 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion channel recordings on an injection-molded polymer chip†
Simone
Tanzi
a,
Marco
Matteucci
a,
Thomas Lehrmann
Christiansen
a,
Søren
Friis
b,
Mette Thylstrup
Christensen
b,
Joergen
Garnaes
c,
Sandra
Wilson
b,
Jonatan
Kutchinsky
b and
Rafael
Taboryski
*a
aDepartment of Micro- and Nanotechnology, Technical University of Denmark, Building 345E, DK-2800 Kongens Lyngby, Denmark. E-mail: rafael.taboryski@nanotech.dtu.dk
bSophion Bioscience A/S, Baltorpvej 154, DK-2750 Ballerup, Denmark
cDanish Fundamental Metrology, Matematiktorvet, Building 307, DK-2800 Kongens Lyngby, Denmark
First published on 23rd September 2013
Abstract
In this paper, we demonstrate recordings of the ion channel activity across the cell membrane in a biological cell by employing the so-called patch clamping technique on an injection-molded polymer microfluidic device. The findings will allow direct recordings of ion channel activity to be made using the cheapest materials and production platform to date and with the potential for very high throughput. The employment of cornered apertures for cell capture allowed the fabrication of devices without through holes and via a scheme comprising master origination by dry etching in a silicon substrate, electroplating in nickel and injection molding of the final part. The most critical device parameters were identified as the length of the patching capillary and the very low surface roughness on the inside of the capillary. The cross-sectional shape of the orifice was found to be less critical, as both rectangular and semicircular profiles seemed to have almost the same ability to form tight seals with cells with negligible leak currents. The devices were functionally tested using human embryonic kidney cells expressing voltage-gated sodium channels (Nav1.7) and benchmarked against a commercial state-of-the-art system for automated ion channel recordings. These experiments considered current–voltage (IV) relationships for activation and inactivation of the Nav1.7 channels and their sensitivity to a local anesthetic, lidocaine. Both IVs and lidocaine dose–response curves obtained from the injection-molded polymer device were in good agreement with data obtained from the commercial system.
Introduction
The patch clamping technique was introduced in 1976 by Neher and Sakmann1 and has become the accepted standard for fundamental studies of ion channel proteins and the discovery of drugs that affect them. Ion channels play a central role in the excitability of nerves and muscles; they underlie the heartbeat, muscle contractions and brain activity as well as many other basic physiological actions.2 Ion channels are macromolecular pores with the ability to regulate the movement of ions across the otherwise impermeable cell membrane.2 Ion channels are also involved in a diverse range of disorders and pathological conditions, called channelopathies.3,4 Mutations in ion channel genes have been associated with diseases such as cystic fibrosis, hypertension, ataxia, arrhythmia and several types of epilepsy.4,5 In pharmacology, ion channels represent highly attractive targets for drug discovery with an estimated market of US $12 billion.6,7Despite their enormous potential as druggable targets, the use of ion channels for drug discovery has been well behind the expectation, mainly due to the lack of adequate screening technology.8 Conventional manual patch clamping delivers high information content, but it is a complex technique that requires skilled personnel and suffers from extremely low throughput, while, on the other hand, all indirect screening methods suffer from low specificity and, thus, the associated risk of generating false negative or positive results.8,9 With the promise of removing this trade-off, the first automated parallel patch clamping system (APC) was made available in 2003.10 Since then, 5 companies (Molecular Devices, Sophion Bioscience, Nanion, Cytocentric, and Fluxion) have entered the market, establishing APC as an essential technology for secondary screening, lead optimization and cardiac safety testing.11 Performances of the different systems have been discussed in a few reviews.11–14 APC systems replace the use of conventional glass micropipettes with single-use, disposable devices fabricated using materials such as quartz15 and silicon/silicon dioxide.16 The majority of the available platforms share the so-called planar approach, where cells in suspension are blindly positioned by suction onto micro-apertures made as through holes in thin membranes.15 Alternatively, a lateral approach has been proposed, where suspended cells are trapped at lateral apertures generated at the junction of two microfluidic channels.17
The ability to form a high electrical resistance seal, preferably in the order of GΩ (the so-called gigaseal) between the aperture and the biological cell membrane, and the fabrication of such micro-sized through holes in thin substrates (10–20 μm) are the two most relevant benchmarks. These constraints have traditionally narrowed the range of usable materials and technologies to almost exclusively micromachining of silicon and glass, at least for commercial use.
