 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic electroporation for cellular analysis and delivery†
Tao
Geng
a and
Chang
Lu
*bc
aDepartment of Agricultural and Biological Engineering, Purdue University, West Lafayette, IN 47907, USA
bDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: changlu@vt.edu; Fax: +1-540-231-5022; Tel: +1-540-231-8681
cSchool of Biomedical Engineering and Sciences, Virginia Tech-Wake Forest University, Blacksburg, VA 24061, USA
First published on 8th July 2013
Abstract
Electroporation is a simple yet powerful technique for breaching the cell membrane barrier. The applications of electroporation can be generally divided into two categories: the release of intracellular proteins, nucleic acids and other metabolites for analysis and the delivery of exogenous reagents such as genes, drugs and nanoparticles with therapeutic purposes or for cellular manipulation. In this review, we go over the basic physics associated with cell electroporation and highlight recent technological advances on microfluidic platforms for conducting electroporation. Within the context of its working mechanism, we summarize the accumulated knowledge on how the parameters of electroporation affect its performance for various tasks. We discuss various strategies and designs for conducting electroporation at the microscale and then focus on analysis of intracellular contents and delivery of exogenous agents as two major applications of the technique. Finally, an outlook for future applications of microfluidic electroporation in increasingly diverse utilities is presented.
 Tao Geng | Tao Geng is currently a postdoctoral scholar at University of California, Berkeley. He received his Ph.D. degree in Biological Engineering in 2012 from Purdue University where he worked on microfluidic electroporation and genetic/epigenetic analysis of cells based on integrated microfluidic systems under the direction of Dr Chang Lu. His primary research interest is focused on miniaturized assays for biomedical applications. |
 Chang Lu | Chang Lu is an associate professor of Chemical Engineering and Biomedical Engineering (by courtesy) at Virginia Tech. Dr Lu obtained his B.S. in Chemistry with honors from Peking University, M.S. and PhD in Chemical Engineering from University of Illinois at Urbana-Champaign. He spent 2 years as a postdoc in Applied Physics at Cornell. He was an assistant and associate professor of Biological Engineering at Purdue University before moving to Virginia Tech. Dr Lu received a Wallace Coulter foundation Early Career Award and NSF CAREER Award, and was named a faculty fellow by the College of Engineering at Virginia Tech in 2012. |
1. Introduction
A cell membrane constitutes the primary barrier for the transport of molecules and ions between the interior and the exterior of a cell. A myriad of methods have been developed to break the cell membrane for either releasing intracellular components out of cells or introducing exogenous molecules into cells.1Electroporation, also known as electropermeabilization, has gained increasing interest in the communities of biophysics, biotechnology, pharmacy and medicine since the early 1980s, owing to its ability to substantially increase the membrane permeability in the presence of a pulsed electric field.2–4 The technique is more reproducible, universally applicable, and efficient than other physical methods and alternative biological and chemical techniques. Conventional electroporation is typically conducted by exerting short electric pulses of defined intensity and duration to a cuvette equipped with embedded electrodes inside.5 The electrodes are commonly fabricated out of aluminum (Al), stainless-steel, platinum (Pt) or graphite, and arranged in a plate-to-plate manner. A pulse generator such as special capacitor discharge equipment is required to generate the high voltage pulses. By tuning the electric parameters, electroporation efficiency and cell viability (for delivery) can be optimized.6 Although the traditional electroporation systems have been widely used, they require a high voltage input and suffer from adverse environmental conditions such as electric field distortion, local pH variation, metal ion dissolution and excess heat generation, resulting in low electroporation efficiency and/or cell viability.The rapidly growing microfluidics-based electroporation overcomes many drawbacks of the bench-scale electroporators owing to its unique characteristics of miniaturization and integration.7–9 First, microfluidic electroporation systems are typically fabricated with standard microfabrication technology such as soft lithography, and a variety of microelectrodes are incorporated into the chips to generate the field necessary for electroporation. By shrinking the distance between electrodes to a few tens of micrometers or creating physical constraints with subcellular dimensions, the required voltage is dramatically decreased to a few volts. The electroporation microchips provide uniform electric field distribution, a favorable fluidic and chemical environment, and rapid heat dissipation in small-volume microchannels. Second, single cells could be manipulated on chips to probe cell heterogeneity. The miniaturization of the systems also makes them very suitable for assays involving rare cells and expensive reagents due to the substantially reduced consumption of samples. Third, the utilization of transparent materials (e.g. polydimethylsiloxane (PDMS) and glass) for microchips allows in situ observation and real-time monitoring of the electroporation process using fluorescent probes, which facilitates the exploration of the electroporation mechanism. Finally, microfluidic electroporation can be integrated with other analytical processes such as dielectrophoresis (DEP), electrophoresis and electroosmosis to implement a total analysis analytical system. This is especially important for applications related to analysis of intracellular contents.
