Lab-on-a-chip technologies for single-molecule studies
Yanhui
Zhao
a,
Danqi
Chen
b,
Hongjun
Yue
b,
Jarrod B.
French
b,
Joseph
Rufo
a,
Stephen J.
Benkovic
*b and
Tony Jun
Huang
*a
aDepartment of Engineering Science and Mechanics, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: junhuang@psu.edu
bDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: sjb1@psu.edu
First published on 14th May 2013
Abstract
Recent developments on various lab-on-a-chip techniques allow miniaturized and integrated devices to perform on-chip single-molecule studies. Fluidic-based platforms that utilize unique microscale fluidic behavior are capable of conducting single-molecule experiments with high sensitivities and throughputs, while biomolecular systems can be studied on-chip using techniques such as DNA curtains, magnetic tweezers, and solid-state nanopores. The advances of these on-chip single-molecule techniques lead to next-generation lab-on-a-chip devices, such as DNA transistors, and single-molecule real-time (SMRT) technology for rapid and low-cost whole genome DNA sequencing. In this Focus article, we will discuss some recent successes in the development of lab-on-a-chip techniques for single-molecule studies and expound our thoughts on the near future of on-chip single-molecule studies.
1. Introduction
The traditional averaged ensemble approach of measuring molecular properties is experiencing a fundamental transition. This change is driven by various single-molecule techniques and miniaturized lab-on-a-chip devices. Single-molecule approaches spatially and temporally isolate individual molecules, such as nucleic acids, lipids, proteins, or dye molecules. In contrast to conventional biological studies that often focus on the averaged behavior of a large number of molecules, single-molecule experiments are capable of distinguishing individual molecular properties from the overwhelming background of the bulk population. In addition, these techniques provide a method for reporting on variations in kinetics and conformational dynamics, existence of rare and transient intermediates and the heterogeneous behavior within a population of molecules of the same type. By providing a level of detail inaccessible by ensemble methods, single-molecule techniques now grace the landscape of a broad range of disciplines in the biosciences and biotechnology.1 Lab-on-a-chip methods, on the other hand, have traditionally been aimed at developing miniaturized and integrated devices capable of one or more bench-top functions.2–10 These on-chip devices, such as those that employ microfluidics, have been well-recognized for their exquisite performance in rapid, sensitive and high-throughput analyses with a low demand for analytes and low cost.11–13 Recently, sophisticated manufacturing of on-chip devices by state-of-the-art micro/nanofabrication techniques has generated a myriad of new scaled down analysis platforms. These chip-like tools are finely tailored for automated, precise manipulation of objects, and the fast detection and analysis of bio-analytes with high sensitivity and high-throughput.14,15Recent years have witnessed the blossoming of a new class of lab-on-a-chip tools specialized in single-molecule analysis, indicative of the emerging integration of the single-molecule and lab-on-a-chip fields. The benefits of this natural progression towards convergent development are clear: the full potential of lab-on-a-chip techniques can only be obtained by further scaling down the detection limit to the single-molecule level, while the challenges inherent in the design and setup of complex instrumentation required by most existing single-molecule techniques can be minimized by adopting on-chip formats. A variety of on-chip devices are now capable of manipulating, trapping, counting and analyzing individual molecules as single-molecule techniques undergo this shift from the conventional serial (one molecule at a time) approaches (e.g., optical tweezers16,17 and atomic force microscopy18,19) to massively-parallel and miniaturized on-chip formats (e.g., microfluidic chips,20,21 nanopores,22–24 and zero-mode waveguide arrays25–27).
In this Focus article, we review the recent progress that has been made in advancing single-molecule studies based on lab-on-a-chip techniques, as well as other innovative techniques with the potential to be integrated with existing lab-on-a-chip platforms for comprehensive single-molecule studies. We will focus our discussion on three aspects of single-molecule on-chip studies, including (1) on-chip single-molecule handling and processing; (2) on-chip single-molecule manipulation; and (3) on-chip high-throughput single-molecule sensing and analysis. In comparison to conventional means, lab-on-a-chip single-molecule techniques provide many advantages. These include facile and inexpensive device fabrication, straightforward experimental setups, minimal bio-reagent requirements, powerful device functionalities, and safe experimental conditions. In addition to a discussion of these aspects of lab-on-a-chip single-molecule technologies along with their recent successes in biological/biochemical studies, especially those involving nucleic acids, we will share our thoughts on the future of this emerging field.
2. On-chip single-molecule handling and processing
A number of microfluidic-based devices allow versatile, precise liquid manipulations and automated liquid compartmentalization. These devices provide the fundamentals for carrying out single-molecule processing and handling on-chip. The implementation of microfluidics for single-molecule studies provides unique advantages, including (1) simplifying and automating the process for single-molecule biochemical assays through precise liquid handling; (2) minimizing expensive biochemical reagent consumption; and (3) maximizing the outputs for biological analysis by automated and high-throughput configurations. Although the application of microfluidic techniques in single-molecule studies is still at its infancy, exciting breakthroughs foreshadow a promising future for the integration of microfluidic devices with single-molecule studies. Our discussion in this section will focus on two cases of utilizing fluidic controls in microfluidic chips to conduct single-molecule sampling and analysis.2.1. On-chip polymerase chain reaction (PCR) based on droplet microfluidics
Droplet microfluidics is an emerging technology that enables individual control of droplets that serve as isolated vessels for the compartmentalization of chemical and biological reagents.28–35 The droplets are usually picoliter to microliter in volume, and can be easily dispensed, transported, merged, and split. Droplets can be used to spatially separate individual biomolecules, allowing for the isolation of single biomolecules in each droplet in order to conduct and observe individual biochemical reactions. Recent methods exploiting the high sensitivity offered by droplet-localized compartmentalization have been developed.36,37 The most important advantages of implementing droplet microfluidics for single-molecule studies are rapid sampling, parallel processing and handling, and high-throughput for the target studies. One example of the application of such a technology is the droplet-based, on-chip emulsion PCR.38,39Water-in-oil emulsion PCR (ePCR) is a useful method to achieve single amplicon trapping and massive parallel sequencing analysis.40–42 To address the technical challenges associated with conventional ePCR (e.g., large droplet size variance and low amplification efficiency due to the utilization of single-primer immobilized microbeads), C. J. Yang et al. developed an agarose droplet microfluidic ePCR method.20 In their design, illustrated in Fig. 1(A), an agarose solution of statistically diluted template DNA is injected by the micro-fabricated nozzle to form aqueous, droplet PCR reactors in the oil phase on the chip. Droplets are uniform in size (3 ± 0.3 nL) and are formed at high speeds of 500 droplets per second. Single-template amplification in the droplet PCR reactors is demonstrated by fluorescent imaging of the stained agarose droplets. This microfluidic ePCR method sidesteps the limitations of conventional ePCR. Because the agarose used in this method has an ultralow gelling temperature of 16 °C, a simple sol-gel switch of aqueous agarose droplets to gelated beads below the temperature helps to facilitate downstream processing of the ePCR products confined in the gelated beads. Also, the liquid phase agarose acts as a crosslinking matrix for the single DNA template of an ePCR droplet reactor, negating the need of microbeads for maintaining monoclonality in each droplet reaction (Fig. 1(B)). In addition, free from the steric hindrance of the microbead surface, this on-chip ePCR has a high amplification efficiency of 95%.
 | ||
| Fig. 1 (A) An agarose emulsion droplet microfluidic method for single-molecule emulsion polymerase chain reactions (PCR). Microfluidic channels are used to isolate single copies of template DNA through droplet formation. Droplets are generated such that each contains either a single copy of DNA template, or none. PCR products are later collected from each droplet separately from the outlet. (B) Sol to gel switch allows ePCR in aqueous droplet and confinement of the PCR product in gelated agarose beads. (C) The optical image and schematic design of the optofluidic-based high-throughput smFRET analyses. Various pumps and valves are used to control the injection and mixing of reagents with different concentrations for sequential and automatic single-molecule analysis. The scale bar is 5 mm. (D) A scheme of the RNA polymerase transcript assay. (E) A heat map of transcription activities at various glutamate and RNA polymerase concentrations obtained directly through programmable control of the optofluidic-based analysis platform. Images are reproduced from ref. 20 and 21. | ||
2.2. Automated single-molecule analysis based on microfluidic large-scale integration (mLSI)
The concept of microfluidic large-scale integration (mLSI), proposed by the Quake group, refers to the development of microfluidic chips with hundreds to thousands of integrated micromechanical valves and other microfluidic components.43–45 The integration of various components, such as valves and pumps, allows precise liquid quantification and mixing on-chip. In addition, hundreds of assays can be performed with multiple reagents in an automated manner. This type of high-level integrated microfluidic chip is expected to be a potential candidate to replace today's conventional biological automation paradigm and has been widely employed in applications such as the protein-interaction network generation, genetic analysis, amino acid analysis, high-throughput screening, bioreactors, and chemical synthesis. The implementation of mLSI allows high-throughput single-molecule analysis.The high level of sophistication in fluid control in mLSI has led to the development of microfluidic-based on-chip single-molecule Forster Resonance Energy Transfer (smFRET) analysis capable of automated data acquisition and sequential measurements. smFRET between donor and acceptor dyes at distances of around 5 nm serve as a molecular ruler capable of reporting biomolecular topology/conformation changes and is usually monitored using sophisticated microscopy methods, such as total internal reflection fluorescence (TIRF) microcopy. This experimental setup, however, demands surface-immobilization of biomolecules on the chamber surface, and hence is limited to one biochemical condition in one reaction chamber, preventing sampling across a wide range of conditions.