The use of polymeric substrates has also been widely investigated, and planar microfluidic chips have been demonstrated in hybrid polydimethylsiloxane–polyimide (PDMS–PI),18 polyimide,19 oxygen plasma-treated PDMS20,21 and polyethylene glycol (PEG)–SU-8 mixture.22 Unfortunately, all the aforementioned polymer devices have so far failed to deliver relevant electrophysiological results. It has been more or less implicitly believed in the community that silicon oxide surfaces possess a unique surface chemistry for forming gigaseals, essentially through van der Waals interactions.23–25 The interaction between lipid membranes and other surfaces is, however, complicated and only poorly understood and, thus, leaves some leverage for challenging the uniqueness of these surfaces26–30 by employing polymers. Ionescu-Zanetti et al. implemented lateral junctions in a microfluidic device fabricated by casting of polydimethylsiloxane (PDMS).17,31 In 2009, this device was eventually commercialized by Fluxion, and it remains the only APC system relying on cornered apertures to date.32
Looking at all of the commercially available APC systems, the manufacturing cost per device is still prohibitive for establishing APC as a routine technique.33 Of the two materials employed nowadays, silicon remains extremely costly to purchase and machine, while PDMS is less suitable for mass production. Moreover, PDMS has issues when used for biological applications such as high permeability for small molecules, non-cross-linked oligomers, and surface diffusion of low molecular weight chains.34
The aim of this work is to propose a method for massive production of extremely cheap APC systems without compromising functionality. The trick lies in understanding the requirements to form tight seals between thermoplastic polymers and the cells. By shaping apertures that allow for a considerably larger effective interface area between low-energy polymer surfaces and the cell membrane, we demonstrate the formation of gigaseals, enabling high-quality ion channel recordings. The proposed device consists of only two parts: an injection-molded part in TOPAS cyclic olefin copolymer (COC) comprising the microfluidics and a polymer film of the same polymer grade, which is thermally bonded to the injection-molded part.35 For comparison, the commercially available QPlate™ from Sophion Bioscience comprises several materials, thermoplastic polymer, elastomer, ceramic, glass, and silicon. Moreover, TOPAS has none of the previously mentioned limitations of PDMS. Furthermore, the materials and fabrication platform fulfills all the requirements for cheap manufacturing in terms of “design for manufacturability”, “economy of scale” and production yield, and it avoids all drawbacks typical of prototyping methods.36 After an initial investment in master mold origination, injection molding delivers a cost per part that exponentially decreases with the number of parts, becoming the cheapest technology when production is greater than ~10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units.36 After assembly into chips, the systems were tested with HEK cells expressing Nav1.7, a voltage-gated sodium channel appropriate for benchmarking studies. Experiments were designed to explore current–voltage (IV) relationships for activation and inactivation of Nav1.7 channels and their sensitivity to a local anesthetic, lidocaine. Both IVs and lidocaine dose–response curves obtained from the injection-molded polymer device were benchmarked against the commercially available QPatch™ system from Sophion Bioscience. In the following section, we provide the details of the device layout and functionality, benchmarking tests against existing state-of-the-art methods, and finally a surface topology characterization of the patching capillaries.
000 units.36 After assembly into chips, the systems were tested with HEK cells expressing Nav1.7, a voltage-gated sodium channel appropriate for benchmarking studies. Experiments were designed to explore current–voltage (IV) relationships for activation and inactivation of Nav1.7 channels and their sensitivity to a local anesthetic, lidocaine. Both IVs and lidocaine dose–response curves obtained from the injection-molded polymer device were benchmarked against the commercially available QPatch™ system from Sophion Bioscience. In the following section, we provide the details of the device layout and functionality, benchmarking tests against existing state-of-the-art methods, and finally a surface topology characterization of the patching capillaries.
Materials and methods
Device fabrication
Chips were manufactured according to the methods recently reported by some of the same authors,37 which in brief consist of lithography, dry etching of the silicon, electroplating in nickel and injection molding of the polymer parts. Two etching processes were employed to form the patching capillaries: Bosch reactive ion etching for sample A and continuous reactive ion etching for sample B. This was to obtain, respectively, rectangular and semicircular profiles. However, in order to replicate patch clamping orifices smaller than the lateral apertures demonstrated for cell trapping,37 the overall process was partially modified. Silicon oxide used as masking material was replaced by standard photoresist, while a thin layer of oxide was applied after the first etching step to prevent damage to the patching channels during the rest of the process. Fabrication was then completed by alternating wet etching steps and oxidation in order to smooth the surfaces around the patching orifices. Details of the full process are given in the ESI.† Parts were molded from COC (TOPAS 5013, Advanced Polymers GmbH). A 100 μm thick extruded polymer film (TOPAS 5013F-04, Advanced Polymers Extrusion Lab) was used as a cover lid. The two parts were bonded by UV-assisted thermal bonding.35Cell culture
Human embryonic kidney 293 cells (HEK 293) expressing the subtype of the voltage-gated sodium channel Nav1.7 were provided by Scottish Biomedical Ltd. The cells were grown and maintained under standard culture conditions at 37 °C and 5% CO2. The cells were cultured in T175 culture flasks (Nunc A/S) to a maximum 80% confluence in high-glucose Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich D0819) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich F2442), 2 μg ml−1 blasticidin S (Sigma-Aldrich 15205) and 600 μg ml−1 geneticin (Sigma-Aldrich G8168). The cells were sub-cultured for two weeks before reaching a stable growth pattern and then used in the experiments. For sub-culture, the culture medium was removed and the cells were washed with phosphate-buffered saline (PBS) without calcium chloride and magnesium chloride (Sigma-Aldrich D8537). Trypsin/EDTA (Sigma-Aldrich T4174) was added to the culture flask, and the flask was incubated at 37 °C for 2 minutes. The medium was added to the flask, and the cells were re-suspended and placed in a new mother flask. Directly before experiments, the culture medium was removed and the cells were washed with PBS without calcium chloride and magnesium chloride. Detachin (VWR) was added to the culture flask, and the flask was incubated at 37 °C for 5 minutes or until the cells showed a round shape indicating detachment from the flask surfaces. The cells were re-suspended in serum-free CHO medium (Sigma-Aldrich C5467) supplemented with 25 mM HEPES (Sigma-Aldrich H0887), 100 μg ml−1 penicillin/streptomycin (Sigma-Aldrich P4333) and 0.04 mg ml−1 soybean trypsin inhibitor (Sigma-Aldrich T6522). Cell density and viability were determined by diluting an aliquot 1:2 in Trypan blue (Sigma-Aldrich T8154) and performing a cell count using the dye exclusion method in a hemocytometer. Cell concentration in the suspension was 2–3 M ml−1.