There have been some excellent reviews on microscale electroporation.7–12 In this review, we begin by describing the fundamentals of the electroporation technique. We then highlight various state-of-the-art microfluidic architectures that were used to perform electroporation on microchips. Based on the strategies used to facilitate electroporation at the microscale, they are sorted into five categories: electrode incorporation and configuration, channel geometry variation and constriction structures, hydrodynamics-enhanced electroporation, compartmentalized electroporation, and miscellaneous methods. Given the distinct advantages offered by these tools, we introduce their enhanced overall performance for intracellular content analysis as well as delivery of exogenous molecules into cells. Finally, the potential challenges and future advances are briefly discussed. We emphasize on electroporation strategies uniquely enabled by microfluidic platforms and summarize guidelines that are generally useful for the design and implementation of microscale electroporation.
2. The basics of electroporation
Although the mechanism underlying electroporation remains not entirely understood yet, two theoretical models were proposed to describe the process: electromechanical instability theory and pore theory.4,13–17 The former theory is a deterministic description based on the mechanical compression of the entire cell membrane by electrical field.18,19 However, it fails to explain a number of experimental observations. The pore model developed by several groups is more prevailing.20–22In an electroporation process, upon the application of short and intense electric pulses to a cell, the cell membrane charges like a capacitor, as the conductivities of the cytoplasm and the extracellular medium are several orders of magnitude higher than that of the membrane. A potential difference is electrically induced across the intact cell membrane within an extremely short charging time (on the timescale of microseconds). The transmembrane potential (ΔψE) induced by an external electric field can be described using the following equation:14,16
| ΔψE (E,M,t) = ψin − ψout = −fgrEcosθ(M) (1 − e−t/τ) | (1) |
| f = l/(l − 0.67r) | (2) |
| g(λ) = λiλe/[λm(λi + 2λe)(r/2dm) + (λi − λm)(λe − λm)] | (3) |
| τ = rCm(λi + 2λe)/[2λiλe + rλm(λi + 2λe)/dm] | (4) |
If the membrane is considered as a pure dielectric, then λm = 0, then
| g = 1 | (5) |
| τ = rCm(λi + 2λe)/(2λiλe) | (6) |
Assuming the cell is a sphere and the charging time is much shorter than the pulse duration, eqn (1) can be simplified as (at steady state):
| ΔψE = −1.5 g(λ)rEcosθ | (7) |
Other than the induced transmembrane potential ΔψE, cells typically also exhibit a resting membrane potential (Δψrest) in the living cell membrane to provide power to cells or to transmit signals. It is conventionally defined as the voltage inside relative to ground outside of the cell, and typically in the range of −40 to −60 mV.16 The total potential difference across the cell membrane is thus expressed as:
| Δψ = ΔψE + Δψrest | (8) |
According to eqn (1), the induced transmembrane potential is dependent on the site of the cell membrane. During the stage of membrane charging, negative and positive charges within the cell accumulate at the regions facing the cathode and anode respectively. Correspondingly, the pole closest to the cathode (θ = π) is depolarized, while the one closest to anode (θ = 0) is hyperpolarized. The Δψ at the hyperpolarized pole is higher due to the negative Δψrest. Therefore, electroporation initially occurs at the pole of the cell closest to the anode, followed by the pole closest to the cathode. The localized and asymmetric electroporation was observed experimentally during the uptake of fluorescence dye into individual CHO-K1 cells, in good agreement with the theoretical prediction from eqn (1) and (8).23
When Δψ exceeds a critical threshold, nanoscale electropores are created within the membrane through the localized structural rearrangements of the lipid molecules, leading to a rapid membrane discharge with a dramatic increase in membrane conductance and a decrease in membrane potential. The critical Δψ value is typically 0.2 to 1 V irrespective of cell types.24 The process of pore formation could be divided into two steps.14 First, hydrophobic pores are formed due to lateral thermal fluctuations of lipid molecules. Second, as the pores expand to a threshold radius, they transit to hydrophilic pores lined by the polar heads of phospholipids in order to minimize the energy of the membrane. It should be noted that although electroporation occurs mainly in the lipid bilayer domains of membranes, the membrane proteins and cytoskeleton also contribute to the process by affecting the behaviors of lipid bilayers.4,14 Accumulation of hydrophilic pores on cell membrane facilitates the exchange of the water-soluble molecules and ions through the cell membrane.
