 Open Access Article
Open Access ArticleA microfluidic device for dry sample preservation in remote settings†
Stefano Begoloa, Feng Shenb and Rustem F. Ismagilov*a
aDivision of Chemistry and Chemical Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, USA. E-mail: rustem.admin@caltech.edu
bSlipChip Corp., 129 N. Hill Ave., Pasadena, CA 91106, USA
First published on 17th September 2013
Abstract
This paper describes a microfluidic device for dry preservation of biological specimens at room temperature that incorporates chemical stabilization matrices. Long-term stabilization of samples is crucial for remote medical analysis, biosurveillance, and archiving, but the current paradigm for transporting remotely obtained samples relies on the costly “cold chain” to preserve analytes within biospecimens. We propose an alternative approach that involves the use of microfluidics to preserve samples in the dry state with stabilization matrices, developed by others, that are based on self-preservation chemistries found in nature. We describe a SlipChip-based device that allows minimally trained users to preserve samples with the three simple steps of placing a sample at an inlet, closing a lid, and slipping one layer of the device. The device fills automatically, and a pre-loaded desiccant dries the samples. Later, specimens can be rehydrated and recovered for analysis in a laboratory. This device is portable, compact, and self-contained, so it can be transported and operated by untrained users even in limited-resource settings. Features such as dead-end and sequential filling, combined with a “pumping lid” mechanism, enable precise quantification of the original sample’s volume while avoiding overfilling. In addition, we demonstrated that the device can be integrated with a plasma filtration module, and we validated device operations and capabilities by testing the stability of purified RNA solutions. These features and the modularity of this platform (which facilitates integration and simplifies operation) would be applicable to other microfluidic devices beyond this application. We envision that as the field of stabilization matrices develops, microfluidic devices will be useful for cost-effectively facilitating remote analysis and biosurveillance while also opening new opportunities for diagnostics, drug development, and other medical fields.
Introduction
Here, we describe a microfluidic device for dry preservation of biological specimens at room temperature. Stabilization of blood, saliva, urine, or other biospecimens is important for bridging on-site analysis in limited-resource settings—where electricity and equipment are non-existent or scarce—with central laboratories and biological archives. For example, while point-of-care assays allow testing of some biomarkers, these assays usually require complementary follow-up tests that are not available in a portable format. In the case of monitoring antiretroviral therapies for HIV and HCV treatments, for instance, while the initial diagnosis can be performed at the point of care, the follow-up assessments to determine the correct treatment are required.1,2 These tests, which may involve sequencing and viral load measurements, currently need to be performed in a centralized laboratory. In addition to facilitating follow-up analysis, stabilization is crucial for the biosurveillance of emerging infectious diseases, since analytes must be stabilized as samples travel from remote locations to central laboratories.3 Stabilization of analytes also aids biobanking and archiving, as biobanks that store large quantities of clinical samples for future study must preserve specimens for extended periods of time.4–6Generally, analytes are kept stable using the “cold chain,” in which samples are transported in dry ice and stored in freezers. The cost and complexity of low-temperature stabilization limit the applications of biosurveillance and follow-up tests.7 Furthermore, freezers that house specimens require electricity, putting samples at risk of destruction in the event of power failure.8,9 And while the cold chain addresses the question of storing specimens, it does not account for other needs of remote analysis. For instance, many biospecimens must be specially prepared at the moment of collection in order to be studied further. In the case of blood biomarkers, analysis is typically performed on serum or cell-free plasma,10 which must be obtained by trained personnel using specific equipment, such as a centrifuge.
Dried blood spots (DBS) have been used as one alternative to the cold chain. This method involves drying whole blood on filter paper to stabilize the sample. Recovery is performed by removing a portion of the blood spot with a hole punch and using a combination of solvents and buffers to elute the sample from the resulting punched-out disc. This method does not require sample preparation or specialized equipment at the moment of sample collection, and has been used for large-scale neonatal screening and biosurveillance.11 However, the use of DBS for quantitative analyses presents some challenges and limitations, as results are dependent on the hematocrit and on where the punch is taken on the paper,12 and samples can be contaminated by open-air exposure or cross-contaminated by the biopsy punch tool. Furthermore, long drying times and external conditions such as humidity may compromise the stability of analytes: for this reason, most protocols require freezing of DBS for long-term storage.13,14 Finally, serum or cell-free plasma, rather than whole blood, are often the matrices of choice for clinical tests; to implement DBS, specific elution procedures need to be optimized for purifying analytes and avoiding interference from residual components in blood. Although dried plasma spots can address some limitations of DBS, they require trained personnel to perform plasma separation.
Chemical stabilization matrices offer an alternative to the cold chain and DBS, but until now, they have not been compatible with devices for resource-limited settings. A number of matrices have been developed, many of which rely on natural chemistries used by living organisms for self-preservation.15 Several commercially available stabilization matrices (Biomatrica, GenTegra, etc.) preserve samples in the dry state. Drying inhibits analyte degradation linked to enzymatic activity and slows down reactions that are activated by light or heat.16,17 These matrices do not necessarily solve the problem of sample preparation, but they do allow long-term storage of DNA and RNA at room temperature. Current protocols for using these stabilization matrices are suitable for laboratory use, but drying the samples typically requires vacuum conditions or external devices (e.g., lyophilizer, pumps, vacuum concentrators) to control humidity, as well as experienced personnel to process the sample.18,19 Portable devices have been proposed,20 but these usually involve electricity and long drying times (from a few hours to overnight). Alternatively, drying can be done without the use of external equipment by exposing the sample to the environment, but airborne contaminants may come into contact with the sample and drying times are dependent on external conditions such as humidity. Moreover, leaving the tubes or titer plates open during drying may lead to cross-contamination between different samples: to solve this problem, one needs to use porous seals (which increase drying times) or a dedicated device20 for preventing aerosol-driven cross-contamination.
An approach that offers reliable preservation, avoids contamination, allows quantitative measurements, is compatible with sample preparation, and can be executed by minimally trained users is needed. Herein, we describe a device that meets these requirements and can be used with commercially available sample preservation matrices.