Solutions and compounds
The intracellular electrolyte solution contained (in mM): 135 CsF, 1/5 ethylene glycol tetraacetic acid (EGTA)/CsOH, 10 mM HEPES and 10 NaCl. The pH was adjusted to 7.3 with KOH and osmolarity to 320 mOsm with sucrose. The extracellular electrolyte solution contained (in mM): 1 CaCl2, 1 MgCl2, 5 HEPES, 3 KCl, 140 NaCl, 0.1 CdCl2 and 20 TEA–Cl. The pH was adjusted to 7.3 with NaOH and osmolarity to 320 mOsm with sucrose. All chemicals were purchased from Sigma-Aldrich. Both solutions were stored in a fridge at 4 °C and vacuum degassed for 20 minutes before use. Lidocaine hydrochloride monohydrate (Sigma-Aldrich L5647) was dissolved in dimethyl sulfoxide (DMSO) to give a 100 mM stock solution kept in the freezer. Subsequent dilutions were performed in an extracellular electrolyte solution.Instrumentation
Recordings were carried out using a HEKA Patch Clamp EPC 9 amplifier (HEKA Electronics) at room temperature. Pulse software (v 8.53, HEKA Electronics) was used for data acquisition. The device was mounted into a customized aluminum box and positioned on the stage of an Olympus IX70 inverted microscope. The two inlet ports connected to the recording patching channel were connected to two reservoirs filled with electrolyte solution and from them to a custom-made pressure controller and controlled with Labview software (National Instruments). Electrodes were electrically connected to the EPC9 head stage mounted on the customized aluminum box, and the chip resistance could be monitored by applying a 10 mV test square pulse for 10 ms. Before cell trapping, a positive pressure of 3–5 mbar was applied to the patching channel to prevent aperture contamination. A cell was captured at the hole after applying suction (negative pressure of 400 mbar) to the patching channel. The amplifier offset potential was zeroed prior to patching the cell, and the holding potential was held at −90 mV. Response currents were sampled at 10 kHz and filtered at 2.9 kHz with a 4-pole Bessel filter. The experiments on the QPatch™ were set up using standard assay settings. In brief, cells were positioned with a negative pressure of −100 mbar until a gigaseal was formed. The cell membrane was then ruptured with a negative pressure pulse of −250 mbar in order to get electrical access to the cell membrane. During this process, the offset potential, the chip capacitance and the cell capacitance were canceled out. The actual experiment protocol with the relevant voltage protocols and compound additions was programmed in the Sophion Assay Software, and it was executed after whole-cell formation was established. QPatch™ data were sampled at 10 kHz and filtered with an 8-pole Bessel filter at 3 kHz.Whole-cell recordings
Once the cells were trapped, a suction of −400 mbar was applied for about 20 seconds and then reduced to −30 mbar. The resistance across the aperture was continuously monitored by applying a 10 mV pulse for 10 ms. At this stage, the resistance was typically between 100 and 200 MΩ. Some of the cells showed whole-cell configuration immediately after patching. Otherwise, the whole-cell configuration was achieved by applying combined suction pulses (from −30 mbar to −400 mbar) and electrical pulses. Achievement of whole-cell configuration was verified by depolarizing the cell. Eventually, the cells reached whole-cell configuration and the resistance gradually increased over minutes during the experiments.Voltage protocols
For IV relationship experiments, a protocol with incremental steps of +10 mV from −90 mV to +70 mV of 1000 ms duration was used. Steady-state inactivation was investigated at +10 mV for 100 ms after the 1000 ms pre-pulse. Each incremental sweep took place with 5 second intervals. For concentration–response experiments, two consecutive depolarizations to +0 mV, respectively of 100 ms and 20 ms duration from a holding potential of −100 mV were used. The two depolarizations were spaced out by 15 ms at the holding potential.Results and discussion
The chip design contains four independent lateral apertures 600 μm apart, which are aligned along a straight channel reserved for extracellular electrolyte solution, as shown in Fig. 1a–c. The side channels are connected at the other end to four separate inlet channels which contain intracellular electrolyte solution. The patching channel is Y shaped and consists of a straight capillary with a rectangular profile and rounded corners (approximately 2 μm × 2 μm) at the end where the cell is captured and a wider opening at the opposing end for the reduction of the total hydraulic resistance. In addition, a second type of device with a semicircular profile was molded and tested. The two groups of devices will be called type A (rectangular profile) and type B (semicircular profile). A cell trapped at one of the patching orifices is shown in Fig. 1d. It was observed that the cell membrane tends to protrude a long distance into the patching capillary, as also previously reported for PDMS devices.17,38 In order to find the optimal length of the patching channels, we made all four channels with different lengths. The optimal length of the straight capillary fraction appeared to be the largest we had (45 μm), while shorter capillaries did not show good sealing properties. This indicates that the gigaseal formation must occur due to interaction between the cell membrane and the polymer along the whole length of the patching capillary and not only in the proximity of the orifice.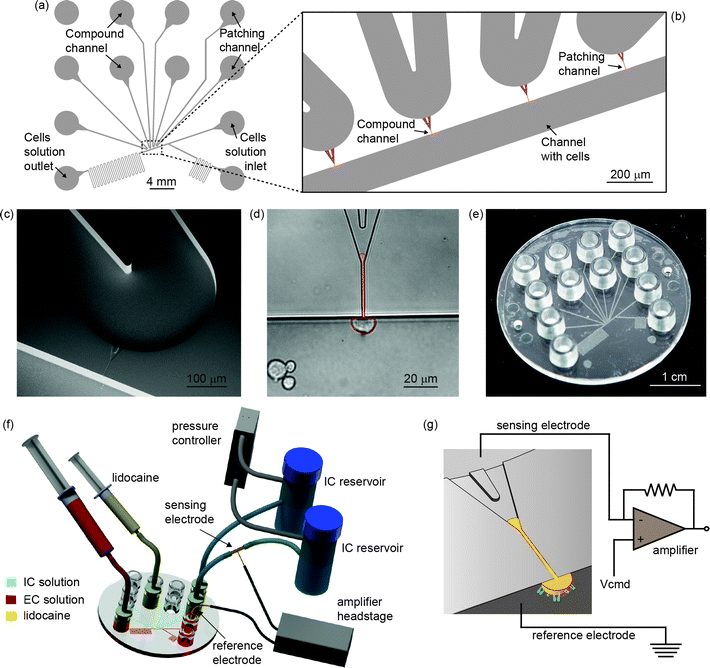 | ||