A promising alternative to overcome this limitation is the optofluidic-based smFRET method developed by the Weiss and Majumdar groups, as shown in Fig. 1(C).21 The device employs digitally controlled valves and pumps to manipulate fluid on the microfluidic mixing ring of the flow channels. In this fashion, reagents can be injected, mixed, and finally flushed away after data is acquired in a sequential, automated manner. Precise liquid handling of sample volumes on the scale of tens of picoliters and the confined observation volume by confocal viewing provide an optofluidic capability of sequential measurements and single-molecule sensitivity. Fig. 1(D) illustrates an RNA polymerase (RNAP) transcription activity assay. In this experiment, RNAP transcribes a DNA template and produces complementary RNA transcripts that hybridize the poly(T) DNA probe whose ends are flanked with donor and acceptor dyes, respectively. Because the efficiency of FRET between the dyes is distance-sensitive (high for relaxed single-stranded (ss) DNA, low for stiffer double-stranded (ds)DNA or DNA/RNA hybrids (D/R)), smFRET readouts allow quantification of poly(T) probe conformation populations (ss vs. D/R), linking hybridization levels with RNAP activities. Notably, sequential measurements by this device permit sampling over a wide range of reaction conditions. The RNAP activity here, for example, can be sampled for each of the 36 combinations of 6 different concentrations of RNAP and glutamate (Fig. 1(E)). This type of high-throughput analysis provides valuable insight into variations of RNAP activity in response to varying biochemical environments. This elegant study strongly demonstrates the profound impact of microfluidic devices on the advance of single-molecule techniques in an automated manner.
A further example of the utility of mLSI for future single-molecule studies was demonstrated recently using an in vitro microfluidic approach to generate protein-interaction networks.46 The platform for protein interaction network generation relies on mechanical trapping of molecular interactions (MITOMI) developed by the same group. This technique allowed parallel and sensitive monitoring and recording of the protein–protein interactions of 43 Streptococcus pneumnoiae proteins through 14,792 on-chip experiments. The results revealed 157 novel interactions, indicating the presence of a large number of undescribed physical interactions between related proteins within biochemical pathways. A similar design concept and platform can be easily transferred to study single protein–protein interactions and their dynamics.
3. On-chip single-molecule manipulation
A number of on-chip devices spatially modulate individual molecules with nanometer or sub-nanometer sensitivities. With the attributes of single-molecule positioning, trapping, and displacement and force measurements, this class of mechanic manipulation tools has enormously facilitated biological studies on the structural dynamics, conformational/topological changes associated with biomolecules and their enzymatic activities. In this section, we will briefly introduce the major on-chip single-molecule manipulation techniques along with their recent applications to biological problems: (1) The trapping of single molecules for biomolecular conformational and photophysical dynamic studies by an Anti-Brownian electrokinetic trap (ABEL) with minimal interference by Brownian motion; (2) single-molecule force spectroscopies, including a dual optical trap and magnetic tweezers in studying DNA hybridization and activities of DNA associating/modifying proteins; (3) and flow-induced stretching and precise alignment of individual DNA strands by the “DNA-curtain” method for visualizing DNA–protein interactions.3.1. Single-molecule trapping in suspensions
A challenge encountered during single-molecule studies in solution is Brownian motions, the random movement of single molecules caused by bombardment from other small molecules in the solution. The Brownian motions make it difficult to track single molecules in solutions, and cause additional challenges for single-molecule-based molecular dynamics experiments that take place over prolonged time periods. One solution to this problem involves the use of a modified molecule that contains a binding site that enables it to be anchored to the substrate. In this case, a steady flow will help to reduce the Brownian motion; however, it does so at the cost of a morphology change of the molecules of interest and disturbances caused by the flow. While this approach is commonly and widely used in various studies in molecular biology and biochemistry, it is not the preferred method for cases when the dynamics of single molecules in suspension are of particular interest.The Anti-Brownian Electrokinetic (ABEL) trap, developed by the Moerner lab, utilizes electrokinetic forces to balance the Brownian motion of the object inside the trapping region.47–50 A schematic representation of the ABEL is shown in Fig. 2(A), where the trapping region is at the center of a four-channel microfluidic chamber that is connected to external electric sources. The tiny Brownian displacements of the trapping object are first measured precisely, and then a feedback signal corresponding to the displacement will be generated and sent to the electrodes. The electric field applied on the conducted solutions will generate electrokinetic forces based on the feedback signal to compensate for the displacement cause by Brownian motion. The feedback and response happens in a short time of ∼150 μs, and this will ensure that the Brownian motion will be confined within a region as small as a single fluorophore, where single-molecule studies can be conducted. The same group has applied this technology to study several interesting and important biological problems. One example is shown in Fig. 2(B), where a mammalian group II chaperonin TRiC/CCT was trapped and studied using the ABEL trap.51 Each trapped TRiC exhibited a stepwise, decreasing fluorescence intensity profile, caused by the photobleaching of Cy3 dyes. Analysis of the traces allows the measurement of the number of nucleotides bound to each chaperonin. Moerner et al. also conducted a study on the conformational- and photo-dynamics of a single fluorescent protein, allophycocyanin. With support from the fluctuations observed in the spontaneous emission lifetime, this study revealed a new biophysical phenomena of light-induced conformational changes for this single protein.52 In an additional study, a principle components analysis on shape fluctuations of single DNA using an ABEL trap indicated nonlinear hydrodynamics that will be complementary to the existing model of polymer dynamics.53
 | ||
| Fig. 2 (A) A schematic representation of the ABEL trap, where a single molecule is trapped in the center of a four-channel microfluidic chamber. Each channel is connected to an electrode to provide a bias electric field generating electric kinetic forces to balance the Brownian motion. A well trapped single fluorescent molecule can provide consistent fluorescent signals to be collected using a confocal fluorescent microscope. (B). The lower spike fluorescent signals are from Cy3–APD interactions. The generations of those signals indicate the interactions between a trapped TRiC using ABEL and the nucleotides entering the trap. Each fluorescent signal spike is then photobleached with a stepwise bleaching event shown in the upper part of the figure. (C) The schematic setup and fluorescent images of dual optical traps with ultra-high precision and resolution. The scale bar is 1 μm. (D) To avoid excessive heat generated during trapping, which could harm the single molecules, trapping lasers and excitation light are sequentially turned “ON” and “OFF” in a programmable manner that is controlled digitally. This trapping method also offers high-resolution detection and a better fluorescence lifetime. Images are reproduced from ref. 51, 52 and 55. | ||
Optical traps are able to offer ultra-high precision and resolution for particle/bead manipulations. It is often argued, however, that their use is limited for biological samples due to the potential damage caused by the excessive heat generated during the trap. Most experiments involving fluorescent dyes also encounter the problem of photobleaching as a result of exposure to trapping lasers.54 These challenges have been successfully circumvented in a recent experimental design, which involves the integration of optical traps with a confocal fluorescent microscope.55 In this setup a single molecule, such as DNA, can be studied in its extended form in suspension by tethering the molecule between two large beads trapped using two optical traps. The optical traps are switched on and off in a sequential manner in a rather short time interval (∼5 μs). The excitation source from the confocal fluorescent microscope is switched on for a short time to collect the fluorescent signals only after the trapping sequence has completed. The whole process is controlled using a field-programmable gate array (FPGA), which provides high-speed synchronous control of the beam manipulation and data acquisition. A schematic representation of the process is shown in Fig. 2(C). The utility of this dual optical trap system was demonstrated by measuring the hybridization of single-stranded DNA oligonucleotides to a complementary sequence. The fluorescent signals of individual fluorophores were measured enabling the investigation of the extension of the DNA strand at sub-nanometer resolution.