Experimental
Structure of devices
The device was based on SlipChip technology.21 Devices described in this paper comprised three different subassemblies (Part 1, Part 2, Part 3, hereafter referred to as P1, P2, P3), made of a combination of poly(methyl methacrylate) and polyethylene terephthalate layers. A full description of the layer configuration is shown in the ESI† (Fig. S1 and S2). The central portion (P2) included a Polypropylene Membrane Filter, 0.45 pore size (Sterlitech Corp., Kent, WA, USA). For plasma experiments, the top part (P1) also included a Vivid Plasma Separation Membrane (Grade GR) (Pall Corp., Port Washington, NY, USA).Production of devices
The devices described in this paper are prototypes, but they were designed in consultation with manufacturing partners to be compatible with mass production (see Fig. S3 in the ESI† for different prototypes). At the device development stage, we used laser cutting since it is suitable for prototyping and allows great flexibility in design and materials selection. This technique allowed us to produce batches on the 100-device scale. However, since laser cutting requires multiple device layers, we also designed the device so that it would be easily adaptable to large-scale manufacturing techniques, such as injection molding. We make the assumption that injection molding would be an inexpensive process to mass-produce these devices, but full economic analysis of the manufacturing process is outside of the scope of this manuscript. The geometry of each layer was designed using CAD software, and the pattern was used to produce through-holes or channels in a polymeric sheet of desired thickness. This design was used by ALine Inc. (Rancho Dominguez, CA, USA) to fabricate devices using laser cutting and lamination for rapid prototyping. Layers were then aligned and bonded by lamination, using pressure sensitive adhesive or thermal adhesives. Clamping systems were custom-designed and produced either by using 3D printing (by Symbient Product Development, Vista, CA, USA) or by laser cutting (by ALine or at the Jim Hall Design and Prototyping Lab at the California Institute of Technology). Pumping lids were produced using laser cutting of poly(methyl methacrylate) in the Jim Hall Design and Prototyping Lab. Parts were bonded together using adhesive transfer tapes (3M 468MP purchased from Uline, Pleasant Prairie, WI, USA).Device assembly
The three layers were held together using a set of custom-made clamps. The final stack was assembled in this order: layers P1, P2, P3, a flexible pad, and a bottom clamp. A layer of lubricant (High vacuum grease, Dow Corning) was used between layers P1, P2, and P3. We did not observe any effects from the presence of this lubricant layer on the biological assays described in this work (such as RNA purification and RT-qPCR). The flexible pad consisted of PDMS (silicone rubber with adhesive back, 1.5 mm thick, McMaster Carr). The same PDMS pad was cut using round punch tools to produce the o-rings that create a tight seal during loading. For more information, please refer to Fig. S1 (ESI†).Device operation
P2 is the moving part of the device: by slipping P2 relative to P1 and P3, the device can be placed in three different positions, each with a specific function. P2 begins in position 1 (“Loading”), in which the sample can be injected into the device. One slip brings P2 to position 2 (“Drying”), in which the sample is split into aliquots, disconnected from the inlet, and exposed to the desiccant that has been pre-loaded into the bottom portion (P3). The slip direction and distance are guided by the clamping system. The user pushes P2 until it is completely embedded in the device, which prevents accidental over-slipping. A second slip brings P2 to position 3 (“Recovery”), in which drying stops and the sample aliquots are connected to through-holes for rehydration and re-collection. This second slip can be performed only with a special “slipping tool,” which was custom-made, in order to avoid accidental slipping during storage and/or transport, or by untrained users. After sample re-collection, the device can be slipped back to position 2 for further storage.Loading experiments
Loading was evaluated by placing a solution at the inlet hole and placing the lid on the device, which created a tight seal. This step generated extra pressure that pushed the sample into the device. The solution was injected into the microwells and channels only due to pressure, and not due to capillary action through the polypropylene membrane: since the membrane’s pores are hydrophobic and small (0.45 μm), only air passes through at the pressures used for loading, allowing the membrane to serve as a barrier between the sample and drying agent. Of course, one may use wicking to fill the channels as well, but this was not the approach we took here. The lid was designed to remain on the device during drying, storage, and transport. The device can also be loaded by using a standard pipettor, but we emphasize that a pipettor is not required. Solutions that were successfully loaded into the device included: DI water; DI water with 0.4 mM of BSA (Sigma Aldrich); solutions of glycerol (Alfa Aesar) in water up to 85% w/w; SsoFast™ EvaGreen® Supermix (Bio-Rad); pooled normal human plasma (George King Biomedical, Inc.); and human whole blood in Acid Citrate Dextrose (ACD) (Zen-Bio Inc.). A green food dye was added to the solutions in some experiments to enhance the contrast in photographs. Videos and pictures of filling experiments were taken with a camcorder (Sony Handycam HDR-CX190). For experiments that estimated volume, images were analyzed using ImageJ Software to approximate the fraction of each well containing the solution. In addition, we implemented a “venting well” strategy in the device through which extra air can evacuate in the event of partial filling (see Fig. S4 in the ESI† for full schematic drawings and mechanism description).Drying experiments
Devices were loaded as described above. After loading, the central portion (P2) was moved to the drying position. In some cases, the device was loaded using a pipettor instead of the lid/o-ring system to allow a better view of the drying dynamics in the channels. We tested both loading strategies and they showed comparable results. For drying experiments, P3 was pre-loaded using a desiccant, typically molecular sieves, 4A, 1–2 mm beads (Alfa Aesar). Other desiccants were successfully tested (CaCl2, CaSO4, and molecular sieves with smaller diameter).Slipping activates drying by putting the sample in vapor contact with the molecular sieves. A polycarbonate porous membrane embedded in P2 prevents contact between sample and desiccant at this stage. Drying is selectively activated: P2 and P3 both have a series of channels that do not overlap in the “Loading” and “Recovery” positions. Only the “Drying” position aligns these channels, thus creating the vapor contact that drives the drying. Drying dynamics and timescale were characterized by taking videos or photographs of the channels using a camcorder or a camera.
Recovery experiments
Samples were loaded and dried in the device using the techniques described above. Both the pipettor and lid strategies were used to load the devices used for the recovery experiments. When a lid was used for the loading, it was removed just before starting the recovery procedure. This ensured that the recovery holes were enclosed during all handling and storage to avoid possible contamination. We designed the device so that slipping to the recovery position aligns the wells with the recovery holes on P1. Each well can be independently controlled, so the recovery can be total (all wells) or partial (only one or more wells). In the experiments below, rehydration was performed by injecting elution buffer (Buffer AVE, Qiagen) into the wells containing dry sample and waiting for it to dissolve. This process can be accelerated by pipetting the solution back and forth in the wells using a pipettor. Sample re-collection was performed by aspiration with a standard pipettor. Washes can be performed by simply re-injecting elution buffer into the wells and repeating the re-collection procedure.The recovered solution can be handled using the pipettor and analyzed with conventional techniques. We tested the recovery efficiency using water containing green food dye, Phosphate buffered saline (PBS) with Alexa Fluor 488 (Sigma Aldrich), or CdTe/Cds core/shell Quantum Dots (QD). QDs were synthesized following a published method,22 with core size between 2.5 and 3 nm and stock concentration on the order of 2.6 μM.