| Fig. 1 (a) Layout of the microfluidic device. (b) Close-up of the key part of the design with patching channel, channel for compound perfusion and channel for cell loading. (c) SEM micrograph of the Y-shaped patching channel connecting extra- and intracellular channels. (d) Optical microscope image of a captured HEK cell. Highlighted in red, the cell membrane is seen to protrude a distance of about 50 μm into the patching capillary shortly after being captured. (e) A single-use injection-molded device used for the experiments. (f) Illustration of the setup used for the recordings. The polymer device is connected to two intracellular solution reservoirs and then to the pressure controller. Ag/AgCl electrodes are connected to the head stage of the amplifier. Syringes are used to transport cells by convective flow from the inlet to the patch zone and to perfuse lidocaine through one of the lateral apertures, respectively. (g) Schematic of the patch clamping recording: a cell trapped to a lateral aperture. Electrodes are located across the orifice. | ||
Our customized polymer chip, 50 mm in diameter and 2 mm in thickness, contained 12 Luer fittings39 used to interface the microfluidic network. A single-use injection-molded device is shown in Fig. 1e. Prior to experiments, the device was primed with the electrolyte solutions. First, the capillary was filled with an intracellular solution, and then, the cell carrier channel was filled with an extracellular solution. The recording channel was connected to an external pressure controller, and reservoirs with the intracellular solution were employed to prevent the formation of air bubbles when applying suction. Ag/AgCl electrodes positioned across the recording channel and connected to the amplifier ensured electrical connection, as shown in Fig. 1f–g.
Cells were introduced into the inlet port and transported by the convective flow induced by a syringe. Before trapping one of the cells, a slight positive pressure was applied to the patching channel to prevent contamination of the aperture. Conveniently, one of the unused patching capillaries could be used for perfusion of lidocaine by using a syringe connected to the corresponding Luer port. During perfusion, the estimated average flow velocity in the carrier channel was approximately 1 mm s−1, and the average flow rate in the perfusion channel was estimated to be about 20% higher than the flow rate in the carrier channel. The lateral flow was applied for less than 10 seconds. A COMSOL® simulation, shown in Fig. 2, supports the premise that 100% of the lidocaine in solution was successfully delivered to the cell. The simulation considered a worst case scenario of lidocaine diffusion when a high lidocaine concentration of 1 mM was perfused.
 | ||
| Fig. 2 Lidocaine (1 mM) delivery to the cell being patched, simulated in COMSOL®. A lateral flow (in red) perfused into the flow in the carrier channel (in blue). The simulation accounted for lidocaine diffusion in the extracellular solution. A sectional view across the channel shows how the concentration corresponds to 100% in the proximity of the cell. | ||
As previously reported,37 injection molding enables high replication accuracy of micro- and nano-features. Fig. 3a–c shows SEM micrographs of the orifice formed at the junction between the patching channel and the carrier channel of a polymer device after the three main steps of the fabrication process: etching, electroplating and molding. The uniformity of the patching channels was confirmed for several devices by the narrow range of chip resistances; the resistances were measured across the 45 μm long patching capillaries with the presence of only the electrolyte solutions. This was 9.4 ± 1.1 MΩ (mean ± standard deviation, n = 33) for type A devices and 10.5 ± 0.7 MΩ (n = 14) for type B devices. The devices with semicircular profile (type B) had a slightly larger chip resistance as expected from their smaller cross-sectional area. Both devices showed comparable resistance variation. Since the chip resistance is mainly a function of the capillary geometry, its variation is a good indication of variations in channel geometry.40 The chip resistances are comparable to those reported by Ionescu-Zanetti17 and Chen;38 however, the variation is significantly smaller, indicating a better reproducibility of the present method. However, the chip resistance is significantly higher than the typical 2–4 MΩ access resistance reported for planar patch clamping devices where the holes are tapered on the back side.41 In addition, the parasitic chip capacitance, that ideally should be as low as possible, was monitored. This was 11.5 ± 0.5 pF for both devices A and B. This capacitance was presumably dominated by the parasitic capacitance of external wiring, as a much lower estimated capacitance of about 10 fF can be obtained for the chip itself by employing a simple model of a pair of parallel wires, being effectively 150 μm long segments of the 50 μm deep micro-channels, separated by a gap of 100 μm (the length of the Y-shaped patching channel), and the dielectric constant of TOPAS (εr = 2.35). For comparison, the chip capacitances for the measurement plates for the QPatch™ are 55 ± 5 pF.