The size and number of pores expands and increases with time as long as the threshold of transmembrane potential is maintained. The size of the pores is governed by the line tension on the pore perimeter, the surface tension of the cell membrane, and the induced transmembrane potential.13 Since different regions on the cell membrane take different times to achieve the threshold transmembrane potential, both the size and density of the pores are spatially heterogeneous at various locations on the cell surface. Based on the pore size, the pores could be divided into two populations: small pores (<2 nm in diameter) and large pores (>2 nm in diameter).22 The highest pore density is created at the poles, but the largest pores are primarily located on the border of the electroporated regions (close to the cell equator). In addition, the hyperpolarized half of the cell has more but smaller pores, whereas the depolarized half of the cell has fewer but larger pores. By modulating cell reorientation by hydrodynamic effects or applying periodic electric pulses with altered polarities and directions, it is possible to extend the pore zone area on the surface of the cells in suspension.25,26
The electroporation process could be modulated by tuning the applied electric parameters (e.g. pulse amplitude, duration, frequency and shape). The pulse amplitude determines the fraction of the electroporated area whereas the pulse duration and number mostly affect the extent of electroporation (the pore density).27 Depending on the degree of the conformational changes in the membrane structure, electroporation can be reversible or irreversible. Under appropriate electric pulses, the pores are transient and can reseal after removal of the field, ensuring the survival of the cells. This sealing process happens in a much longer timeframe (over a range of minutes) than pore formation and is strongly dependent on the electroporation conditions and temperature.16 Reversible electroporation is widely applied to the delivery of a myriad of molecules into cells or tissues, such as fluorescent dyes, DNA, RNA, oligonucleotides, proteins, peptides, drugs, and nanoparticlesetc.21,28–30 It has been proposed that many transport mechanisms including diffusion, convection, electrophoresis, electroosmosis, endocytosis and macropinocytosis may account for the molecular transport across an electroporated membrane.13,15,17 The size, electrical charge and shape of the molecules have a substantial influence on their transport.15 In the case of electroporation-based transfection, electrophoretic force is one of crucial mechanisms for the translocation of polyanionic DNA molecules into cytoplasm.31,32 Hence, the combinations of short high-voltage and long low-voltage pulses could enhance transfection efficiency while maintaining cell viability.33,34 It is suggested that high-voltage pulses contribute to electropermeabilization, while low-voltage pulses provide an electrophoretic force to drag DNA towards the cell membrane and/or insert it into the cells. A successful electrotransfection process involves several steps: DNA migration and interaction with the electroporated membrane to form complexes during electroporation, uptake into cytoplasm, intracellular trafficking, and entry into the nucleus.35 DNA molecules need to overcome multiple obstacles including the extracellular matrix, cell membrane, cytoplasm (especially the cytoskeletal network), nuclear envelope and even the cell wall (for bacteria, algae, fungi, and plant cells) to achieve ultimate gene expression. Irreversible electroporation occurs under intensive electric parameters and typically leads to cell death after the procedure. Weaver13 proposed that it is likely caused either by the generation of permanent pores in a portion of the membrane region after extensive electropermeabilization or by chemical stress resulting from molecular transport through transient pores. Although the exact mechanism is still unclear, the process of irreversible electroporation allowed lysing cells to effectively extract intracellular materials (e.g. DNA, RNA, enzymes, and cytoplasmic proteins, recombinant proteins) for cellular analysis or producing therapeutic proteins.36–38 It was also used to selectively kill cells for hematopoietic cell selection and tumor cell purging39,40 as well as non-thermal inactivation of microorganisms in food products.41
Cells undergo electroporation only with proper “local” field intensity and duration. Thus the device design and protocol development always need to revolve around creation of these favorable conditions at the microscale and exposure of cells to the electric field.