3.2. A fluidic-based “DNA Curtain” for visualizing DNA–protein interactions
Nucleic acid associating/processing proteins play a key role in the function and regulation of nucleic acids and have received a great deal of attention as potential drug targets. Intriguingly, a number of these proteins are found to interact with their nucleic acid substrates in a sequence-specific manner: DNA transcription factors regulate gene expressions by binding to their cognate DNA motifs, known as transcription promoters. DNA repair machineries target certain types of DNA damage and restriction endonucleases locate and cut (nick) at specific DNA sequences. Because the size of the genome, even in simple life forms (e.g., E. coli), is in the millions of base pairs range, efficient “target-searching” on DNA is expected to facilitate the functions of these proteins. Classic biochemical methods have provided detailed mechanistic and kinetic insights into various aspects of sequence-dependent DNA–protein interactions leading to models portraying the sliding, hopping and colliding behaviors that are possibly involved in the “target-searching”. Validations of these models, however, are best achieved by visualizing the interactions in real-time at single-molecule resolution.The fluidic-based “DNA curtain” developed by the Greene group has recently emerged as an elegant on-chip single-molecule tool for the investigation of DNA–protein interactions.56–60 As shown in Fig. 3(A), when driven by flow, DNA (lambda DNA, 48 kbps, ∼16 μm) molecules that are tethered to a fluidic lipid bilayer on the surface drift downstream until they reach a thin layer of metal as a diffusion barrier, and thus align with each other forming a “DNA curtain”.58 In another approach, unattached, free ends of aligned DNA molecules can also be fixed to metal anchors deposited by lithography. The DNA, thus, remains stretched as a “DNA rack” even in the absence of flow. With appropriate fluorescent labeling of the DNA, using intercalating dyes (e.g., sytox orange and Yo-Pro-1), and of the enzyme, using quantum dots, this precise alignment greatly improves the output of a single experiment by allowing simultaneous observation of individual DNA–protein interacting events on hundreds of DNA substrates in a single field of view. This fluidic-chip setup has been successfully utilized to elucidate the searching modes of a DNA repair complex to a DNA damage site (Fig. 3(B)), and the disruption of a transcription complex by a DNA translocase at the single-molecule level.60
 | ||
| Fig. 3 The fluidic-based “DNA curtain”. (A) The schematic design of DNA alignment along a diffusion barrier by hydrodynamic flow. (B) Immobilization of the free ends of the aligned DNA molecules on anchors forms a DNA rack, where DNA remained stretched in the absence of flow. This setup has been used to elucidate the searching modes (e.g., 3D collision or 1D sliding) of the DNA repair complex (purple) for a DNA damage site (red star). (C) A schematic representation of single-molecule replication by the E. coli replisome on a circular DNA template aligned along a barrier etched by a diamond-tip scribe. The growing DNA curtain is stained by Yo-Pro-1, a DNA intercalating fluorescent dye, as the templates are extended. Images are reproduced from ref. 58, 60 and 61. | ||
An additional example of the versatility of the DNA curtain technique was demonstrated by Georgescu et al. in single-molecule studies on DNA replication. In this case, the DNA diffusion barrier was used to align circular DNA replication substrates and monitor the real-time extension of the substrates by the E. coli replisome, the DNA replication machinery. The growing DNA curtain employed in this study is shown in Fig. 3(C).61 This single-molecule approach to DNA replication allowed for real-time measurements of replication speed, processivity of the individual replisomes, and notably, the variations in behavior within a population of replisomes under investigation that are obscured by conventional ensemble-averaging techniques.
3.3. Magnetic tweezers in single-molecule DNA replication and transcription studies
DNA associating/processing reactions are often accompanied by topological changes of DNA substrates (e.g., DNA twists, bends, supercoils, DNA replication loops, transcription bubbles and highly packed DNA in the form of nucleosomes) that alter their end-to-end distance. Conventional techniques have limited ability to characterize these structurally significant, but dimensionally subtle changes due to their lack of sensitivity. Therefore, comprehensive kinetic and mechanistic studies of these reactions and the associated DNA topological changes demand nanometer/sub-nanometer scale manipulation techniques, one example of which is magnetic tweezers.Magnetic tweezers typically employ a pair of magnets to apply forces to a single biomolecule, most often DNA, which is tethered between a magnetic bead and a glass slide surface (Fig. 4(A)).62,63 In a typical experimental setup, magnetic force from the Z-direction positions the bead under constant tension, while protein-induced, end-to-end displacement of the DNA molecule can be measured as a function of time by tracking the 3D position of the bead using high-speed video. A spatial resolution of ∼1 nm (equivalent to just ∼3 base pairs) and a temporal resolution in milliseconds are achievable with this method.
 | ||
| Fig. 4 Magnetic tweezers used for single-molecule studies of DNA replication and transcription. (A) A schematic illustration of the experimental setup of magnetic tweezers. (B) Experimental traces of the activities of T4 gp41 helicase (green) and primosome (red) in the absence of rNTPs on the hairpin DNA substrate. (C) A schematic presentation of two possible models for the T4 primase and helicase interactions and the expected traces for each model (left), and two experimental traces of primosome activities on the hairpin DNA substrate in the presence of rNTPs (right). The disassembly mechanism (top): gp61 during primer synthesis dissembles with gp41, forming the primase–primer complex on DNA that “blocks” rezipping of the hairpin after being unwound by gp41. The priming loop mechanism (bottom): gp41 and gp61 remain associated and the DNA, when continuously unwound by gp41 during primer synthesis, forms a loop that gives a “jump” extension after it collapses. The features of “block” and “jump” in the experimental traces provide evidence for the operations of disassembly and priming loop mechanisms, respectively (right). (D) Unwinding of one turn of the promoter by RNA polymerase with a positively (top) or negatively (bottom) superhelical substrate gives rise to a drop or an increase of the position of magnetic beads in the Z-direction. (E) The experimental setup of a hybrid system of magnetic tweezers and epifluorescence microscopy. Rotation of the bead forms the supercoiled DNA, which is subsequently pulled sideways into the focal plane of an aperture objective for fluorescence imaging. Images are reproduced from ref. 63, 64, 69 and 70. | ||
Magnetic tweezers have been widely recognized as an ideal micromechanical manipulation platform for high-throughput single-molecule studies of DNA processing reactions. A look through the recent literature is sufficient to appreciate the broad scope of the application of this technology for the investigation of biochemical reactions involving nucleic acids. Manosas et al. have recently used magnetic tweezers to study the dynamics of the T4 replisome, a model system for the study of DNA replication machinery, and its subassemblies on hairpin DNA substrates.64 The experimental design for this system is shown in Fig. 4(A), and an example of a real-time data trace is given in Fig. 4(B). This trace (green) shows an increase in signal resulting from DNA extension by the T4 helicase (gp41) unwinding, followed by a decrease for rezipping of the hairpin as gp41 travels to the duplex end and then translocates the single-stranded region. The real-time observation of individual reactions by gp41 provides detailed insights into its step size, unwinding rate under various tensions, and the unwinding mechanism of this helicase. Following this innovative use of magnetic tweezers, the same group also studied the coupling mechanisms between the unwinding of the helicase gp41 and the primer synthesis activity of the primase gp61, the two components of the primosome subassembly of the T4 replisome.64 The trace corresponding to the primosome activity is almost indistinguishable from that of gp41 alone in the absence of ribonucleotides (rNTPs) with no primer synthesis (Fig. 4(B), red). In the presence of rNTPs, there are two new features observed, a burst increase in rezipping followed by blocking, and a sudden increase in extension, or jump, as shown in Fig. 4(C), reflecting the operation of two coupling mechanisms in connecting helicase and primase activities during the T4 replication. Recently, more challenging questions related to DNA replication, such as the coupling behaviors between subassemblies of the T4 replisome, and the restart of a stalled replication fork-by-fork regression in the T4 system, have been elegantly studied using magnetic tweezers.65,66