 | ||
| Fig. 3 (a–c) SEM micrographs of the patching orifice after the main three steps of the fabrication process for type A devices: silicon master, nickel insert, and replicated polymer part (from left to right). (d) 3D AFM image of the patching orifice (type A). (e) 3D AFM image of the sidewall of the patching capillary (type A). (f) 3D AFM image of the semicircular patching orifice (type B). (g) 3D AFM image of the sidewall of the semicircular patching capillary (type B). | ||
In order to test the devices, whole-cell recordings were performed with human embryonic kidney (HEK) cells. Of 47 cells captured, 19 cells (40% of the total) allowed for whole-cell access with seal resistance of at least 100 MΩ, and of those, 7 (15%) were gigaseals. A success rate of 15% is thus attributed to our experiments. The rest of the cells showed either no whole-cell achievement or seal resistance below 100 MΩ. More precisely, tests were carried out on devices both having patching capillaries with rectangular (type A, n = 33) and semicircular (type B, n = 14) profiles. As shown in Table 1, both device types showed very similar sealing capability and ability to achieve the whole-cell configuration in a range of seal resistances. Both profiles also have similar gigaseal frequencies. The exact confidence interval for gigaseal frequency was calculated using the Clopper–Pearson method42 and is given at a confidence level of 90%. The confidence intervals for gigaseal frequency were also comparable for the two profiles. During cell experiments, it was noted that high-resistance seals could only be achieved on longer channels, and shorter channels were also tested with cells, but they all failed to deliver quality seals (data not reported). Importantly, for the 45 μm long channels, each cell showing a seal resistance above 250 MΩ also allowed for whole-cell recordings. It was observed that tight seals appeared gradually after the whole-cell configuration was established, eventually reaching gigaseal during a period of 10–20 minutes. Achievement of the whole-cell configuration was verified by activation of sodium currents through a depolarization of the cell membrane. Seals had an average lifetime of 30 minutes. Furthermore, it was also observed that whole-cell configuration with rupture of the patched cell membrane was obtained spontaneously upon cell capture without an intermediate cell-attached state, where electrical access to the interior of the cell goes through the patched piece of cell membrane inside the pipette in traditional patch clamping experiments. This seems to differ from other systems, both those using traditional pipettes and planar chips, where a drop in resistance when breaking the cell membrane is normally observed.
| Device | Total of cells tested | R < 100 MΩ | R > 100 MΩ | R > 250 MΩ | R > 1000 MΩ | Gigaseal frequency | Confidence interval for gigaseal |
|---|---|---|---|---|---|---|---|
| Rectangular profile (A) | 33 | 14 (9) | 20 (13) | 8 (8) | 5 (5) | 15% | 6%–29% |
| Semicircular profile (B) | 14 | 6 (4) | 8 (6) | 3 (3) | 2 (2) | 14% | 3%–38% |
In the literature, numerous discussions regarding the importance of various device parameters for high-quality seal formation exist, such as the aperture diameter,43 length of membrane protrusion into the microhole,44 smooth edges of the microholes,45 and hydrophilicity of the substrate.43 Additionally, these evaluations were done with different substrate materials and capturing geometries and none with thermoplastic polymers. Although the proposed technology allows for a valid device trade-off strategy based on shorter patching channels having lower seal and series resistance, but relying more on leak subtraction (see ESI†), our experiments indicate that long and smooth patching channels and apertures of approximately 4 μm2 with clean and smooth orifice edges allow for good seal quality. Furthermore, these parameters contribute to an increase in total contact area between the cell membrane and the polymer surface, and therefore, the effective contact area seems to be the most critical prerequisite to look at. To support this hypothesis, the topography of the orifices and the inside of the patching capillaries were thoroughly characterized by atomic force microscopy (AFM). AFM data from channels with rectangular and semicircular profiles are compared in Table 2 and discussed here. The profiles across the patching channels revealed the presence of scallops with an average periodicity of approximately 180 nm for type A profile chips with scallops generated by the Bosch-type reactive ion etching process employed for master mold formation.46 The amplitude and periodicity of the scallops defines the waviness of the surface and dominates the calculated roughness value. To overcome the dominating effect of the scallops and evaluate the surface roughness Rq between the scallops, images were filtered using a cutoff wavelength shorter than the average periodicity between the scallops. After filtering, the surface roughness of the sidewalls of the rectangular and semicircular channels are comparable and in the range of 1 to 2 nm. Therefore, the surface roughness Rq of the sidewall for both samples seems to be determined primarily by the smoothening steps that were the same for both. In general, the inside of the patching channel, including the bottom of the channel and the foil, exhibits very low surface roughness Rq of 1 to 2 nm, and it is also here, between the cell membrane and the polymer, that the seal is assumed to form. These findings suggest that the surface roughness inside the patching channel is a second prerequisite for good seal formation. In contrast, the waviness originating from the scallops does not seem to influence the seal quality. The cell membrane, which has a thickness of only 5–10 nm, is likely to conform to the shape of the surface waviness, which has a periodicity much larger than the thickness of the cell membrane. This effect has been studied before for pipette tip roughness.47 Hence, one can hypothesize that low surface roughness in addition to the patch channel length increases the effective contact area between the cell membrane and the polymer surface and, thus, facilitates the formation of tight seals. The shape of the cross section, either rectangular or semicircular, did not influence the capability of seal formation.