3. The various strategies for microfluidic electroporation
The extensive research in the area of microfluidics increasingly enables the implementation of electroporation in microchip formats. To date, a number of innovative strategies have been developed to electroporate cells on microfluidic platforms. We divide prior work on these technologies into five categories.3.1. Electrode incorporation and configuration
The most straightforward way to implement electroporation at the microscale is having fabricated microelectrode structures inside microchannels or microchambers. The design of the microelectrodes is of crucial importance for the electroporation process because the geometry defines the electric field distribution and uniformity and thus greatly affects the electroporation efficiency (Fig. 1).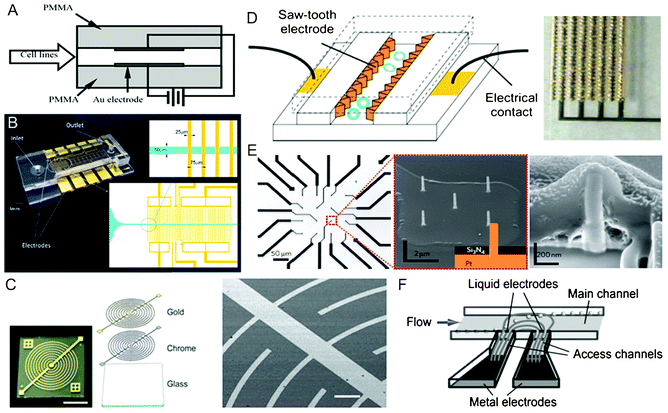 | ||
| Fig. 1 Electrode incorporation and configuration. (A) Schematics of parallel Au plate electrodes patterned in a PMMA straight channel for continuous flow-through electroporation.42 Reproduced with permission from ref. 42. Copyright 2001 Elsevier. (B) A PDMS/glass straight channel patterned with planar Au interdigitated electrodes with rectangular strips.71 Reproduced with permission from ref. 71. Copyright 2013 American Chemical Society. (C) Optical and SEM images of Au interdigitated electrodes with curved strips. The electrodes were fabricated by sputtering two metal layers (chrome and Au) on a glass substrate.77 Reproduced with permission from ref. 77. Copyright 2011 The Royal Society of Chemistry. (D) Schematics and image of 3D Au saw-tooth vertical sidewall electrodes embedded in a straight channel.89 Reproduced with permission from ref. 89. Copyright 2005 The Royal Society of Chemistry. (E) Optical and SEM images of a four-by-four nanopillar (1.5 μm tall and 150 nm in diameter) platinum electrode array, and SEM image of cell-nanopillar interactions.99 Reproduced by permission from ref. 99. Copyright 2012 from Macmillan Publishers Ltd. (F) Schematics of the principle of “liquid electrodes”.106 Reproduced with permission from ref. 106. Copyright 2007 The Royal Society of Chemistry. | ||
The simplest layout for coplanar electrodes was composed of one pair of straight electrode strips arranged in parallel to serve as the anode and the cathode.53–59 In a more complex configuration, the electrodes were designed as curved strips with sharp tips to concentrate the electric field for cell trapping.60 Although a single pair of electrodes were used for electroporation of adherent cells,53 suspended cells60 and single cardiomycytes,55,56 the electric field generated by only two electrodes lacked uniformity and was spatially limited in the thin layer adjacent to the electrode surface. In comparison, interdigitated electrodes containing an array of parallel electrode strips were introduced to provide relatively homogeneous electric field to cells over a larger area. The high-density arrays could also significantly decrease the gap between electrodes, and thus minimized the overall voltage needed. The shape of the electrode array was arranged as a square or angular to match the configuration of the microchannel or microchamber. Each electrode strip could be constructed in a rectangular,61–71 castellated,72,73 circular,74–76 curved77 or saw-tooth78–80 formats. Fig. 1B and C show two interdigitated electrodes with rectangular and curved strips, respectively. The saw-tooth shape was one of the most efficient structures.78–80 The short distance between the electrode pairs of the saw-tooth shape significantly decreased the required voltage. If the electrode structure was perpendicular to the cell suspension flow, cells would be exposed to periodic variation in electric field strength. Lee and Tai78 performed earlier research by developing a microchip with interdigitated saw-tooth shaped Au electrodes where the electrode pairs were coated by a polymer to electroporate various types of cells using various sets of electric parameters.