In addition to mechanical stretching and the measurement of real-time displacement, another beneficial function of magnetic tweezers is the ability to apply torque to the bead-tethered DNA by simply rotating the bead. With this capability, magnetic tweezers have been used to probe the effects of torsional constraints on the dynamics of DNA–protein reactions.67 Recently, an intriguing use of magnetic tweezers, in which the magnetic bead acts as both torque transducer and displacement probe, was developed by the Ebright and Strick groups for single-molecule studies on transcription initiation by RNA polymerase.68 In their experimental setup (Fig. 4(D)), a dsDNA molecule with a single transcription promoter is twisted under torsion to either right-handed (negative) or left-handed (positive) plectoneme, a supercoiled state of DNA. Using careful calibration, displacement of the bead from the Z-direction can readily report on the number of the superhelical turns of the DNA substrate at a value of 56 nm per turn. During transcription initiation on these DNA substrates, E. coli RNA polymerase, upon binding to the promoter, would unwind one turn of the promoter DNA. The unwinding would give rise to a compensating loss of one turn with the negatively supercoiled DNA or one turn increase with the positively supercoiled substrate. In either case, the local geometry change of the unwinding, catalyzed by RNA polymerase, is translated to an increase or a drop of the bead position in tens of nanometers in the Z-axis. This clever experimental design has proven successful in elucidating the kinetics of promoter unwinding and clearance during transcription initiation, and the “scrunching” mechanism underlying transcription initiation by RNA polymerase.69
Despite the strengths and versatilities of magnetic tweezers, end-to-end displacement measurements only provide indirect information as to enzymatic activity on a tethered molecule, and fail to capture the details regarding the precise topological changes and location of the enzymatic reaction. A straightforward means to overcome this limitation is the integration of magnetic tweezers technology with imaging tools, such as epifluorescence microscopy. One representative example of this type of hybrid system was developed by the Dekker group. As shown in Fig. 4(E), supercoiled DNA, formed by rotating a pair of magnets, is pulled in a perpendicular direction by an additional magnet. The 21 Kb-long supercoiled DNA is pulled into the focal plane of an aperture objective and can then be fluorescently imaged. This example demonstrates the advantages that the marriage of micromanipulation and an imaging platform can provide: the position, movement, and hence dynamics of an individual plectoneme is not only directly visualized, but also analyzed under well-controlled mechanical stretching forces.70
Magnetic tweezers have not only rapidly gained popularity, but have also become a fertile ground for technology innovations towards a highly sensitive (sub-nanometer),71 parallelized (up to ∼300 beads simultaneously)72, adaptable (integration with a microfluidic chip)73 on-chip instrument with multi-dimensional outputs (both position and fluorescence as readouts).74
4. On-chip high-throughput single-molecule sensing and analysis
The adaptation of bench-top instruments to lab-on-a-chip formats has proven successful in the development of miniaturized tools with scaled-down dimensions and impressive detection sensitivities to single-molecule levels. One example of these “shrunken” instruments is a “nanopore”, a microchip format of a “Coulter counter” used for high-throughput single-molecule detection. Moreover, miniaturized chips fabricated with arrays of nanometer-sized reaction vessels precisely constructed by novel material processing/fabrication techniques, as in the cases of zero-mode-waveguides, have been developed with superior signal-to-noise ratio for detection and high-throughputs. Also, nano-fabricated solid-state nanopore arrays further improve the throughputs of nanopore-sensing analysis by using massively parallel measurements. In this section, we will focus on these two novel on-chip single-molecule platforms and their applications in high-throughput biomolecule sensing and biochemical analysis.4.1. Zero-mode waveguide-assisted real-time translation dynamics of ribosomes
Typically, single-molecule studies of biological problems use light spectroscopy with observation volumes in the attoliter (10−18 L) range (e.g., TIRF), and are carried out in sub-nanomolar concentrations, above which the background noise from unbound dye-labeled species becomes overwhelming and single-molecule resolution cannot be achieved as illustrated in the left panel of Fig. 5(A). Zero-mode waveguides (ZMWs), nanofabricated wells with a diameter 50–200 nm in metal film deposited on glass/quartz coverslips, are an emerging tool to allow reactions to increase sample concentrations to the physiological relevant micromolar range.25–27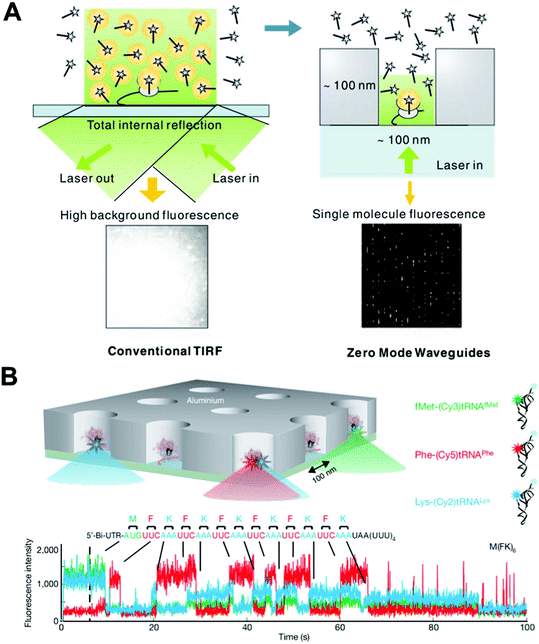 | ||
| Fig. 5 (A) A comparison of the illumination of a TIRF experiment and one employing ZMWs. The ZMW overcomes the high fluorescent background that is problematic in TIRF by decreasing the illumination volume by ∼3 orders of magnitude. (B) Real-time single-molecule ribosome translation reaction on a single mRNA copy in ZMW arrays. Time-resolved fluorescent spectra of fluorescently labeled aminoacylated-tRNAs provide information on the temporal order and identity of the aminoacylated-tRNAs, and thus the cognate codons of the mRNA template. Images are reproduced from ref. 75 and 77. | ||
The sub-wavelength scale architecture of ZMWs shown in the right panel of Fig. 5(A), when coupled with the incident excitation laser, confines the resulting evanescent excitation field to obtain a zeptoliter (10−21 L) observation volume. By scaling down the volume by ∼3 orders of magnitude, ZMW-based studies can thus be carried out in the micromolar range, breaking through the concentration barrier encountered by traditional techniques. This distinctive feature has been exploited in real-time, single-molecule DNA sequencing. In the ZMW sequencing setup, a single polymerase deposited in each ZWM well incorporates complementary dNTPs on the single DNA template. ZMWs allow the reaction to occur at up to 10 μM dNTPs, a concentration that is optimal for polymerase activities in terms of incorporation rate and fidelity. By labeling the 4 dNTPs with spectrally distinct fluorophores, the stepwise incorporation by a single polymerase generates fluorescent signatures that unveil the identities of cognate bases of the DNA template. This sequencing-by-synthesis method not only utilizes the astounding speed and processivity of polymerase to rapidly obtain sequence information in real-time and with long sequence reads, but also allows massively parallel sequencing.75 This seminal work has led to the commercial development of an on-chip sequencing platform with the trade name “SMRT”.76
ZMWs have also recently been used to investigate challenging biochemical questions, such as how DNA translation occurs at the ribosome.77 Extensive biochemical and structural studies on the ribosome have resulted in a wealth of information on the underlying mechanism for the transit of transfer RNA (tRNA) through the A, P, and E sites of the ribosome during translation. A comprehensive dynamic characterization, however, requires the real-time observation of the transit process using the physiological concentration of tRNAs. Uemura et al. did just that and applied ZMWs to a translation study using micromolar tRNA ligand concentrations. As shown in Fig. 5(B), each ZMW well harbors a single ribosome-catalyzed translation reaction of a messenger RNA (mRNA) that carries alternating codons for phenylalanine (F) and lysine (K). As the ribosome travels along the template and decodes codons, the time-resolved burst and drop in fluorescence unveils the arrival/departure of fluorescently labeled aminoacylated/unacylated tRNA, and also the identity and order of the tRNAs. This study elegantly demonstrates that up to two tRNAs can briefly, but simultaneously orient in a ribosome and that the release of uncharged tRNA is not coupled to the binding of a new aminoacylated tRNA coming to the A site. As well as being suited for single-molecule DNA sequencing and the study of biochemical reactions on nucleic acids, the versatility of ZMWs has been demonstrated in studies on biomolecular binding/interaction, and protein receptor diffusion and oligomerization on living cell plasma membranes.78,79
4.2. Nanopore-based single-molecule sensing and detection
As detailed in previous sections, fluorescence is advantageous and widely used for single-molecule studies due to its high sensitivity. However, in addition to complicated and bulky optical setups, utilizing fluorescence requires the dye labeling of molecules of interest. This raises issues, such as labeling site specificity, labeling efficiency, and most importantly the perturbation of biomolecular activity caused by the presence of the label. The development of “nanopores”, inspired by natural ion channels that act as portals for selective ion sensing and transportation through membranes, introduces a new type of single-molecule platform that circumvents these issues by producing electric readouts for stochastic single-molecule events.Nanopores can be described as a miniaturized chip version of a “Coulter counter”. A signal is observed when biomolecules are electrophoretically driven through a nanoscopic pore deposited on a membrane that separates two electrolyte filled chambers and briefly block the ionic current. Resistive-pulse measurement across the nanopore yields characteristic modulation of an ionic current (e.g., amplitude and duration of the current block) that is related to the properties of the molecule present in the nanopore at a given time. Theoretically, by fine-tuning the recognition elements of the nanopore, biomolecules that vary in length, size, and structure can be reliably sensed and identified by their specific electric signatures, all without requiring sample labeling or amplification. Indeed, biological nanopores, particularly α-hemolysin, and certain genetically or chemically modified variants are now tailored for sensing a variety of bioanalytes; such as the detection of neurotransmitters, as well as the discrimination of enantiomers of amino acids by introducing a chiral selection environment to α-hemolysin;80,81 the protein detection or characterization of protein–aptamer interactions using a nanopore/aptamer hybrid;82 and protein kinase detection with α-hemolysin modified by kinase inhibitor peptides.83 As such, precise protein engineering guided by detailed structural information of biopores is expected to bring new application opportunities that utilize nanopore sensing technology.