| Device | Rq (nm) | Rq (nm) | Rq (nm) |
|---|---|---|---|
| Bottom of patching capillary | Sidewall patching capillary | Foil | |
| a The image was line-wise corrected by a second-order least mean squares fit in order to remove the (semicircular) shape of the profile. b The surface roughness Rq was calculated from an image after it was filtered so that only shortwave profile components with a cutoff wavelength of λ = 0.18 μm are included. The filtering was done to avoid the influence of the Bosch process-induced scallops on the calculated surface roughness (only relevant for profile A devices). | |||
| Rectangular profile | 2.0 ± 0.3 | 6.5 ± 1.3a | 1.2 ± 0.2 |
| (2.4 ± 0.4)b | |||
| Semicircular profile | 0.7 ± 0.2 | 1.0 ± 0.2a | 1.2 ± 0.2 |
| (0.8 ± 0.1)b | |||
Electrophysiological cell experiments were designed to record activation and inactivation currents in whole-cell voltage clamp mode from voltage-gated sodium ion channels Nav1.7. Sodium channels play an essential role for the generation of action potentials in excitable cells, and they have a key role in pain perception.48 In order to benchmark the performance of the polymer devices, electrophysiology data taken using the devices were compared with data taken using the commercially available QPatch™ system. The cells, the electrolyte solutions and the compounds used for the QPatch™ recordings and for the polymer chip recordings were exactly the same. Raw data were collected and subsequently leak subtracted in order to compensate for the capacitance; Fig. 4a shows an example of a family of sodium activation currents. A +10 mV square pulse of 10 ms from −90 mV prior to depolarization was used to calculate the leak current that was subtracted from the recorded data.49 A description of leak subtraction is provided in the ESI.† Automated P/n leak subtraction50 was employed in the QPatch™ system. Activation currents were obtained after the start of depolarization pulses from −90 mV to 70 mV, while the holding potential was kept at −90 mV for all the experiments. Steady-state inactivation currents were investigated at +10 mV after conditioning potentials ranging from −90 mV to 70 mV were applied for 1000 ms. The resulting current amplitude represents the portion of sodium channels in the activated and inactivated states, respectively. For the polymer devices, the activation threshold was found to be between −50 and −40 mV, and the maximal current amplitude was obtained for voltages between −30 and −20 mV. At positive potentials, the current amplitude gradually decreased as the electrochemical driving force disappeared. The IV curves show good agreement with those obtained using the QPatch™ system. However, IV curves from the polymer device had a tendency to shift towards more negative potentials. Fig. 4b shows two representative IV curves for both systems. The inactivation graph in Fig. 4d shows the gradual transition from a state where the sodium channels were predominantly closed to a state where they were predominantly inactivated, above −50 mV for the polymer devices. The cutoff at −90 mV does not show at which potential the transition starts but is presumed to be at about −100 mV. As mentioned for the activation currents, the IV relationships obtained for the polymer devices, when compared to IVs from the QPatch™ system, had the tendency to shift towards more negative potentials. This shift was attributed to voltage drops across the higher access resistance of the polymer device, which was not compensated for, while series resistance compensation was employed for the QPatch™ recordings. Series resistance compensation for the used HEKA amplifier is tailored to the electrical properties of glass pipettes. Due to their different impedance properties, we did not expect the usual dynamic series resistance compensation to work properly for the polymer devices reported on here. Hence, in order to avoid amplifier oscillations, we did not employ the series resistance compensation for the polymer chip. An additional explanation for the shape of the IV relationship could also be rooted in the expected difference in the mechanical state of the cell membrane in the polymer chip and the QPatch™. Such effects are known to cause shifting of the IV relationships.51 As we shall see in the following, this difference did not influence the pharmacological studies.
 | ||
| Fig. 4 (a) A family of Na+ activation currents in response to depolarization pulses from −90 mV to 70 mV, measured on the polymer device. The protocol used for determining both activation and inactivation is also shown. The membrane potential was held at a holding potential of −90 mV, subsequently shifted to potentials ranging from −90 to 70 mV for 1000 ms, and finally to 10 mV. To the left, the raw current responses are shown, and to the right, the data after leak subtraction. (b) The resulting IV relationship for peak Na+ channel currents. The activation threshold was −50 mV, and the maximal current amplitude was obtained at −30 mV for the polymer device. The threshold was −30 mV with a maximum at −10 mV for the QPatch™. (c) A family of Na+ inactivation currents from the same recording upon the step to 10 mV. To the left, the raw current responses are shown, and to the right, the data after leak subtraction. (d) The inactivation graph for the Na+ channel. At potentials more negative than −80 mV, the channels were predominantly closed, whereas at potentials above −30 mV, they were predominantly inactivated for the QPatch™. For the polymer device, the channels were predominantly inactivated at potentials above −50 mV. | ||
Dose–response experiments were carried out to further benchmark the polymer device with a pharmacological application. Lidocaine inhibition of whole-cell sodium currents was explored in voltage-clamped mode by application of lidocaine concentrations ranging from 30 μM to 1 mM. The ability of lidocaine to bind to sodium channels is state dependent, as lidocaine binds to the sodium channel in the inactivated state only.52 To explore state dependency, the cell was depolarized twice at 0 mV for 100 ms and 20 ms, respectively, from a holding potential of −100 mV with a temporal separation of 15 ms, see Fig. 5a. Depolarization was repeated every 5 seconds. Lidocaine response was studied at the start of the second pulse after the resting interval, during which only a portion of the sodium channels was able to recover from inactivation. Fig. 5b shows a typical recording in the absence of compound where the activation peak 2 was about 80% of the size of the activation peak 1, used as a reference. When lidocaine was applied, reference peak 1 exhibited a current that was almost unchanged, while peak 2 was substantially reduced. Fig. 5c shows activation currents from the same cell before and after 100 μM lidocaine was delivered to the cell. The current–time relationship for the peak sodium currents recorded in response to the first (empty circle) and the second (full circle) depolarization is shown in Fig. 5d. Inhibition is plotted against lidocaine concentration in Fig. 5e. The amplitudes of sodium currents immediately prior to lidocaine application were set to 100%. Complete inhibition was observed at 1 mM lidocaine, while the inhibition was only 20% at 30 μM lidocaine which was also the lowest concentration applied. From Fig. 5d, it can be observed that the inhibition produced by lidocaine was reversible within 60 seconds after its perfusion was stopped. For the polymer device, the half-blocking concentration IC50 was 119 ± 11 μM. This value is in excellent agreement with the IC50 of 152 ± 11 μM found with the QPatch™. The Hill coefficients were also very similar for the two systems.