Another commonly used design was circular/square-shaped microelectrodes.81–88 Two circles were employed to mimic the two-needle array used in clinical electroporators, but the effective electroporation area was limited and the field intensity was inhomogeneous.81 Circular/square electrode arrays were used to expand the effective area. Combined with other forms of counter electrodes, each electrode could be individually addressed to achieve site-specific electroporation.82–86 Single cell electroporation was also accomplished by reducing the circle diameter to be comparable to the cell size.87,88
It is noteworthy that spatially non-uniform electric field of the microelectrodes and the arrays also results in DEP during which dielectric cells could be attracted to the electrode tips and give rise to a pearl-chain-like alignment. A common set of electrode patterns can be implemented to initially trap and concentrate target cells in regions of interest by dielectrophoretic force and then perform subsequent electroporation while cells are exposed to different electric pulsing protocols.60,67–69,72–76,78–80,82,83,89 Therefore, this technique, adopted in either a static or flow-through manner, is particularly suitable for applications combining electrical cell lysis with downstream intracellular content analysis.72–76,79,80,82
Vertical sidewall electrodes represent one type of 3D electrodes because they provide a great spatial uniformity in electric field distribution along the width, depth, and length of a microchannel. By precisely constructing the electrode configurations, various electric field distributions could be achieved for electrical manipulation of cells. Lu et al.89 designed a saw-tooth electrode array embedded within vertical sidewalls of a straight channel in order to provide a more uniform electric field to continuously flowing cells (Fig. 1D). Alternatively, a serpentine channel was fabricated out of Al (with the metal serving as both electrodes and sidewalls), where cells were processed in a semicontinuous mode.90,91 In addition, thick Au electrodes (∼25 μm) were used to construct the half height of the sidewalls in order to homogenously deliver the maximum energy of nanosecond electric pulses to the suspended cells across the straight microchannel.92 By shortening the space between the electrode pair, the thick sidewall electrodes allowed electroporation of a single cell, either suspended or adherent, under a uniform electric field.93–96 Besides metals, a doped silicon wafer (∼200 μm thick) containing the microfluidic network was sandwiched between two glass wafers for electroporation-processing of cells.97
Other types of 3D electrodes have been developed for electroporation in the form of pillars,98,99 needles100,101 or nails.102,103 For instance, 3D cylindrical microelectrodes (50 μm in diameter, 50 μm in height with 20 μm gap among them) were built by fabricating protruded copper pillars inside the channel.98 The comparison to 2D electrodes of similar size revealed that 3D cylindrical electrodes exhibited promoted electroporation efficiency than 2D planar electrodes. It was also observed that multiple pores could be generated in the cell membrane with 3D electroporation while only a single pore with 2D electrodes, which apparently resulted from enhanced uniformity in electric field distribution. More recently, chips featuring high-density subcellular-sized or nanoscale electrode arrays have been developed to accomplish localized and selective electroporation of single cells growing on the electrode arrays (Fig. 1E).99,102,103
In addition, some novel forms of 3D electrodes originally developed for DEP were employed in the electroporation area.104–107 “Liquid electrodes” were patterned in an array format to perform electroporation under ac electric fields.104 The system was composed of three units: (1) a main fluidic channel for cell flowing, (2) two distant planar Pt electrodes of large surface area deposited on the bottom of enclosed chambers on both sides of the main channel, and (3) relatively narrow access channels connecting main channel and electrodes perpendicularly (Fig. 1F). The term “liquid electrode” referred to the vertical equipotential plane at the interface between the access and main channels. It behaved as a vertical sidewall electrode in the main channel, and thereby provided a relatively homogenous electrical field across the depth of the total channel. The distribution of the electric field was affected by the geometry of the access channel rather than the metal electrode. In another example, a pair of channels were incorporated on the two sides of a main fluidic channel and separated with a thin (20 μm) PDMS membrane.105 The side channels were filled with a conductive material such as indium solder and connected to a power supply via copper wires inserted into the solder to serve as the electrodes, while the main channel contained cell trapping sites. Except improved electric field homogeneity within the region surrounding the cell surface, the noncontact between the electrodes with the samples eliminated the adverse heating shock, chemical contamination, biomolecule fouling and electrode damage.