On the basis of the finding that single-stranded DNA (ssDNA), that is some tens of Kb in length, can be threaded through α-hemolysin, it has been envisioned that characteristic modulations of ionic current by sequential occupancy of each nucleotide of the DNA strand in the nanopore would read off the strand's sequence. In practice, however, the realization of nanopore-based DNA sequencing has been hampered by several technical issues. The translocation speed of DNA through the nanopore (>1 nucleotide μs−1) is too fast to resolve the ionic current modulations at the single-nucleotide level.84 Moreover, the simultaneous occupancy of multiple nucleotides in the electric “reading” region of a nanopore prevents sequential discrimination of individual nucleotides.
These roadblocks, however challenging, have not deterred scientists from attempting nanopore-based DNA sequencing. With this goal in mind, two alternative strategies have been proposed. One is de novo DNA sequencing by slowing down the transportation speed of DNA within nanopores. Currently, immobilization of a DNA strand to a nanopore is used to achieve this purpose. Fig. 6(A) shows an electrophoretically stretched DNA strand immobilized in an α-hemolysin by the interaction of the biotinylated end of the DNA and streptavidin.85 By mutating amino acid residues at the R1 site (see Fig. 6(A)) of the biopore, the single nucleobase at this position, including those that may be epigenetically modified, can now be distinguished.86,87 The alternate strategy, as shown in Fig. 6(B), is “exo-sequencing”, in which an exonuclease digests a DNA strand and releases nucleoside monophosphates that are subsequently identified by a nanopore in the order of release.22 To this end, a key technical requirement for nanopores to distinguish different monophosphates in solution has been recently met by the Bayley group. By covalently attaching a bulky cyclodextrin molecule as an adapter to α-hemolysin, the resulting biopore is shown to discriminate the binding events of each of dAMP, dTMP, dCMP and dGMP with 99% accuracy (right panel of Fig. 6(B)). Although other issues still persist, these advances demonstrate the strength of nanopore technology and have brought great promise to the goal of label-free and amplification-free DNA sequencing.
 | ||
| Fig. 6 (A) Immobilization of ssDNA (blue) in an α-hemolysin nanopore (grey/black) by the interaction between the biotinylated (yellow) end of the DNA strand and streptavidin (red). The intensity of the nucleotide (orange) within the strand that resides at the R1 position (green) can be identified by its characteristic electric fingerprint. (B) A scheme of the exo-sequencing and residual pore current recording from the α-hemolysin nanopore attached to a cyclodextrin adapter. The 4 nucleoside monophosphates in the solution can be discriminated by the characteristic residual current levels. (C) A scheme of the ultra-thin nanopore-based miRNA sensing and quantification method. The miRNA enriched sample (RL) gave comparable spike-like pulse signals to those of the positive control (PC). Images are reproduced from ref. 22, 85 and 89. | ||
Nanopore sensing was initially carried out using protein ion channels deposited on a phospholipid bilayer (i.e., using hemolysin and MspA porin).24 The past decade has witnessed the emergence of several different types of synthetic nanopores, including nanofabricated solid-state, biological/solid-state hybrids, biomimetic, and more recently, DNA origami scaffolding nanopores.22,23,88 In particular, solid-state nanopores deliver the advantages of low cost, exceptional durability, precise tuning of the size and structure, and ease in fabricating high-density nanopore arrays. These synthetic nanopores are rapidly becoming a promising alternative to their biological counterparts. Although single-nucleotide discrimination has not been achieved using solid-state nanopores, with advances in nanofabrication the list of biomolecules that are able to be sensed by solid-state nanopores has successfully expanded to include short nucleic acids of some ten base pairs. This is opening new opportunities for the detection of microRNA (miRNA), a cancer biomarker made up of small RNA molecules that participate in RNA-interfering gene silencing by hybridizing to messenger RNA. Detection and quantification of miRNAs and other cancer biomarkers using conventional methods remains challenging and laborious due to their low cellular abundance. Solid-state nanopores arrayed on an ultra-thin (∼10 nm) silicon nitride membrane have recently been developed and used by Wanunu et al. to meet this challenge.89Fig. 6(C) illustrates the method used to detect miRNA (i.e., miR122a) using solid-state nanopores. A miRNA-specific probe is first introduced to the whole RNA extract from rat tissue to hybridize miR122a to a double-stranded form. The resulting probe:miR122a duplex is then isolated by magnetic beads coated with p19 protein via the specific interaction of p19 with the probe:miR11a duplex. After elution, the duplex is subjected to nanopore analysis. The sample, enriched with miR11a, gives the spike-like signals with amplitudes matching those of the positive control, while negative controls show no signal or signals lower than the threshold for confirmative detection of the miRNA. Remarkably, this nanopore-based method permits measurements of miRNA at sub-fmol μL−1 concentrations.
With a short history of less than two decades, nanopore sensing has evolved tremendously; it has been transformed from a single-pore made up of pore-forming protein to massive pore arrays constructed from diverse materials, with structures of nanoscale uniformity that are tailored for a broad scope of applications.90 Moreover, the innovative integration of nanopores with other single-molecule instruments (e.g., TIRF and optical tweezers) demonstrates the adaptability and versatility needed for future multiplexed on-chip formats of nanopores.91,92 These and other advances in this technology suggest that nanopores will serve as low-cost, label-free single-molecule platforms for diagnostics and biomolecular detection/analysis in years to come.
5. Conclusions and perspectives
A broad range of bioscience and biotechnology is marching toward the goal of achieving single-molecule precision and resolution in analysis. This has been enormously facilitated by the convergent development of lab-on-a-chip and single-molecule techniques, which give birth to a novel class of on-chip single-molecule tools. Using these tools, biomolecular studies can now be carried out in chip-like devices with molecular precision, high-throughput and efficiency, and a level of automation unrivaled by the conventional ensemble approaches. In this section, we will share our opinions on the future development, innovation, optimization and commercialization of lab-on-a-chip techniques for single-molecule studies.5.1. Developing integrated microfluidic platforms for automated, high-throughput, and parallel single molecule studies
Microfluidic devices provide powerful miniaturized versions of their bulky, bench-top counterparts with comparable or even more impressive performance. The miniaturized platform allows for more and more possibilities and opportunities to carry out single-molecule studies, especially for automated single-molecule studies to provide sufficient data for statistical analysis. With the existing barrier between the microscale and nanoscale, directly implementing current microfluidic techniques into looking at the single-molecule is still challenging. However, the handling and processing of single molecules do not require direct contact and precise positioning of the handling target. Because those bioassays have to maintain their viability in an aqueous environment, they can be readily handled by novel microfluidic techniques, such as microfluidic large-scale integration and various droplet-based microfluidic techniques, as we discussed in a previous section. Developing microfluidic-based techniques for single-molecule studies possess advantages, such as: (1) Simple and precise device fabrication processes, including soft-lithography-based techniques that have now matured with strong support from researchers all over the world. (2) A fully automated chip design for sophisticated and high-throughput bio-reagent processing, where chip functions can be programmed to handle liquid compartments confining single molecules. (3) The minimum requirement for the amount of biomolecule analytes, because microfluidic devices inherently require small volume samples. (4) The design of microfluidic devices, flow patterns and behaviors can be precisely controlled and finely tuned for the particular biomolecules/biochemical reactions in question. For example, using bubble mixers93-95 to mix different reaction agents aids the sample preparation process and provides controllable bio-reactions. (5) The safe and environmentally friendly platform where all the biochemical reactions are sealed inside a chamber so the risks of direct exposure and inter-contaminations are greatly reduced. Because minimum bio-waste is generated during reactions in a sealed chamber, waste handling is made simple and straightforward by directing all of the waste to waste containers without human intervention.We envision that microfluidic-based devices will become a major research platform for single-molecule studies in the near future. Two exciting examples are the protein–interaction networks based on MITOMI discussed in section 2,46 and a PDMS microreactor for DNA sequencing,96 although neither are now applied to single-molecule investigations. How to fully take advantage of liquid behavior on the microscale for single-molecule studies is an interesting, challenging, and rewarding question. In addition, most microfluidic devices discussed here still require separate apparatus for detection, monitoring, and analysis. This demands on-chip imaging and detection methods to realize fully functional microfluidic devices for single-molecule studies. Fortunately, various on-chip imaging techniques are in development.97–99 Promising imaging devices, such as a lens-free on-chip microscope100 and a lens-free optical tomographic microscope,101 and their derivatives would certainly help to achieve completely functional on-chip devices for high-throughput single-molecule analysis without the constraints of technically demanding equipment.