Plasma experiments
Human whole blood in Acid Citrate Dextrose (ACD) (Zen-Bio Inc.) was spiked with inactivated HIV-1 viral particles (AcroMetrix® HIV-1 Panel copies per mL, Life Technologies). For each experiment, a 200 μL drop of blood was placed on the plasma filtration membrane, and the lid was placed on the device. The pressure generated by the lid was enough to filter the plasma and to load the separated plasma into the device wells. To help visualization of the separated plasma, in some experiments green food dye or Alexa Fluor 488 (20 mM final concentration) were added to the starting blood sample. After complete loading, the plasma was either re-collected directly (to evaluate the filtration process) or treated using the same process as was used for the other samples (dried down, rehydrated, and re-collected). After re-collection, HIV-1 RNA was purified from the plasma samples using QIAamp MinElute Virus Spin Kit (Qiagen).PCR amplification
RNA concentration was measured using reverse transcriptase quantitative polymerase chain reaction (RT-qPCR). Primer sequences for HIV-1 viral RNA were selected from a previous publication: GRA ACC CAC TGC TTA ASS CTC AA; GAG GGA TCT CTA GNY ACC AGA GT.23 To amplify HIV-1 viral RNA, each 100 μL of RT-PCR mix contained the following: 50 μL of 2× EvaGreen SuperMix, 1 μL of each primer (10 μmol L−1), 2.5 μL of SuperScript® III Reverse Transcriptase, 37.5 μL of RNA template solution, and 0.5 μL of nuclease-free water. The amplifications were performed using an Eco Real-Time PCR System (Illumina). To amplify the RNA, an initial 15 min at 50 °C was applied for reverse transcription, then 2 min at 95 °C for enzyme activation, followed by 40 cycles of 30 s at 95 °C, 30 s at 55 °C, and 45 s at 72 °C. Melting curves were acquired after the final cycle by bringing the samples to 95 °C for 15 s, then to 53 °C for 15 s, and finally, back to 95 °C.RNA storage experiments
Two different classes of purified RNA samples were used for storage tests: Control RNA 250 (Ambion) and HIV-1 RNA purified from the AcroMetrix® HIV-1 Panel copies per mL (Life Technologies). Purification was performed using the QIAamp MinElute Virus Spin Kit (Qiagen).Samples were diluted to the desired concentration (80 ng μL−1 for control RNA, 375 copies per microliter for HIV-1 RNA) and mixed with a chemical stabilization matrix for RNA (RNAStable, purchased from Biomatrica, Inc.).
Several aliquots of these solutions were injected into the device and dried for stabilization. The devices were then stored at 50 °C for up to 5 weeks. In parallel, several aliquots of the same solution were used as controls. Controls included aliquots stored in a −80 °C freezer and aliquots stored in the liquid state at 50 °C in microcentrifuge tubes. Each storage condition was performed in triplicate. At each time point, the sample was recovered from the devices using the rehydration and re-collection procedures, and three washes were performed for each well. All solutions were then diluted so that the total volume of each aliquot was 100 μL.
Evaluation of RNA stability
Stability of the control RNA samples was tested by electrophoresis using a 2100 Bioanalyzer (Agilent Technologies, Inc.) with the RNA 6000 Nano Kit. Stability of the HIV-1 RNA samples was tested using RT-qPCR as described above.Results and discussion
The general approach we pursued in this work was to bridge limited-resource settings and laboratories by creating a device that preserves samples that have been collected by untrained users and allows reliable recovery of these samples in a central facility. To satisfy the need for devices to be operated by untrained users, we developed a device with three simple steps (Fig. 1A–C) for handling and storage of samples: first, the user places the collected sample at the device inlet; second, the user places the lid on the device, which initiates automated loading of the sample; and third, the user slips the device, which splits the sample into aliquots and activates drying (Fig. 1D). At this stage, the device is ready for shipment or storage, and no further manipulation of the device by the untrained user is required.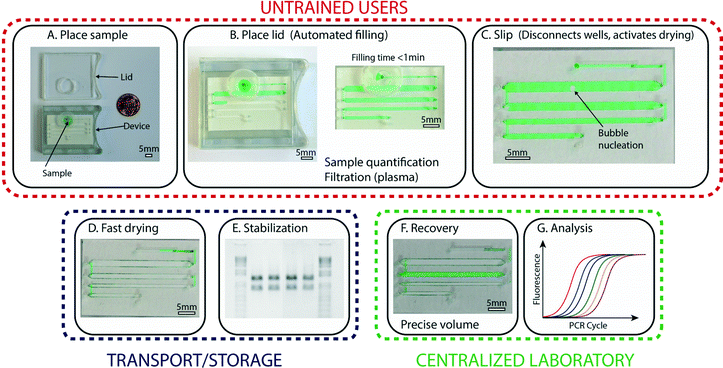 | ||
| Fig. 1 An overview of the operations and capabilities of the device. (A) A photograph representing the first step, in which an untrained user places the collected sample (here, a green aqueous solution) at the device inlet. (B) A photograph of the device after the lid has been placed (left), which activates the automated filling process that enables precise volume quantification (right). Sample preparation, such as plasma filtration, is integrated in this step. (C) A zoomed-in photograph of the device after the user has performed the third step: a slip that disconnects the aliquots and activates drying. (D) A photograph showing the device after complete drying. (E) A photograph of an agarose gel (inverted intensity) showing clear bands and no degradation, indicating that samples stored in the device remain stable without the need for refrigeration. (F) A photograph of the device with one rehydrated well (green). Aliquots can be re-collected independently, so part of the sample may be kept dried for further storage. (G) A graph showing a standard qPCR profile; recovered samples can be analyzed using traditional methods and instruments. Scale bars are 5 mm in all photographs. The loading process is also shown in Video S1, ESI.† | ||
The sample, which stays fully enclosed in the device, remains stable and in the dry state during transport and/or storage (Fig. 1E). At the desired time, a trained user can perform a second slip of the device with a special “slipping tool” and rehydrate one or more of the aliquots by injecting water into the wells (Fig. 1F) as detailed below. These rehydrated aliquots can then be re-collected using a standard pipettor and are ready for further analysis or purification (Fig. 1G). If not all aliquots are needed for analysis, aliquots that are not rehydrated can be stored long-term (e.g., for archival purposes). Although the devices described in this paper are prototypes, they were designed in consultation with manufacturers to ensure compatibility with mass production.