3.2. Channel geometry variation and constriction structures
A high local electric field can be generated by having constriction segments or structures in microchannels or microchambers. Electroporation occurs when the cells of interest flow through or are positioned within the constricted regions. Due to the high electrical resistance of the small constriction regions, cells experience a high field intensity that is confined in such configurations even when the overall input voltage is low. Fig. 2 illustrates some representative designs of constriction structures for electroporation.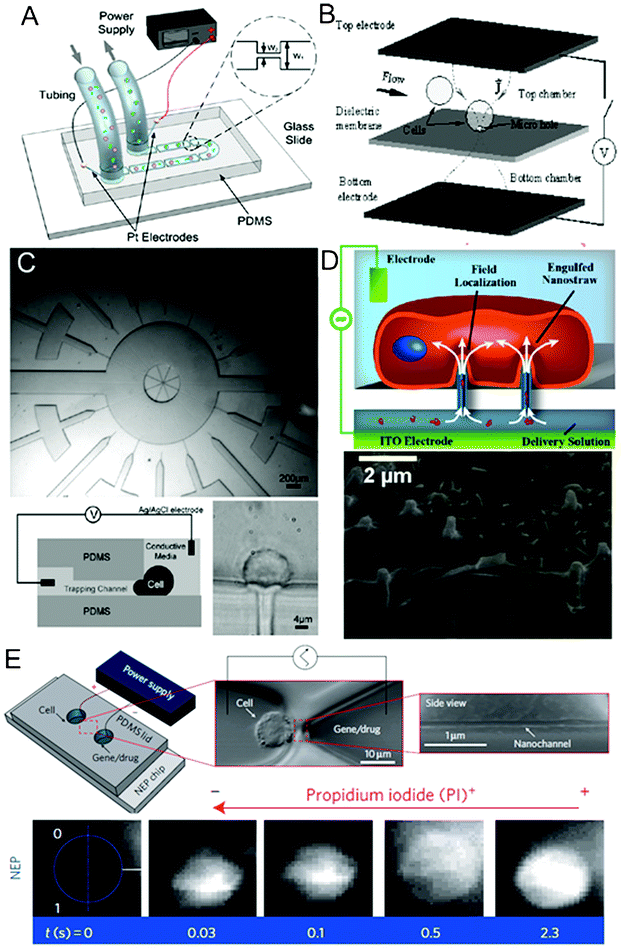 | ||
| Fig. 2 Channel geometry variation and constriction structures. (A) Schematics of a volume-scalable flow-through electroporation chip containing a number of alternating wide and narrow sections based on a constant voltage.129 Reproduced with permission from ref. 129. Copyright 2010 Elsevier. (B) Schematics of a microhole structure in a silicon nitride dielectric membrane located between two electrodes for flow-through single-cell electroporation.141 Reproduced with permission from ref. 141. Copyright 2003 Elsevier. (C) Images and schematics of an array of narrow lateral channels for cell trapping and localized single-cell electroportion.147 Reproduced with permission from ref. 147. Copyright 2005 The Royal Society of Chemistry. (D) Schematics and SEM image of the intimidate contact between a cell and nanostraws.155 Reproduced with permission from ref. 155. Copyright 2013 American Chemical Society. (E) Top: schematics and images of a nanochannel electroporation chip and a cell positioned at the tip of the nanochannel. Bottom: fluorescence images of cell uptake of PI dye after nanochannel electroporation. The rapid increase in fluorescence indicated that dye transport was not dominated by diffusion.153 Reproduced by permission from ref. 153. Copyright 2011 from Macmillan Publishers Ltd. | ||
Our group has been developing flow-through electroporation techniques for a number of years. We used a simple fluidic channel that consisted of a number of alternating wide and narrow sections for flow-through electroporation based on a constant voltage (Fig. 2A).39,116–119,121,122,125–132 Given a uniform depth of the channel, the electrical field strength in each segment was inversely proportional to the width of the section when a constant voltage was established across the channel. The geometric variation not only allowed electroporation to occur exclusively in a defined (narrow) section(s), but also effectively decreased the overall voltage required for electroporation. While flowing through the channel, cells experienced pulse-like electrical field variation(s) with a common power supply providing constant voltage. The electroporation parameters (e.g. pulse width, intensity, and pattern) could be easily adjusted by changing the overall voltage