 | ||
| Fig. 5 (a) Protocol used for determining the concentration–response relationship of lidocaine inhibition on Nav1.7 channels consisting of two depolarizations to 0 mV from a holding potential of −100 mV with an interval of 15 ms. (b) Raw current response from a recording on the polymer device before lidocaine application. The amplitude of peak 2 is about 80% of the amplitude of peak 1. (c) Raw current response for peak 1 and peak 2 before (black) and after (red) 100 μM lidocaine was applied to the cell. (d) Activation currents for Nav1.7 channels for determining the lidocaine inhibition on Nav1.7 channels. The graph shows values of the current amplitudes of peaks 1 and 2 before and after 100 μM lidocaine was applied to the cell. Peak 1 is not affected, and peak 2 is reduced by 50%. (e) Concentration–response relationships of lidocaine inhibition on Nav1.7 channels. Data were fitted with a Hill-type equation. IC50 was 119 ±11 μM (Hill coefficient n = 1) for the polymer device and 152 ±12 μM (Hill coefficient n = 0.9) for the QPatch™. | ||
Conclusions
We have demonstrated electrophysiological recordings on an injection-molded polymer device platform for the first time. The results demonstrate that polymer APC devices can provide whole-cell current responses from voltage-gated sodium channels and permit accurate analysis of drug potency for state-dependent inhibitors such as for the local anesthetic lidocaine. Moreover, the devices exhibit excellent data quality when benchmarked against the commercially available QPatch™ system. Formation of gigaseals (15% of the total cells) between the polymer surfaces and the cell membranes was achieved by making patching capillaries that allowed for a large sealing area. The length of the patching channel together with the low surface roughness on the inside of the patching capillaries were identified as the most important parameters for good seal formation. We attribute this observation to indicate that a large effective sealing area between the cell membrane and the polymer surface is required to obtain tight seals. Most likely, the exceptionally large sealing area required for the polymer patching channels when compared to silica-based channels is necessary to assist sealing due to slow van der Waals type interactions between the relatively low-energy polymer surface and the cell membrane. However, the long patching channels pose an engineering challenge for proper series resistance compensation when compared to traditional glass pipette recordings. Finally, we have successfully combined the simplicity of lateral cornered apertures with the use of injection molding to demonstrate the most cost-effective production and materials platform for APC systems to date. The technological readiness level of the reported material and production platform is high, while the specific design of the intra- and extracellular channels (Fig. 1a) is only aimed at prototyping. Thus, the Y-shaped patching channel constitutes the only essential feature of our microfluidic design, and channels for extra- and intracellular electrolyte solutions could be designed differently for a much smaller unit footprint. For maintaining exactly the same functionality per functional chip unit, six inlet/outlet wells would still be required. However, if we allow shared cell and compound inlets, one chip unit will require only four ports.Acknowledgements
We gratefully acknowledge financial support from the Danish Advanced Technology Foundation through the Advanced Technology Project PILOC (grant no. 061-2010-1) and from the Danish Council for Strategic Research through the Strategic Research Center PolyNano (grant no. 10-092322/DSF).References
- E. Neher and B. Sakmann, Nature, 1976, 260, 799–802 CrossRef CAS.
- B. Hille, Ion Channels of Excitable Membranes, Sinauer Associates, Inc., Sunderland, MA, USA, 2001 Search PubMed.
- F. Lehmann-Horn and K. Jurkat-Rott, Physiol. Rev., 1999, 79, 1317–1372 CAS.
- M. Ackerman and D. Clapham, N. Engl. J. Med., 1997, 336, 1575–1586 CrossRef CAS PubMed.
- F. Ashcroft, Nature, 2006, 440, 440–447 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- A. Wickenden, B. Priest and G. Erdemli, Future Med. Chem., 2012, 4, 661–679 CrossRef CAS PubMed.
- J. Xu, X. Wang, B. Ensign, M. Li, L. Wu, A. Guia and J. Xu, Drug Discovery Today, 2001, 6, 1278–1287 CrossRef CAS.
- W. Zheng, R. Spencer and L. Kiss, Assay Drug Dev. Technol., 2004, 2, 543–552 CrossRef CAS PubMed.
- J. Xu, A. Guia, D. Rothwarf, M. Huang, K. Sithiphong, J. Ouang, G. Tao, X. Wang and L. Wu, Assay Drug Dev. Technol., 2003, 1, 675–684 CrossRef CAS PubMed.
- J. Dunlop, M. Bowlby, R. Peri, D. Vasilyev and R. Arias, Nat. Rev. Drug Discovery, 2008, 7, 358–368 CrossRef CAS PubMed.
- C. Wood, C. Williams and G. Waldron, Drug Discovery Today, 2004, 9, 434–441 CrossRef CAS.
- C. Farre, M. George, A. Bruggemann and N. Fertig, Drug Discovery Today: Technol., 2008, 5, e23–e28 CrossRef PubMed.
- C. Farre and N. Fertig, Expert Opin. Drug Discovery, 2012, 7, 515–524 CrossRef CAS PubMed.
- N. Fertig, R. Blick and J. Behrends, Biophys. J., 2002, 82, 3056–3062 CrossRef CAS.
- M. Asmild, N. Oswald, K. Krzywkowski, S. Friis, R. Jacobsen, D. Reuter, R. Taboryski, J. Kutchinsky, R. Vestergaard, R. Schroder, C. Sorensen, M. Bech, M. Korsgaard and N. Willumsen, Recept. Channels, 2003, 9, 49–58 CrossRef CAS.
- C. Ionescu-Zanetti, R. Shaw, J. Seo, Y. Jan, L. Jan and L. Lee, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9112–9117 CrossRef CAS PubMed.
- D. Martinez, C. Py, M. W. Denhoff, M. Martina, R. Monette, T. Comas, C. Luk, N. Syed and G. Mealing, Biomed. Microdevices, 2010, 12, 977–985 CrossRef CAS PubMed.
- A. Stett, V. Bucher, C. Burkhardt, U. Weber and W. Nisch, Med. Biol. Eng. Comput., 2003, 41, 233–240 CAS.