Device loading
In order to ensure simple and robust loading of the sample, we implemented two main design features that enable several capabilities. The first design feature is a “pumping lid,” which is a combination of an o-ring and lid that limits the maximum pressure applied during loading (Fig. 2A). The pumping mechanism used for sample loading is based on the use of a top lid that contains a cavity. When the lid is placed on the device, a tight seal is created with the flexible o-ring. This action compresses the air in the cavity and produces extra pressure, which serves as the driving force for loading the sample into the device. The maximum applied pressure is dependent only on the lid geometry, and can be described as: ΔPMAX = VO-RING/VCAVITY. This feature prevents the user from accidentally applying excessive pressure, and may be useful for a wide range of microfluidic devices. In the experiments described in this paper, we tested various lid geometries, and the applied pressures ranged between 20 and 76 mbar.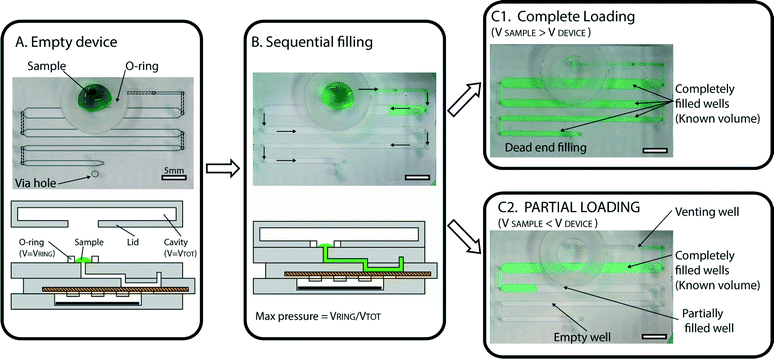 | ||
| Fig. 2 Automated loading with precise volume quantification. (A) A photograph (top) and a schematic drawing (bottom) of an empty device, with a sample (green solution) placed at the inlet, with a flexible o-ring surrounding the inlet. Lines have been added to the photograph to illustrate the geometry (dashed lines outline ducts in P1 (one of the device’s three major parts, or subassemblies), and solid, light gray lines outline wells in P2). A light gray circle was used to indicate the via hole used for dead-end filling, described in more detail in the ESI† (Fig. S5). (B) Photograph and schematic demonstrating that as the lid is placed on the device, it forms a tight seal with the o-ring, creating pressure and initiating loading. Wells are loaded sequentially (following the path indicated by arrows) so that each well is completely filled before the solution enters the next well. (C1) A photograph of a fully loaded device, in which the sample volume (55 μL in this case) is larger than the total device capacity (50 μL). All wells are filled, and loading has stopped automatically. (C2) A photograph of a partially loaded device, in which the sample volume (25 μL in this case) is lower than the total device capacity. Loading stops automatically as soon as air enters the first well (“venting well”), allowing quantitative measurements to be obtained by counting full wells. | ||
The second design feature crucial for loading involves a combination of dead-end and sequential filling that together allow precise volume quantification. Dead-end filling is a strategy previously applied for the loading of SlipChip devices,24 in which the fluid (e.g., oil or air) originally present in the wells or channels of the device is evacuated through the gap between the device layers as the sample is entering the wells or channels. In the cases previously reported, the “sealing pressure” (defined as the maximum pressure that can be used for loading the device without the sample entering the gap) is inversely proportional to the gap between the device layers (see model described in ref. 24).
In the experiments described in this work, the air originally present in the wells of the device was pushed through the membrane pores and evacuated through a via hole placed in the proximity of the last well. A schematic description of this mechanism is provided in the ESI† (Fig. S5). We emphasize that the liquid sample is injected into the device wells and channels only, and does not penetrate the porous membrane that serves as the bottom of the device’s wells. This configuration increases the range of loading pressures, as the sealing pressure24 in this case is controlled by the size of the pores in the membrane. As an example, when loading blood plasma (surface tension 50 mN m−1) into a well filled with air and using a membrane with a pore size of 0.45 μm, the sealing pressure for the membrane is ~2 bar (roughly 4 times more than the sealing pressure for a 1 μm gap between plates). This value is significantly higher than the pressures generated by the pumping lid, and we did not observe any permeation of liquid through the porous membrane. Choosing a membrane with an even smaller pore size would further increase the sealing pressure, making the device resistant to even higher loading pressures.
Filling stops automatically when all the wells are filled with the sample, and any extra volume placed on the device inlet is not injected into the device, providing an effective control on the maximum loaded volume. With sequential filling (Fig. 2B), each well fills completely before the liquid reaches the following well. If the input volume is lower than the device capacity, loading stops automatically as soon as the entire sample has been injected (Fig. 2C). Any extra air that follows the end of the liquid sample slug can be vented through an extra “venting well” in the storage layer to stop filling. The original volume of sample can then be quantified by counting the number of full wells or by taking a picture of the device after filling and analyzing it using ImageJ Software to approximate the fraction of each well containing the solution. Repeated loading experiments demonstrated that filling is consistent with these predictions (Fig. 3).
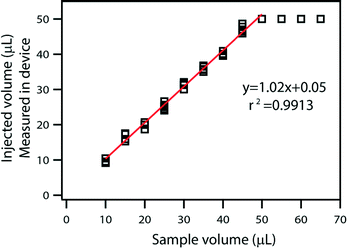 | ||
| Fig. 3 A plot comparing injected volume as measured by the device with the original sample volume. The relation is linear below 50 μL, which is the maximum device capacity. | ||
These design features obviate the need for users to precisely control pressure and input volume, both of which are challenging operations without proper training and equipment. Knowledge of the input volume is critical for calculating concentrations of analytes in the biofluid being preserved and then analyzed. Moreover, we demonstrated that a wide range of solutions can be loaded using this approach (see Table S1 and Video S1 in the ESI†). The large flexibility in pressures, ensured by the modified dead-end filling mechanism described above, made it possible to rapidly drive fluids with the viscosity of water (with velocities up to 10 μL s−1) through the device. This loading approach was also suitable for loading samples with a wide range of properties, including viscosities up to 110 mPa s (85% w/w of glycerol in water) and surface tensions down to 7 mN m−1 (0.4 mM BSA). For this BSA solution, the calculated sealing pressure was 0.31 bar.
In this work, the matrix was pre-mixed with the sample prior to injection into the device. The device has been designed to be compatible with a variety of matrices, even those not yet developed. The preferred method of incorporating a specific preservation matrix into the device would depend on the properties of the particular matrix and its dissolution kinetics.