5.2. Exploring new techniques and unlocking the potential of existing single-molecule techniques
State-of-the-art on-chip single-molecule techniques with better overall performance and multi-functionalities are expected to emerge with the promise of broader applications in biological and biochemical studies. Conventional magnetic tweezers, for example, have limitations of weak torque, and medium-to-low resolution (1–10 nm) in tracking. However, the new generation of magnetic tweezers are now capable of exerting >100 pN force, tracking with high-resolution at sub-nanometer distances and providing fluorescent readouts when integrated with a TIRF microscope. This high force enables direct measurement of the applied torque on biomolecules, while the high resolution in tracking and fluorescent readout gives more precise information, as well as direct measurement of the location/position of conformational changes of a biomolecule. Nanopore-based sensing is another promising single-molecule approach that is continuously evolving: from a biological nanopore to solid-state nanopore made of ultra-thin metal membranes, graphene or more recently DNA origami through the help of material-processing techniques. The evolution of the nanopore field shows no signs of slowing down, and as analyte-specific pore shape designs become feasible, the field moves closer and closer to single-nucleotide sensing and DNA sequencing. In addition, “mix and match” hybrid single-molecule platforms, such as nanopore/magnetic tweezers102 and nanopore/TIRF,92 microfluidic/optical tweezers/TIRF hybrids,103 will lead to the emergence of novel on-chip devices with multidimensional single-molecule outputs. Together, we expect that the single-molecule tools in hand will continuously progress to an increasingly important role in biological/biochemical research and biotechnology.Recent breakthroughs on new techniques, such as plasmonic nano-antennas104 and surface acoustic wave (SAW)-based acoustic tweezers,105,106 also offer alternative solutions for single-molecule sensing and analysis. Plasmonic nano-antenna pairs utilize advanced nanofabrication techniques to precisely position nanostructures with controllable gaps. These nanoscale gaps will create localized “hot spots” that have enhanced local field intensity through the confinement of incident light energy (Fig. 7(A)).The “hot spots” from nano-antenna pairs will help to capture and confine single molecules inside or close to the “hot spot” regions, due to the drag force generated from the local intensity field difference. This allows single-molecule studies with enhanced signal readouts. Moreover, the concentrated energy field will also enhance the signals carried by the single molecule, and the signals can be directed through nano-antenna to external collectors for monitoring and analysis. SAW-based acoustic tweezers are another newly developed technique for particle/cell manipulation and positioning (Fig. 7(B)). Although SAW-based microfluidic techniques105,107–112 are now mainly used in cell-based biological studies at the current stage, their potential for single-molecule studies is promising. A recent report has achieved PCR reactions on-chip113 by combing a SAW technique with phononic crystals,114–116 and this is just one tentative exploration of the SAW technique for molecular studies. We believe that the future development of SAW-based microfluidic techniques on new transducer design and phase control will eventually achieve manipulation of single molecules.
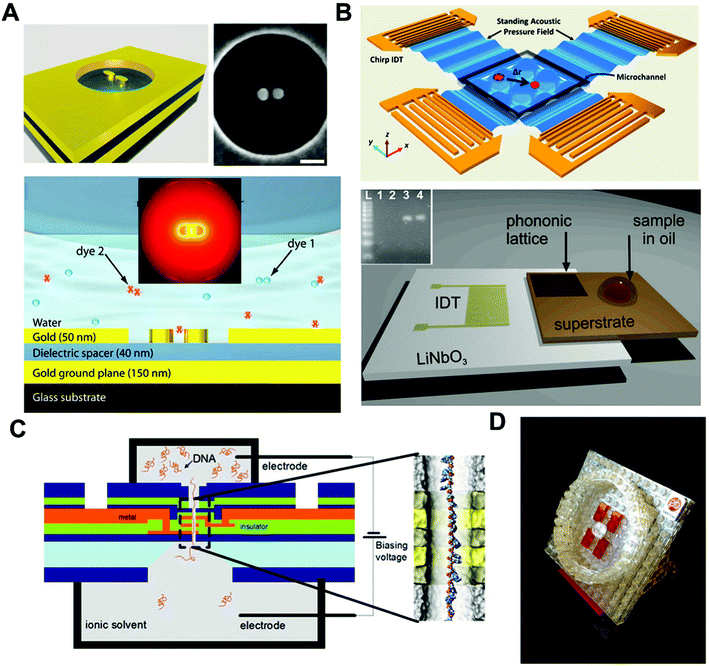 | ||
| Fig. 7 (A) Nano-antenna pair for single-molecule surface enhanced Raman scattering (sm-SERS). The scale bar is 90 nm. The molecule can be trapped at the “hot spot” of the nano-antenna and SERS signals can be enhanced via the directionality of the antenna to achieve single-molecule SERS detection. (B) Surface acoustic waves (SAW)-based acoustic tweezers. Acoustic tweezers can manipulate single particles and cells in a biocompatible manner (top). SAW-based PCR utilizing photonic crystals to modulate SAWs for real-time PCR (bottom). The inset is an electrophoretic gel analysis of the amplicon from the real-time PCR on SAW. (C) “DNA transistor” for a next-generation genomic reader. A single-stranded DNA is passing through the nanopore under the influence of an electric field. Bias voltages are applied through the nanopore structure to control the speed of ssDNA travelling inside the nanopore. The precision of the signal readout is expected to reach single-nucleotide level. (D) A schematic representation of the commercial “SMRT” cell product developed by Pacific Bioscience for DNA sequencing using ZMW technology. Images are reproduced from ref. 76, 104, 106, 113 and 117. | ||
5.3. Pushing lab-on-a-chip techniques out of the lab
To make an impact, the lab-on-a-chip techniques for single-molecule studies will have to be transformed into tangible, real-world applications and products. In this regard, IBM is working on a project called the “DNA transistor” as a genetic reader for DNA sequencing and personalized genome analysis.117 The whole device utilizes IBM's strength in semiconductor fabrication techniques, building a multi-layer nanopore structure that sits on a silicon substrate. With precise control of the bias voltage applied onto the “DNA transistor”, precise control of the DNA's position inside the nanopore is achievable, making readouts of DNA information at single-nucleotide resolution possible (Fig. 7(C)). Once accomplished, IBM claims the entire device will cost under $100. It will be a revolutionary breakthrough in both healthcare and semiconductor industries. In the near future, DNA sequencing and genome analysis may be as simple as pushing several buttons over the course of a few hours. In contrast, the Human Genome Project (HGP) cost $3 billion, took more than 10 years to finish, and required highly skillful scientists working together from all over the world. Another example is Pacific Bioscience, a company targeting technological innovation for biological sciences. They invented and marketed the Zero-Mode-Waveguide discussed in our previous section as a sequencing platform, termed the “SMRT” cell. A model for SMRT is shown in Fig. 7(D). The SMRT cell, together with other techniques developed by the same company, can achieve DNA sequencing with long reads up to 20,000 base pairs with high accuracy and sensitivity.76 We expect to see more techniques like those commercialized, making the best use of the developed technology to improve human health. Other than “DNA transistor” and “SMRT” chips, there are many other techniques such as on-chip flow cytometry for single-molecule studies,118,119 and laser scanning analysis of blood using commercial DVD drives,120 with promising potential as single-molecule analyzing tools for commercialization. At the current rate of technological progress, the goal of achieving commercially available lab-on-a-chip single-molecule analysis devices with enhanced functionalities and low cost is a realistic hope.Acknowledgements
This research was supported by National Institutes of Health (NIH) Director's New Innovator Award (1DP2OD007209-01).References
- X. S. Xie, P. J. Choi, G. W. Li, N. K. Lee and G. Lia, Annu. Rev. Biophys., 2008, 37, 417–444 CrossRef CAS.
- A. J. de Mello, Lab Chip, 2003, 3, 29N–34N RSC.