- K. Klemic, J. Klemic, M. Reed and F. Sigworth, Biosens. Bioelectron., 2002, 17, 597–604 CrossRef CAS.
- X. Li, K. Klemic, M. Reed and F. Sigworth, Nano Lett., 2006, 6, 815–819 CrossRef CAS PubMed.
- B. Xu, Z. Liu, Y. Lee, A. Mak and M. Yang, Sens. Actuators, A, 2011, 166, 219–225 CrossRef CAS PubMed.
- T. M. Suchyna, V. S. Markin and F. Sachs, Biophys. J., 2009, 97, 738–747 CrossRef CAS PubMed.
- C. Miller, Biophys. J., 2009, 97, 687–687 CrossRef PubMed.
- A. Priel, Z. Gil, V. T. Moy, K. L. Magleby and S. D. Silberberg, Biophys. J., 2007, 92, 3893–3900 CrossRef CAS PubMed.
- J. Israelachvili and H. Wennerstrom, Nature, 1996, 379, 219–225 CrossRef CAS PubMed.
- M. Schulz, A. Olubummo and W. H. Binder, Soft Matter, 2012, 8, 4849–4864 RSC.
- V. Tandon, S. K. Bhagavatula and B. J. Kirby, Electrophoresis, 2009, 30, 2656–2667 CrossRef CAS PubMed.
- V. Tandon, S. K. Bhagavatula, W. C. Nelson and B. J. Kirby, Electrophoresis, 2008, 29, 1092–1101 CrossRef CAS PubMed.
- V. Tandon and B. J. Kirby, Electrophoresis, 2008, 29, 1102–1114 CrossRef CAS PubMed.
- J. Seo, C. Ionescu-Zanetti, J. Diamond, R. Lal and L. Lee, Appl. Phys. Lett., 2004, 84, 1973–1975 CrossRef CAS.
- A. P. Golden, N. Li, Q. Chen, T. Lee, T. Nevill, X. Cao, J. Johnson, G. Erdemli, C. Ionescu-Zanetti, L. Urban and M. Holmqvist, Assay Drug Dev. Technol., 2011, 9, 608–619 CrossRef CAS PubMed.
- B. Fermini, Top. Med. Chem., 2008, 3, 1–25 CrossRef.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- M. Matteucci, T. L. Christiansen, S. Tanzi, P. F. Ostergaard, S. T. Larsen and R. Taboryski, Microelectron. Eng., 2013, 111, 294–298 CrossRef CAS PubMed.
- H. Becker, Lab Chip, 2009, 9, 2759–2762 RSC.
- S. Tanzi, P. F. Ostergaard, M. Matteucci, T. L. Christiansen, J. Cech, R. Marie and R. Taboryski, J. Micromech. Microeng., 2012, 22, 115008 CrossRef CAS.
- C. Chen and A. Folch, Lab Chip, 2006, 6, 1338–1345 RSC.
- K. O. Andresen, M. Hansen, M. Matschuk, S. T. Jepsen, H. S. Sorensen, P. Utko, D. Selmeczi, T. S. Hansen, N. B. Larsen, N. Rozlosnik and R. Taboryski, J. Micromech. Microeng., 2010, 20, 055010 CrossRef.
- W. Ong, K. Tang, A. Agarwal, R. Nagarajan, L. Luo and L. Yobas, Lab Chip, 2007, 7, 1357–1366 RSC.
- J. Kutchinsky, S. Friis, M. Asmild, R. Taboryski, S. Pedersen, R. Vestergaard, R. Jacobsen, K. Krzywkowski, R. Schroder, T. Ljungstrom, N. Helix, C. Sorensen, M. Bech and N. Willumsen, Assay Drug Dev. Technol., 2003, 1, 685–693 CrossRef CAS PubMed.
- C. J. Clopper and E. S. Pearson, Biometrika, 1934, 26, 404–413 CrossRef PubMed.
- K. Klemic, J. Klemic, M. Reed and F. Sigworth, Biosens. Bioelectron., 2002, 17, 597–604 CrossRef CAS.
- T. Sordel, F. Kermarrec, Y. Sinquin, I. Fonteille, M. Labeau, F. Sauter-Starace, C. Pudda, F. d. Crecy, F. Chatelain, M. D. Waard, C. Arnoult and N. Picollet-D'hahan, Biomaterials, 2010, 31, 7398–7410 CrossRef CAS PubMed.
- F. Sigworth and K. Klemic, Biophys. J., 2002, 82, 2831–2832 CrossRef CAS.
- R. Abdolvand and F. Ayazi, Sens. Actuators, A, 2008, 144, 109–116 CrossRef CAS PubMed.
- M. Malboubi, Y. Gu and K. Jiang, Microelectron. Eng., 2010, 87, 778–781 CrossRef CAS PubMed.
- F. Yu and W. Catterall, Genome Biol., 2003, 4, 207 CrossRef PubMed.
- J. Dempster, in, ed. Anonymous, Academic Press, London, 2001, pp. 172–225.
- F. Bezanilla and C. M. Armstrong, J. Gen. Physiol., 1977, 70, 549–566 CrossRef CAS.
- D. Schmidt and R. MacKinnon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19276–19281, DOI:10.1073/pnas.0810187105.
- B. Bean, C. Cohen and R. Tsien, J. Gen. Physiol., 1983, 81, 613–642 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Description of the leak current subtraction applied to the raw data obtained from the polymer devices; description of the simulation for the perfusion of lidocaine from one of the lateral channels into the deep channel during the dose–response experiments; description of the fabrication process (table and illustrations); description of the challenges faced during the development of the presented polymer device (and figures). See DOI: 10.1039/c3lc50760b |
| This journal is © The Royal Society of Chemistry 2013 |