Aliquoting and drying
Once loading is complete, aliquoting and rapid drying are initiated by a single slip that is controlled by an internal mechanism to prevent over-slipping. We designed the device so that slipping to the drying position (i) disconnects the wells from the inlet and isolates all the wells from one another, and (ii) activates drying by exposing the sample in the wells to the pre-loaded desiccant in P3, one of the device’s three major parts or subassemblies (see Experimental). This slip also embeds the central layer (P2) in the device so that it cannot be inadvertently un-slipped during shipment or storage. The slipping step is shown in Video S1, ESI.†After the user has performed the slip, the drying process in each well begins with the nucleation of an air bubble that grows as evaporation occurs (Fig. 4). We relied on a well-understood idea25–28 that by reducing length scales over which evaporation takes place, vapor transfer can be accelerated. This process is rapid also because evaporation takes place through the porous membrane that serves as the bottom layer of the wells. The surface to volume ratio in this case was 3.3 mm2 for each μL. Drying times for the different solutions tested were typically on the order of 30–45 min for a total volume of 50 μL (Fig. 4E) (see Fig. S6 and Video S2 in the ESI† for drying dynamics). As a comparison, the same volume in a microcentrifuge tube required up to 12 hours to be evaporated in a laboratory environment. These timescales were suitable for the storage experiments shown in this paper, but this framework offers a wide range of drying times that can be adjusted by changing device parameters such as the volume of wells, surface-to-volume ratio, type of desiccant, and membrane properties (such as pore size, fractional open area of the membrane, and membrane thickness). After drying is complete, the presence of sample in each well can be confirmed by the presence of solid residuals (Fig. 4C and D), which can be used as a strategy to detect empty wells in the event of partial filling.
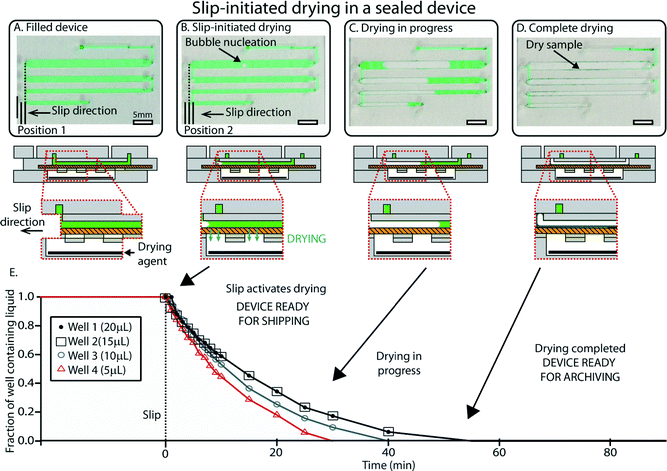 | ||
| Fig. 4 An overview of slip-initiated drying. (A) A photograph (top) and schematic drawing (bottom) showing the device after loading. In this case, the device was loaded with a pipettor instead of using the “pumping lid” to ensure unobstructed visualization of the sample wells. (B) A photograph and a schematic drawing of the device once it has been slipped. The slip disconnects wells and allows vapor contact between the sample and the desiccant (black). Evaporation starts by nucleation of an air bubble in each well. (C) A photograph and a schematic drawing showing drying in progress. The bubbles grow due to progressive loss of water caused by evaporation. (D) A photograph and a schematic drawing of the device once drying is complete and the sample is stabilized (green sample residuals are visible in the wells). (E) A plot showing the fraction of the wells containing liquid over time for a representative device. See ESI† (Fig. S6) for full schematic drawings and operation of the device. | ||
Recovery
A second slip allows a trained user to selectively recover stored aliquots through the device’s sterile pathways. As mentioned above, the device is designed so that the central layer is completely embedded in the device during storage or transport to prevent accidental slipping during transit or by untrained users. A special slipping tool (Fig. 5A and B) is required to perform this second slip. When the device is in the recovery position, vapor contact with the drying agent in P3 is blocked; in our experiments, no evaporation was observed. In addition, each well is connected with two through-holes that have not been used during loading and are thus free from contaminants. These holes were designed to match the size of pipettor tips, ensuring robustness of the recovery step. A pipettor can be used to inject water into the wells to dissolve the dry sample (Fig. 5C). Rehydration times for standard solutions ranged from 1 to 20 min, depending on the solubility of the solid residual and on the channel geometry. These timescales can be further reduced by using the pipettor to perform back-and-forth injection of the solution, thus speeding up the dissolution (see Video S3 in the ESI†).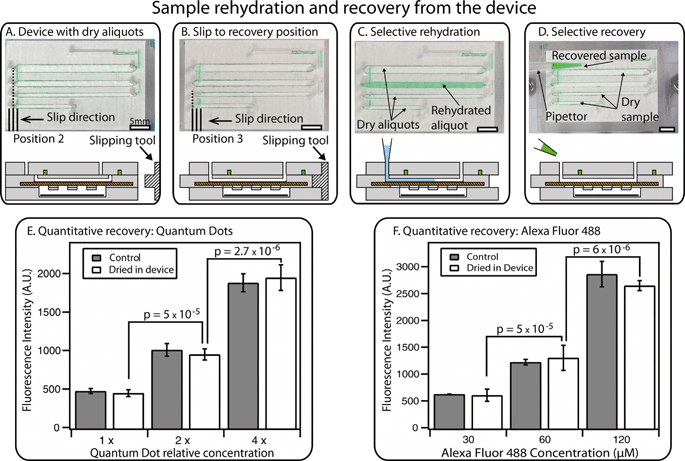 | ||
| Fig. 5 An overview of sample rehydration and recovery. (A) A photograph (top) and a schematic drawing (bottom) of the device containing dry sample after 12 hours, along with a drawing of a separate “slipping tool” used for slipping to the recovery position. (B) A photograph and a schematic drawing showing a device that has been slipped to the recovery position using the special slipping tool. (C) A photograph and a schematic drawing of the device after selective rehydration of a single well with a pipettor. (D) A photograph and a schematic drawing showing selective recovery of the sample from a single rehydrated well (other aliquots remain dry for further storage). Full schematics and operation of the device in A–D are available in the ESI† (Fig. S7). (E) A graph showing quantitative recovery of quantum dot solutions after drying has taken place in the device. (F) A graph showing quantitative recovery of Alexa Fluor 488 solutions after drying has taken place in the device, compared to the original solution (“control”). In E and F, brackets indicate p values, the null hypothesis being that the two concentrations were the same in each case. Error bars represent the 95% confidence interval (n = 3 for each condition). See Fig. S8 and S9† for calibration curves, Tables S2 and S3† for statistical calculations, and Video S3† for demonstration of rehydration and recovery in the ESI†. | ||
After rehydration, the sample can be retrieved from each well by aspirating with a standard pipettor (Fig. 5D). Using this procedure, we re-collected liquid solutions with the same concentration as the injected sample, demonstrating that quantitative measurements can be obtained from stored aliquots (Fig. 5E and F). We evaluated the reliability of recovery by injecting solutions containing either fluorescent molecules (Alexa Fluor 488) or nanoparticles (CdTe/Cds quantum dots) (Fig. 5E and F). After drying and rehydration, the solutions were re-collected from the wells of the device. This method allowed quantitative measurements: solutions with different concentrations were reliably distinguished, showing clear difference between solutions with a 2-fold difference in dilution (all p values were 5 × 10−5 or less, the null hypothesis being that the two concentrations were the same in each case). No more than ±10% difference was observed when comparing the signal from the rehydrated aliquots with the starting stock solution (see Tables S2 and S3 in the ESI†). If needed, liquid residuals in the channel can be washed out by injecting more water into the well and re-collecting the solution; the washing step can be repeated additional times if necessary.