applied and channel geometry. Since the physics of our flow-through electroporation did not depend on the absolute dimensions of the device, the device could be scaled up easily by increasing the cross-sectional areas of different sections of a channel proportionally for processing large-volume cell samples (up to 20 mL min−1).129 We also showed that similar devices also worked under a constant low-frequency (10–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz) ac voltage.132 Our flow-through electroporation did not require a pulse generator and thus drastically simplified the equipment required for the delicate procedure of cell electroporation. A dc-biased ac electric field was used in similar channel to combine DEP with electroporation for integrated cell trapping and electroporation on a single chip.133 Target cells were selectively accumulated in the front entrance of the narrow channel and subsequently electroporated within the channel. The switching between the two processes could be achieved by simply varying the dc component of the total voltage.
000 Hz) ac voltage.132 Our flow-through electroporation did not require a pulse generator and thus drastically simplified the equipment required for the delicate procedure of cell electroporation. A dc-biased ac electric field was used in similar channel to combine DEP with electroporation for integrated cell trapping and electroporation on a single chip.133 Target cells were selectively accumulated in the front entrance of the narrow channel and subsequently electroporated within the channel. The switching between the two processes could be achieved by simply varying the dc component of the total voltage.
Microchannels with alternating wide/narrow sections were also adapted to conduct flow-through single-cell electroporation by reducing the dimensions of the narrow section to cellular or subcellular sizes.118,134 An ac electric field was employed to prevent the bubbles electrolytically generated at the electrodes from blocking the extremely narrow channel and the electric circuit.134 A single rod-shaped large cardiomyocyte was also spanned across a constriction between two wide open channels while being sealed with mineral oil to enable regional electroporation of the cell.135
Besides varying the width of the channel which was easy to implement given the planar nature of typical microfabrication, alternatively the constriction segments of a channel could also be constructed by reducing the depth of a segment of the channel. Fox et al.136,137 created two constriction segments in a microfluidic channel with platinum microelectrodes at the two ends of each constriction to continuously electroporate cells with high throughput. Although the microelectrode fabrication procedure was relatively complex, placing the electrodes close to the constrictions greatly reduced the undesired energy consumption.