- D. Spetzler, J. York, C. Dobbin, J. Martin, R. Ishmukhametov, L. Day, J. Yu, H. Kang, K. Porter, T. Hornung and W. D. Frasch, Lab Chip, 2007, 7, 1633–1643 RSC.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- P. Neuzi, S. Giselbrecht, K. Lange, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef CAS.
- X. Mao and T. J. Huang, Lab Chip, 2012, 12, 4006–4009 RSC.
- S. Yang, F. Guo, B. Kiraly, X. Mao, M. Lu, K. W. Leong and T. J. Huang, Lab Chip, 2012, 12, 2097–2102 RSC.
- A. A. Nawaz, X. Mao, Z. S. Stratton and T. J. Huang, Lab Chip, 2013, 13, 1457–1463 RSC.
- P. Li, Z. S. Stratton, M. Dao, J. Ritz and T. J. Huang, Lab Chip, 2013, 13, 602–609 RSC.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS.
- X. Mao and T. J. Huang, Lab Chip, 2012, 12, 1412–1416 RSC.
- F. B. Myers and L. P. Lee, Lab Chip, 2008, 8, 2015–2031 RSC.
- H. Craighead, Nature, 2006, 442, 387–393 CrossRef CAS.
- W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12596–12602 CrossRef CAS.
- B. Shergill, L. Meloty-Kapella, A. A. Musse, G. Weinmaster and E. Botvinick, Dev. Cell, 2012, 22, 1313–1320 CrossRef CAS.
- C. Cecconi, E. A. Shank, S. Marqusee and C. Bustamante, Methods Mol. Biol., 2011, 749, 255–271 CAS.
- A. Borgia, P. M. Williams and J. Clarke, Annu. Rev. Biochem., 2008, 77, 101–125 CrossRef CAS.
- C. Veigel and C. F. Schmidt, Nat. Rev. Mol. Cell Biol., 2011, 12, 163–176 CrossRef CAS.
- X. Leng, W. Zhang, C. Wang, L. Cui and C. J. Yang, Lab Chip, 2010, 10, 2841–2843 RSC.
- S. Kim, A. M. Streets, R. R. Lin, S. R. Quake, S. Weiss and D. S. Majumdar, Nat. Methods, 2011, 8, 242–245 CrossRef CAS.
- J. Clarke, H. C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS.
- S. Garaj, W. Hubbard, A. Reina, J. Kong, D. Branton and J. A. Golovchenko, Nature, 2010, 467, 190–193 CrossRef CAS.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647–20652 CrossRef CAS.
- T. Miyake, T. Tanii, H. Sonobe, R. Akahori, N. Shimamoto, T. Ueno, T. Funatsu and I. Ohdomari, Anal. Chem., 2008, 80, 6018–6022 CrossRef CAS.
- P. Zhu and H. G. Craighead, Annu. Rev. Biophys., 2012, 41, 269–293 CrossRef CAS.
- M. J. Levene, J. Korlach, S. W. Turner, M. Foquet, H. G. Craighead and W. W. Webb, Science, 2003, 299, 682–686 CrossRef CAS.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem. Int. Edit., 2010, 49, 5846–5868 CAS.
- R. R. Pompano, W. S. Liu, W. B. Du and R. F. Ismagilov, Annu. Rev. Anal. Chem., 2011, 4, 59–81 CrossRef CAS.
- S. Y. Teh, R. Lin, L. H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- A. Huebner, S. Sharma, M. Srisa-Art, F. Hollfelder, J. B. Edel and A. J. Demello, Lab Chip, 2008, 8, 1244–1254 RSC.
- X. C. I. Solvas and A. deMello, Chem. Commun., 2011, 47, 1936–1942 RSC.
- H. N. Joensson and H. Andersson-Svahn, Lab Chip, 2011, 11, 4144–4147 RSC.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2155 RSC.
- F. Guo, M. I. Lapsley, A. A. Nawaz, Y. Zhao, S. C. Lin, Y. Chen, S. Yang, X. Z. Zhao and T. J. Huang, Anal. Chem., 2012, 84, 10745–10749 CrossRef CAS.
- T. D. Rane, C. M. Puleo, K. J. Liu, Y. Zhang, A. P. Lee and T. H. Wang, Lab Chip, 2010, 10, 161–164 RSC.
- S. H. Kim, S. Iwai, S. Araki, S. Sakakihara, R. Iino and H. Noji, Lab Chip, 2012, 12, 4986–4991 RSC.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. L. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS.
- R. Tewhey, J. B. Warner, M. Nakano, B. Libby, M. Medkova, P. H. David, S. K. Kotsopoulos, M. L. Samuels, J. B. Hutchison, J. W. Larson, E. J. Topol, M. P. Weiner, O. Harismendy, J. Olson, D. R. Link and K. A. Frazer, Nat. Biotechnol., 2009, 27, 1025-U1094 CrossRef.
- M. Nakano, J. Komatsu, S.-i. Matsuura, K. Takashima, S. Katsura and A. Mizuno, J. Biotechnol., 2003, 102, 117–124 CrossRef CAS.
- K. Shao, W. Ding, F. Wang, H. Li, D. Ma and H. Wang, PLoS One, 2011, 6, e24910 CAS.
- R. Williams, S. G. Peisajovich, O. J. Miller, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 545–550 CrossRef CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS.
- I. E. Araci and S. R. Quake, Lab Chip, 2012, 12, 2803–2806 RSC.
- D. Gerber, S. J. Maerkl and S. R. Quake, Nat. Methods, 2009, 6, 71–74 CrossRef CAS.
- A. E. Cohen and W. E. Moerner, Appl. Phys. Lett., 2005, 86 Search PubMed.
- Q. Wang and W. E. Moerner, Appl. Phys. B: Lasers Opt., 2010, 99, 23–30 CrossRef CAS.
- Q. Wang and W. E. Moerner, ACS Nano, 2011, 5, 5792–5799 CrossRef CAS.
- Q. Wang, R. H. Goldsmith, Y. Jiang, S. D. Bockenhauer and W. E. Moerner, Acc. Chem. Res., 2012, 45, 1955–1964 CrossRef CAS.
- Y. Jiang, N. R. Douglas, N. R. Conley, E. J. Miller, J. Frydman and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16962–16967 CrossRef CAS.
- R. H. Goldsmith and W. E. Moerner, Abstr. Pap. Am. Chem. S., 2010, 239 Search PubMed.
- A. E. Cohen and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12622–12627 CrossRef CAS.
- C. Joo, H. Balci, Y. Ishitsuka, C. Buranachai and T. Ha, Annu. Rev. Biochem., 2008, 77, 51–76 CrossRef CAS.
- M. J. Comstock, T. Ha and Y. R. Chemla, Nat. Methods, 2011, 8, 335-U382 CrossRef.
- R. E. Georgescu, N. Y. Yao and M. O'Donnell, FEBS Lett., 2010, 584, 2596–2605 CrossRef CAS.
- T. Fazio, M. L. Visnapuu, S. Wind and E. C. Greene, Langmuir, 2008, 24, 10524–10531 CrossRef CAS.
- A. Graneli, C. C. Yeykal, T. K. Prasad and E. C. Greene, Langmuir, 2006, 22, 292–299 CrossRef CAS.
- M. L. Visnapuu and E. C. Greene, Nat. Struct. Mol. Biol., 2009, 16, 1056–1062 CAS.
- J. Gorman, F. Wang, S. Redding, A. J. Plys, T. Fazio, S. Wind, E. E. Alani and E. C. Greene, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3074–3083 CrossRef CAS.
- R. E. Georgescu, I. Kurth and M. E. O'Donnell, Nat. Struct. Mol. Biol., 2012, 19, 113–116 CAS.
- I. De Vlaminck and C. Dekker, Annu. Rev. Biophys., 2012, 41, 453–472 CrossRef CAS.
- M. Manosas, A. Meglio, M. M. Spiering, F. Ding, S. J. Benkovic, F. X. Barre, O. A. Saleh, J. F. Allemand, D. Bensimon and V. Croquette, Methods Enzymol., 2010, 475, 297–320 CAS.
- M. Manosas, M. M. Spiering, Z. Zhuang, S. J. Benkovic and V. Croquette, Nat. Chem. Biol., 2009, 5, 904–912 CrossRef CAS.
- M. Manosas, S. K. Perumal, V. Croquette and S. J. Benkovic, Science, 2012, 338, 1217–1220 CrossRef CAS.
- M. Manosas, M. M. Spiering, F. Ding, V. Croquette and S. J. Benkovic, Nucleic Acids Res., 2012, 40, 6187–6198 CrossRef CAS.