Since each well is individually retrievable, a trained user can rehydrate one or more wells and perform either partial or total recovery of the original sample. If only a portion of the total number of wells is recovered, the device can be slipped back to the drying position for further storage.
Integration with plasma processing
We have next shown that this device can be integrated with a plasma separation module without changes to the device’s architecture or operation. We introduced a porous membrane (Vivid Plasma Separation Membrane, Grade GR) into the P1 subassembly to filter whole blood and obtain cell-free plasma (Fig. 6A). After filtration, separated plasma flowed directly into the device wells (in practice, this eliminates the need for involvement from untrained users). The same “pumping lid” approach described earlier (Fig. 2) was used here both for plasma separation and device loading. The device is designed to process as much as 200 μL of whole blood, a volume that can reasonably be collected by finger pricks. This input volume was suitable for complete loading of all the devices that were tested (Fig. 6B). To aid visibility of plasma, dye can be added to whole blood, as was done in Fig. 6B with Alexa Fluor 488, and in Fig. S10 in the ESI,† in which we used a green food dye. In the future, the membrane itself may be treated with a visible dye. In addition, visualization and thus quantification may also be enabled by the use of commercially available “indicating drying agents” that change color in the presence of humidity. In this approach, only the drying agent in close contact with the wells containing sample would change color, allowing one to count the number of filled wells. While we did not use such a drying agent in this work, we expect this approach should be compatible with this device. Additionally, the plasma filtration membrane may eventually be treated directly with a preservation matrix so that the matrix will automatically release during the filtration step. | ||
| Fig. 6 Integration of the sample preservation device with a plasma filtration module. (A) Schematic drawings that show the components of the plasma filtration module (top) and the process for obtaining cell-free plasma with this module (bottom). The pumping lid is used both to obtain plasma and to load the device. (B) Two photographs of the device after the lid has been closed and plasma has been successfully obtained from whole blood and loaded into the wells. Whole blood was spiked with 20 mM of Alexa Fluor 488 to aid visualization of the separated plasma. Left: Bright-field image, showing the red and white blood cells (residuals) at the device inlet and the plasma loaded in the wells. Right: Fluorescence image of the device (for improved visualization of the plasma within the wells). (C) A graph comparing quantification results of HIV-1 RNA from plasma extraction with a traditional centrifuge (left) and extraction using the device’s plasma filtration module (right). p Values are in the same range for both techniques: *** indicates a p value of less than 0.001, and n.s. indicates a p value higher than 0.05 (no significant difference) (see Table S4 in the ESI† for full details of statistical analysis). The null hypothesis for p values was that both concentrations were equivalent. Error bars represent the 95% confidence interval (n = 3 for all experiments, except for the centrifugation experiment at the lower concentration, for which n = 5). | ||
Using this filtration approach, we successfully separated plasma from whole blood that was spiked with HIV-1 viral particles. We were able to purify HIV-1 RNA from these plasma samples. Compared to plasma controls separated by centrifugation, the recovery rate of HIV-1 RNA after plasma filtration with this device was always more than 60%. We used different concentrations of HIV-1 particles and found that the p values were comparable for the control samples and the device (1.2 × 10−4 for the centrifuge and 1.6 × 10−4 for the device) (Fig. 6C) (see Table S4 in the ESI† for p value calculations).
Since there is currently no chemical matrix available for stabilization of HIV-1 RNA in plasma, we did not test the stability of preserved RNA in plasma. However, we verified that the device can successfully dry normal plasma. We demonstrated that injected plasma can be dried down, stored, and re-collected as described earlier (see Fig. S10 in the ESI† for details). Rehydration times for plasma were longer (on the order of 15–20 min) than for the previous experiments, so we typically injected the water back and forth with a pipettor to speed up the dissolution. One critical parameter is the speed at which the plasma samples are dried. Since, in the dry state, blood plasma produces more solid residuals than purified solutions, there is a risk that the wells may get clogged when the sample dries. The device’s fast drying times avoided this problem by distributing residuals across the walls of the wells, allowing successful rehydration of all wells (Fig. S10 in the ESI†). In contrast, when we tested the drying of plasma through the porous membrane using the P2 subassembly alone (not integrated with the device) and no desiccant to drive the drying process, times for drying were on the order of 1 to 2 hours, and the solid residuals tended to accumulate in one spot in the wells and block the fluidic path for rehydration.
Sample stabilization
We validated the preservation and recovery capabilities of the device and its compatibility with commercially available matrices by using control RNA and HIV-1 RNA. We chose RNA because it is less stable than DNA29 and because it is relevant for quantifying HIV-1 viral load. Recent work30 has shown that purified RNA stored dry in a solid-phase extraction cartridge (without stabilization matrices) remained stable for about one week at 37 °C, and degradation was observed for longer storage times. In the following experiments, we introduced RNA buffer into the device using the pumping lid mechanism, slipped to initiate drying, and placed the device in an incubator at the test temperature.To test general RNA stability, we used a mixture of control RNA 250 (Ambion) and a stabilization matrix (RNAStable, Biomatrica). Electrophoresis experiments performed with an Agilent 2100 Bioanalyzer showed that the 80 ng μL−1 of control RNA aliquots stored dry in the device for 4 days at 50 °C did not show any detectable degradation, indistinguishable from the controls stored in a freezer at −80 °C (see Table S5 and Fig. S11 in the ESI† for full details, evaluated semi-quantitatively using an approach similar to the one described previously).31,32 As a comparison, degradation of control RNA was obvious in aliquots stored in the liquid state at 50 °C, evidenced by the presence of short products that formed a smear pattern (Fig. 7A).
