Chem., 2006, 78, 3472–3477 CrossRef CAS PubMed.
- B. Kuczenski, W. C. Ruder, W. C. Messner and P. R. Leduc, PLoS One, 2009, 4, e4847 Search PubMed.
- D. Ahmed, C. Y. Chan, S. C. S. Lin, H. S. Muddana, N. Nama, S. J. Benkovic and T. J. Huang, Lab Chip, 2013, 13, 328–331 RSC.
- P. H. Huang, Y. L. Xie, D. Ahmed, J. Rufo, N. Nama, Y. C. Chen, C. Y. Chan and T. J. Huang, Lab Chip, 2013, 13, 3847–3852 RSC.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5, 401–406 RSC.
- K. Gupta, D. H. Kim, D. Ellison, C. Smith, A. Kundu, J. Tuan, K. Y. Suh and A. Levchenko, Lab Chip, 2010, 10, 2019–2031 RSC.
- P. Agarwal, S. Zhao, P. Bielecki, W. Rao, J. Choi, Y. Zhao, J. Yu, W. Zhang and X. He, Lab Chip, 2013, 13, 4525–4533 RSC.
- X. Y. Ding, J. J. Shi, S. C. S. Lin, S. Yazdi, B. Kiraly and T. J. Huang, Lab Chip, 2012, 12, 2491–2497 RSC.
- X. Y. Ding, S. C. S. Lin, B. Kiraly, H. J. Yue, S. X. Li, I. K. Chiang, J. J. Shi, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11105–11109 CrossRef CAS PubMed.
- X. Ding, P. Li, S. C. Lin, Z. S. Stratton, N. Nama, F. Guo, D. Slotcavage, X. Mao, J. Shi, F. Costanzo and T. J. Huang, Lab Chip, 2013, 13, 3626–3649 RSC.
- J. J. Shi, D. Ahmed, X. Mao, S. C. S. Lin, A. Lawit and T. J. Huang, Lab Chip, 2009, 9, 2890–2895 RSC.
- Y. C. Chen, X. Y. Ding, S. C. S. Lin, S. K. Yang, P. H. Huang, N. Nama, Y. H. Zhao, A. A. Nawaz, F. Guo, W. Wang, Y. Y. Gu, T. E. Mallouk and T. J. Huang, ACS Nano, 2013, 7, 3306–3314 CrossRef CAS PubMed.
- A. Gunther, S. Yasotharan, A. Vagaon, C. Lochovsky, S. Pinto, J. L. Yang, C. Lau, J. Voigtlaender-Bolz and S. S. Bolz, Lab Chip, 2010, 10, 2341–2349 RSC.
- M. M. G. Grafton, L. Wang, P. A. Vidi, J. Leary and S. A. Lelievre, Integr. Biol., 2011, 3, 451–459 RSC.
- I. Wong and C. M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS PubMed.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- J. W. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS PubMed.
- J. W. Zhou, D. A. Khodakov, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2012, 33, 89–104 CrossRef CAS PubMed.
- M. J. Powers, K. Domansky, M. R. Kaazempur-Mofrad, A. Kalezi, A. Capitano, A. Upadhyaya, P. Kurzawski, K. E. Wack, D. B. Stolz, R. Kamm and L. G. Griffith, Biotechnol. Bioeng., 2002, 78, 257–269 CrossRef CAS PubMed.
- A. Sin, K. C. Chin, M. F. Jamil, Y. Kostov, G. Rao and M. L. Shuler, Biotechnol. Prog., 2004, 20, 338–345 CrossRef CAS PubMed.
- C. H. M. Jacques, H. B. Hopfenberg and V. Stannett, J. Appl. Polym. Sci., 1974, 18, 223–233 CrossRef CAS.
- N. H. Sung, R. E. Gahan and R. E. Haven, Polym. Eng. Sci., 1983, 23, 328–336 CAS.
- P. K. Yuen, H. Su, V. N. Goral and K. A. Fink, Lab Chip, 2011, 11, 1541–1544 RSC.
- P. K. Yuen and M. E. DeRosa, Lab Chip, 2011, 11, 3249–3255 RSC.
- K. Perez-Toralla, J. Champ, M. R. Mohamadi, O. Braun, L. Malaquin, J.-L. Viovy and S. Descroix, Lab Chip, 2013, 13, 4409–4418 RSC.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Y. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef CAS PubMed.
- R. Derda, S. K. Tang, A. Laromaine, B. Mosadegh, E. Hong, M. Mwangi, A. Mammoto, D. E. Ingber and G. M. Whitesides, PLoS One, 2011, 6, e18940 CAS.
- G. G. Lewis, M. J. DiTucci, M. S. Baker and S. T. Phillips, Lab Chip, 2012, 12, 2630–2633 RSC.
- Q. S. Chen, J. Wu, Y. D. Zhang and J. M. Lin, Anal. Chem., 2012, 84, 1695–1701 CrossRef CAS PubMed.
- D. Gao, H. F. Li, N. J. Wang and J. M. Lin, Anal. Chem., 2012, 84, 9230–9237 CAS.
- S. F. Mao, D. Gao, W. Liu, H. B. Wei and J. M. Lin, Lab Chip, 2012, 12, 219–226 RSC.
- T. C. Chao and N. Hansmeier, Proteomics, 2013, 13, 467–479 CrossRef CAS PubMed.
- C. G. Anene-Nzelu, K. Y. Peh, A. Fraiszudeen, Y. H. Kuan, S. H. Ng, Y. C. Toh, H. L. Leo and H. Yu, Lab Chip, 2013, 13, 4124–4133 RSC.
- J. P. Wikswo, E. L. Curtis, Z. E. Eagleton, B. C. Evans, A. Kole, L. H. Hofmeister and W. J. Matloff, Lab Chip, 2013, 13, 3496–3511 RSC.
- A. Williamson, S. Singh, U. Fernekorn and A. Schober, Lab Chip, 2013, 13, 3471–3480 RSC.
| This journal is © The Royal Society of Chemistry 2013 |