- K. C. Neuman and A. Nagy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS.
- A. Revyakin, R. H. Ebright and T. R. Strick, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4776–4780 CrossRef CAS.
- A. Revyakin, C. Y. Liu, R. H. Ebright and T. R. Strick, Science, 2006, 314, 1139–1143 CrossRef CAS.
- M. T. van Loenhout, M. V. de Grunt and C. Dekker, Science, 2012, 338, 94–97 CrossRef CAS.
- K. Kim and O. A. Saleh, Nucleic Acids Res., 2009, 37 CAS.
- M. Kruithof, F. Chien, M. de Jager and J. van Noort, Biophys. J., 2008, 94, 2343–2348 CrossRef CAS.
- C. Lavelle, J. M. Victor and J. Zlatanova, Int. J. Mol. Sci., 2010, 11, 1557–1579 CrossRef CAS.
- X. Long, J. W. Parks, C. R. Bagshaw and M. D. Stone, Nucleic Acids Res., 2013, 41, 2746–2755 CrossRef CAS.
- A. Petrov, G. Kornberg, S. O'Leary, A. Tsai, S. Uemura and J. D. Puglisi, Curr. Opin. Struct. Biol., 2011, 21, 137–145 CrossRef CAS.
- http://www.pacificbiosciences.com/products/smrt-technology/ .
- S. Uemura, C. E. Aitken, J. Korlach, B. A. Flusberg, S. W. Turner and J. D. Puglisi, Nature, 2010, 464, 1012–1017 CrossRef CAS.
- T. Sameshima, R. Iizuka, T. Ueno, J. Wada, M. Aoki, N. Shimamoto, I. Ohdomari, T. Tanii and T. Funatsu, J. Biol. Chem., 2010, 285, 23157–23162 CrossRef.
- C. I. Richards, K. Luong, R. Srinivasan, S. W. Turner, D. A. Dougherty, J. Korlach and H. A. Lester, Nano Lett., 2012, 12, 3690–3694 CrossRef CAS.
- A. J. Boersma and H. Bayley, Angew. Chem., Int. Ed., 2012, 51, 9606–9609 CrossRef CAS.
- A. J. Boersma, K. L. Brain and H. Bayley, ACS Nano, 2012, 6, 5304–5308 CrossRef CAS.
- D. Rotem, L. Jayasinghe, M. Salichou and H. Bayley, J. Am. Chem. Soc., 2012, 134, 2781–2787 CrossRef CAS.
- S. Cheley, H. Z. Xie and H. Bayley, ChemBioChem, 2006, 7, 1923–1927 CrossRef CAS.
- A. Meller, L. Nivon, E. Brandin, J. Golovchenko and D. Branton, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1079–1084 CrossRef CAS.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. H. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS.
- E. V. B. Wallace, D. Stoddart, A. J. Heron, E. Mikhailova, G. Maglia, T. J. Donohoe and H. Bayley, Chem. Commun., 2010, 46, 8195–8197 RSC.
- D. Stoddart, A. J. Heron, J. Klingelhoefer, E. Mikhailova, G. Maglia and H. Bayley, Nano Lett., 2010, 10, 3633–3637 CrossRef CAS.
- S. Garaj, W. Hubbard, A. Reina, J. Kong, D. Branton and J. A. Golovchenko, Nature, 2010, 467, 190-U173 CrossRef.
- M. Wanunu, T. Dadosh, V. Ray, J. Jin, L. McReynolds and M. Drndic, Nat. Nanotechnol., 2010, 5, 807–814 CrossRef CAS.
- H. Bayley, Nature, 2010, 467, 164–165 CrossRef CAS.
- U. F. Keyser, B. N. Koeleman, S. Van Dorp, D. Krapf, R. M. M. Smeets, S. G. Lemay, N. H. Dekker and C. Dekker, Nat. Phys., 2006, 2, 473–477 CrossRef CAS.
- B. McNally, A. Singer, Z. L. Yu, Y. J. Sun, Z. P. Weng and A. Meller, Nano Lett., 2010, 10, 2237–2244 CrossRef CAS.
- D. Ahmed, X. Mao, B. K. Juluri and T. J. Huang, Microfluid. Nanofluid., 2009, 7, 727–731 CrossRef CAS.
- D. Ahmed, X. Mao, J. Shi, B. K. Juluri and T. J. Huang, Lab Chip, 2009, 9, 2738–2741 RSC.
- X. Mao, B. K. Juluri, M. I. Lapsley, Z. S. Stratton and T. J. Huang, Microfluid. Nanofluid., 2009, 8, 139–144 CrossRef.
- P. A. Sims, W. J. Greenleaf, H. Duan and X. S. Xie, Nat. Methods, 2011, 8, 575–580 CrossRef CAS.
- Y. Zhao, Z. S. Stratton, F. Guo, M. I. Lapsley, C. Y. Chan, S. C. Lin and T. J. Huang, Lab Chip, 2013, 13, 17–24 RSC.
- J. Wu, G. Zheng and L. M. Lee, Lab Chip, 2012, 12, 3566–3575 RSC.
- L. Pang, H. M. Chen, L. M. Freeman and Y. Fainman, Lab Chip, 2012, 12, 3543–3551 RSC.
- X. Cui, L. M. Lee, X. Heng, W. Zhong, P. W. Sternberg, D. Psaltis and C. Yang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10670–10675 CrossRef CAS.
- S. O. Isikman, W. Bishara, S. Mavandadi, F. W. Yu, S. Feng, R. Lau and A. Ozcan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7296–7301 CrossRef CAS.
- H. B. Peng and X. S. S. Ling, Nanotechnology, 2009, 20 CAS.
- A. L. Forget, C. C. Dombrowski, I. Amitani and S. C. Kowalczykowski, Nat. Protoc., 2013, 8, 525–538 CrossRef CAS.
- A. Ahmed and R. Gordon, Nano Lett., 2012, 12, 2625–2630 CrossRef CAS.
- J. Shi, D. Ahmed, X. Mao, S. C. Lin, A. Lawit and T. J. Huang, Lab Chip, 2009, 9, 2890–2895 RSC.
- X. Ding, S. C. Lin, B. Kiraly, H. Yue, S. Li, I. K. Chiang, J. Shi, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11105–11109 CrossRef CAS.
- J. Shi, X. Mao, D. Ahmed, A. Colletti and T. J. Huang, Lab Chip, 2008, 8, 221–223 RSC.
- J. Shi, H. Huang, Z. Stratton, Y. Huang and T. J. Huang, Lab Chip, 2009, 9, 3354–3359 RSC.
- J. Shi, S. Yazdi, S. C. Lin, X. Ding, I. K. Chiang, K. Sharp and T. J. Huang, Lab Chip, 2011, 11, 2319–2324 RSC.
- X. Ding, S. C. Lin, M. I. Lapsley, S. Li, X. Guo, C. Y. Chan, I. K. Chiang, L. Wang, J. P. McCoy and T. J. Huang, Lab Chip, 2012, 12, 4228–4231 RSC.
- X. Ding, J. Shi, S. C. Lin, S. Yazdi, B. Kiraly and T. J. Huang, Lab Chip, 2012, 12, 2491–2497 RSC.
- S. C. Lin, X. Mao and T. J. Huang, Lab Chip, 2012, 12, 2766–2770 RSC.
- J. Reboud, Y. Bourquin, R. Wilson, G. S. Pall, M. Jiwaji, A. R. Pitt, A. Graham, A. P. Waters and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15162–15167 CrossRef CAS.
- J. Shi, S.-C. S. Lin and T. J. Huang, Appl. Phys. Lett., 2008, 92, 111901 CrossRef.
- S.-C. Lin, T. Huang, J.-H. Sun and T.-T. Wu, Physical Review B, 2009, 79 CAS.
- S.-C. S. Lin and T. J. Huang, Physical Review B, 2011, 83 Search PubMed.
- http://researcher.watson.ibm.com/researcher/view_project.php?id=1120 .
- J. M. Emory, Z. Peng, B. Young, M. L. Hupert, A. Rousselet, D. Patterson, B. Ellison and S. A. Soper, Analyst, 2012, 137, 87–97 RSC.
- J. R. Krogmeier, I. Schaefer, G. Seward, G. R. Yantz and J. W. Larson, Lab Chip, 2007, 7, 1767–1774 RSC.
- H. Ramachandraiah, M. Amasia, J. Cole, P. Sheard, S. Pickhaver, C. Walker, V. Wirta, P. Lexow, R. Lione and A. Russom, Lab Chip, 2013, 13, 1578–1585 RSC.
| This journal is © The Royal Society of Chemistry 2013 |