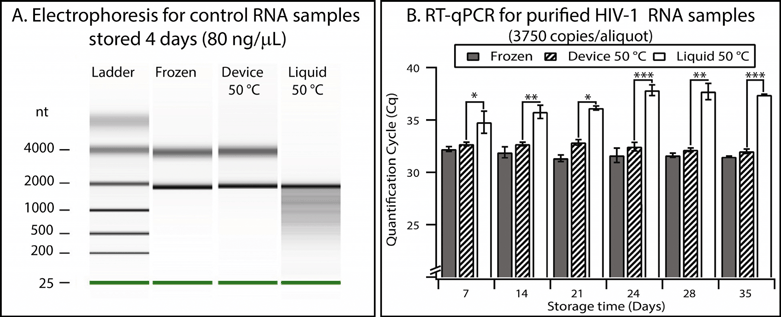 | ||
| Fig. 7 Validation of the device using control RNA and HIV-1 RNA. (A) Densitometry plot showing electrophoresis results from control RNA (80 ng μL−1) mixed with a stabilization matrix (RNAStable, Biomatrica) and stored for four days under different conditions. (B) A graph showing the results of quantitative analysis of purified HIV-1 samples performed with RT-qPCR. Error bars represent the 95% confidence interval (n = 3). Stars are used to indicate p values: * if the p value is less than 0.05, ** if less than 0.01, and *** if less than 0.001. See Table S7 in the ESI† for calculations. | ||
Next, we showed that the device also allowed stabilization and quantitative recovery of HIV-1 RNA. RNA was purified from inactivated HIV-1 viral particles (see Experimental). Aliquots (10 μL) containing ~3750 copies of RNA and mixed with the preservation matrix were stored in one of three conditions: (i) frozen at −80 °C, (ii) in the device in the dry state at 50 °C, or (iii) in the liquid state at 50 °C in microcentrifuge tubes. Experiments at elevated temperatures are typically used to evaluate long-term stability of samples in a reasonable amount of experimental time; furthermore, such experiments reflect temperature fluctuations to which samples may be exposed during shipping when temperature is not controlled. Since this RNA was at a low concentration (375 copies μL−1), RT-qPCR was used to evaluate stability and recovery rate. Quantification cycles (Cq) were measured for each storing condition and were compared with controls (aliquots stored at −80 °C) (Fig. S12 and S13 and Table S6†). Liquid samples stored at 50 °C showed significant degradation after as few as 7 days (p value = 0.04), and this degradation became more prominent over time (p value after 35 days was 6.1 × 10−8) (see Fig. 7B and Table S7 in the ESI†). When comparing the frozen controls and those samples stored in the device at 50 °C, we observed a minor difference in Cq values (0.5 cycles on average, n = 18), but no substantial change (always less than 0.3 cycles) in Cq was observed over a 5-week period. Stability of the RNA at 50 °C over a 5-week period in this “accelerated aging” test17,33 implies that the RNA may be stable at room temperature in the device for as long as 8 months.17,33
Conclusions
We developed and validated a device for using chemical stabilization matrices to preserve samples in the dry state. This device is portable, compact, and self-contained, so it can be transported and operated by minimally trained users even in low-resource settings. An integrated plasma separation approach further increases the device’s relevance to resource-limited settings. In addition, while so far we have tested prototypes manufactured on the scale of 100 devices, the device architecture, structure, and operation are compatible with mass production to enable large-scale manufacturing. The features of this device, including dead-end and sequential filling, driven by the pumping lid, as well as the modularity of this platform (which facilitates integration and simplifies operation) would be applicable to other microfluidic devices beyond this application. The described device allows for subsequent recovery of preserved samples in a centralized laboratory, enabling quantitative measurements and volume quantification over a wide range (5–50 μL in the current version, and scalable to up to 1 mL). We have demonstrated that this device is compatible with standard laboratory techniques, and we anticipate that it will be made compatible with SlipChip-based microfluidic strategies for biological analysis to enable rapid point-of-use sample processing and analysis with preservation of the excess sample.34–36Now that we have demonstrated and validated the features of the device such as pumping, modular design, integration of plasma filtration, and aliquoting, the next step is to develop and test new matrices to stabilize desired targets, such as HIV-1 RNA in plasma. We emphasize that in this work, we did not create new chemical stabilization matrices and used only those commercially available. The range of applications of this device will expand as additional matrices are being developed in this active field of research. We envision that the device will be used for remote analysis, allowing quantitative tests such as quantifying viral load or genotyping for viral infection (of HIV, HCV, etc.). While we focused on preservation and recovery of nucleic acids, we have demonstrated that this approach is also compatible with recovery of small molecules and particles. Recovery of small molecules is attractive for studying pharmacokinetics in clinical trials of drug candidates.10,11 Preservation of proteins and live organisms using this approach is of active interest to us as well.
Examples of other potential applications include the testing of different analytes from a single sample (multiplexing) by using more than one stabilization matrix, as well as multi-patient testing achieved by the storage of more than one sample in the same device. In addition, sample stabilization during transport could also facilitate large-scale, remote clinical trials by allowing subjects to collect samples at home in order to minimize the costs of traveling to a centralized facility, sample collection, and analysis. Another potential application of analyte stabilization is the ability to track the clinical history of a patient by preserving samples collected regularly over a long period of time. This would dramatically increase the amount of available information for personalized medicine, allowing one to trace the temporal evolution of disease and biomarkers not only after the diagnosis, but potentially over a patient’s lifetime. This approach is currently unfeasible due to the high costs of cold-chain stabilization and biobanking, but it would be enabled by further developments of bio-preservation reagents and of integrated, simple-to-use microfluidic devices of the type described here.
Acknowledgments
This work was funded in part by DARPA Cooperative Agreement No. HR0011-11-2-0006 (for plasma filtration and integration) and by NIH grant No. R01EB012946 administered by the National Institute of Biomedical Imaging and Bioengineering (for initial concept development). This paper does not necessarily reflect the position or policy of the U.S. government or these agencies, and no official endorsement should be inferred. The authors would like to thank the Millard and Muriel Jacobs Genetics and Genomics Laboratory for access to its bioanalyzer, Liang Li for discussions about pumping and device fabrication, Qichao Pan for work on the bibliography, discussions about stabilization, and initial tests of drying on a previous version of the device, Bing Sun and Stephanie McCalla for their help in setting up RT-PCR quantification, Yu-Hsiang Hsu, Liang Ma and Alexander Tucker-Schwartz for discussions about device design and properties, Rolf Muller and Judy Muller-Cohn at Biomatrica Inc. for useful discussions about sample preservation, and Whitney Robles for contributions to writing and editing this manuscript. Disclosure: R.F.I. and F.S. have a financial interest in SlipChip Corp.References
- US National Institute of Health, Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents, http://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf, Accessed June 12, 2013 Search PubMed.
- US Department of Veteran Affairs, Update on the Management and Treatment of Hepatitis C Virus Infection, http://www.hepatitis.va.gov/provider/guidelines/2012HCV-pretreatment-assessments.asp, Accessed June 12, 2013 Search PubMed.
- R. Burke, K. Vest, A. Eick, J. Sanchez, M. Johns, J. Pavlin, R. Jarman, J. Mothershead, M. Quintana, T. Palys, M. Cooper, J. Guan, D. Schnabel, J. Waitumbi, A. Wilma, C. Daniels, M. Brown, S. Tobias, M. Kasper, M. Williams, J. Tjaden, B. Oyofo, T. Styles, P. Blair, A. Hawksworth, J. Montgomery, H. Razuri, A. Laguna-Torres, R. Schoepp and D. Norwood, BMC Public Health, 2011, 11, S6 CrossRef PubMed.
- M. Baker, Nature, 2012, 486, 141–146 CrossRef CAS PubMed.
- R. E. Hewitt, Curr. Opin. Oncol., 2011, 23, 112–119 CrossRef PubMed.
- P. H. J. Riegman, M. M. Morente, F. Betsou, P. de Blasio and P. Geary, Mol. Oncol., 2008, 2, 213–222 CrossRef PubMed.
- D. Jensen, Room Temperature Biological Sample Storage, Stanford University Pilot, http://sustainablestanford.stanford.edu/sites/sem.stanford.edu/files/documents/Stanford_Room_Temp_Pilot_May09.pdf, Accessed June 12, 2013 Search PubMed.
- B. Borrell, Nature, 2012, 491, 169 CrossRef CAS PubMed.
- R. Dalton, Nature, 2005, 437, 300–300 CrossRef CAS PubMed.
- P. Timmerman, S. White, S. Globig, S. Ludtke, L. Brunet and J. Smeraglia, Bioanalysis, 2011, 3, 1567–1575 CrossRef CAS PubMed.
- P. A. Demirev, Anal. Chem., 2012, 85, 779–789 CrossRef PubMed.
- M. Holub, K. Tuschl, R. Ratschmann, K. A. Strnadová, A. Mühl, G. Heinze, W. Sperl and O. A. Bodamer, Clin. Chim. Acta, 2006, 373, 27–31 CrossRef CAS PubMed.
- S. Uttayamakul, S. Likanonsakul, R. Sunthornkachit, K. Kuntiranont, S. Louisirirotchanakul, A. Chaovavanich, V. Thiamchai, S. Tanprasertsuk and R. Sutthent, J. Virol. Methods, 2005, 128, 128–134 CrossRef CAS PubMed.
- M. F. Pirillo, P. Recordon-Pinson, M. Andreotti, M. G. Mancini, R. Amici and M. Giuliano, J. Antimicrob. Chemother., 2011, 66, 2823–2826 CrossRef CAS PubMed.
- J. H. Crowe, F. A. Hoekstra and L. M. Crowe, Annu. Rev. Physiol., 1992, 54, 579–599 CrossRef CAS PubMed.
- E. Wan, M. Akana, J. Pons, J. Chen, S. Musone, P. Y. Kwok and W. Liao, Curr. Issues Mol. Biol., 2010, 12, 135–142 CAS.
- R. Muller, J. Muller-Cohn, M. Munson and O. Clement, Scientific evaluations of Biomatrica’s technologies for room temperature storage and stabilization of biological samples, http://www.biomatrica.com/downloads/Biomatrica_Technology_Validation_Studies.pdf, Accessed June 12, 2013 Search PubMed.
- IntegenX, GenTegra™ RNA—User Manual, http://www.mygentegra.com/images/stories/literature/gentegra%20rna%20user%20manual.pdf, Accessed May 5, 2013 Search PubMed.
- B. Inc., RNAstable®/RNAstable LD® Handbook, 2012.
- M. Saghbini, J. Venneri, T. Carrico, H. McMahon, K. McWeeny and R. Nuňez, Aerosol and Cross-Contamination Study of the IntegenX FastDryer for Drying GenTegra DNA Tubes, http://integenx.com/wp-content/uploads/2013/03/aerosol-and-cross-contamination-study-of-the-integenx-fastdryer-for-drying-gentegra-dna-tubes.pdf, Accessed June 12, 2013 Search PubMed.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 8, 2286–2292 RSC.
- G. Zhenyu, Z. Lei, F. Zheng, Z. Weihong and Z. Xinhua, Nanotechnology, 2008, 19, 135604 CrossRef PubMed.
- S. McBreen, S. Imlach, T. Shirafuji, G. R. Scott, C. Leen, J. E. Bell and P. Simmonds, J. Virol., 2001, 75, 4091–4102 CrossRef CAS PubMed.
- L. Li, M. A. Karymov, K. P. Nichols and R. F. Ismagilov, Langmuir, 2010, 26, 12465–12471 CrossRef CAS PubMed.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS PubMed.
- J.-U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. Jia, K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129, 8825–8835 CrossRef CAS PubMed.
- X. Casadevall i Solvas, V. Turek, T. Prodromakis and J. B. Edel, Lab Chip, 2012, 12, 4049–4054 RSC.
- L. Daubersies, J. Leng and J.-B. Salmon, Lab Chip, 2013, 13, 910–919 RSC.
- R. H. Garrett and C. M. Grisham, Biochemistry, Brooks/Cole Publishing Company, 2010 Search PubMed.
- S. Byrnes, A. Fan, J. Trueb, F. Jareczek, M. Mazzochette, A. Sharon, A. F. Sauer-Budge and C. M. Klapperich, Anal. Methods, 2013, 5, 3177–3184 RSC.
- E. G. Zoetendal, C. C. Booijink, E. S. Klaassens, H. Heilig, M. Kleerebezem, H. Smidt and W. M. de Vos, Nat. Protoc., 2006, 1, 954–959 CrossRef CAS PubMed.
- T. A. Van de Goor, PharmaGenomics, 2003, 3, 16–18 Search PubMed.
- K. J. Hemmerich, Medical Plastics and Biomaterials Magazine, 1998, 16–23 Search PubMed.
- F. Shen, W. Du, E. K. Davydova, M. A. Karymov, J. Pandey and R. F. Ismagilov, Anal. Chem., 2010, 82, 4606–4612 CrossRef CAS PubMed.
- F. Shen, W. Du, J. E. Kreutz, A. Fok and R. F. Ismagilov, Lab Chip, 2010, 10, 2666–2672 RSC.
- W. Liu, D. Chen, W. Du, K. P. Nichols and R. F. Ismagilov, Anal. Chem., 2010, 82, 3276–3282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50747e |
| This journal is © The Royal Society of Chemistry 2013 |