Constriction structures such as microscale holes were used for electroporation of single or multiple cells.138–146 In their pioneering work, Hang and Rubinsky138,139 developed the first microfabricated single-cell electroporation chip containing two facing n+ polysilicon electrodes with two fluidic chambers separated by a silicon nitride dielectric membrane in between (Fig. 2B). A microhole with subcellular dimensions was etched through the membrane to connect the two chambers. Individual suspended cells could be captured at the microhole due to the pressure gradient between the two chambers. The trapped intact single cell serves as an insulator in the electrical circuit, whereas the electrical current increased once the cell becomes permeabilized by electroporation. This characteristic enabled real-time monitoring of the electroporation process at the single-cell level using the electrical signal. In spite of the precision, the low processing rate of microhole devices limited their practical applications. There were two approaches to improve the throughput for single-cell analysis. First, the authors constructed a microchannel that continuously transported cells to the microhole on a one by one basis for processing.141 After exposure to electric pulses, the electroporated cell was released and another cell was loaded onto the electroporation hole. Second, a high-density array of microholes (or orifices) was used to allow a number of cells to be treated in parallel.143,144 This microhole structure was expanded to be applied to electroporation of biomimetic vesicles with similar membrane (phospholipid) and size (10–25 μm) to those of living cells.145,146
Channels with subcellular sizes were also used for trapping and electroporating cells.147–150 Khine et al.147 reported a simple PDMS microchip which used multiple narrow lateral channels (4 μm × 3.1 μm in the cross section) for cell trapping and Ag wire electrodes in reservoirs for localized single-cell electroporation in parallel (Fig. 2C). Individual cells extended and deformed into the trapping channels when negative pressure was applied and this was suggested to contribute to membrane rupture under electric pulses. Similar to the work by Huang and Rubinsky,138 the system also measured the electrical resistance to monitor membrane permeabilization. They later developed a computer program to achieve the real-time feedback control of electroporation processes.148 A similar approach was also implemented in a silicon/glass device with multiple parallel capillary channels (4 μm wide and 15 μm deep) serving as the trapping sites for single cells. These features connected two parallel channels where platinum surface electrodes were positioned.150
Nanoscale structures such as nanopores, nanostraws, and nanochannels provide even smaller constriction features compared to the microscale ones and offer high precision and efficiency for electroporation.151–156 Lee and co-workers developed electroporation based on nanoscale structures over the years.151–153 In their early work, they applied porous poly(ethylene terephthalate) track-etched membranes to locally electroporate cells.151 Cells were sandwiched between two membranes that had an average pore size of 400 nm (bottom) and 3 μm (top), respectively. The nanoscale tunnels focused the electric field due to the constriction effect. Although the electroporation efficiency was fairly low, the device had the capacity of treating hundreds to millions of cells in each batch. In their later work, to promote the uniformity of the electric field distribution on cells and thus the electroporation efficiency, the porous membrane on the bottom was modified by gelatin coating and drilled to form uniform micronozzle arrays by laser ablation technique.152 A similar strategy has also been implemented by another group using porous alumina cell culture membranes (with 20 nm pores) combined with a thin PDMS film having well-defined hole structures (100 to 600 μm in diameter) to conduct localized in situelectroporation of adherent cells.154 More recently, protruded alumina nanostraw (i.e. hollow nanowire; 250 nm) arrays were constructed on a polycarbonate track-etched membrane to improve the electroporation performance due to more intimate interactions between the cell membrane and the nanostructure (Fig. 2D).155 In their later effort, the Lee group used nanoscale channels for electroporation and delivery with subcellular resolution and high efficiency.153 They created an array of nanochannels (∼90 nm in diameter and ∼3 μm long) connecting two microchannels at two ends (Fig. 2E).153 An individual cell was precisely positioned at the tip of the nanochannel by moving the cell inside the connected microchannel using an optical tweezers system, while the other microchannel was filled with the solution containing genes or other biomolecules for delivery. Electric pulses were then applied via palladium wire electrodes placed in the microchannel reservoirs, and electropermeabilization occurred in the tiny area of cell membrane defined by the nanochannel dimensions due to the high localized field strength. Nelson et al.156 also demonstrated single-cell electroporation through a nanopore by directly positioning a cell over the structure. Such a method offered a highly precisely delivered quantity and minimal cellular damage.
In addition, silica microbead arrays packed in microchannels by pneumatically actuated elastomeric valves or a weir structure also served as an efficient platform for physical cell capture and electroporation-based lysis of bacterial cells.59,123,124 We used these matrix-like structures formed by the microscale beads as a filter to capture tiny bacterial cells while allowing the liquid to flow though. A large number of bacterial cells could be stuck in the gaps among the beads. While exerting the electric voltage at the two ends of the bead array, the electric field was significantly concentrated in these small gaps to enhance cell electroporation.
3.3. Hydrodynamics-enhanced electroporation
Microfluidics allows the creation of favorable hydrodynamic conditions for cell manipulation in fluidic networks, and this effect has been harnessed to enhance the performance of microscale electroporation (Fig. 3).26,131,157–159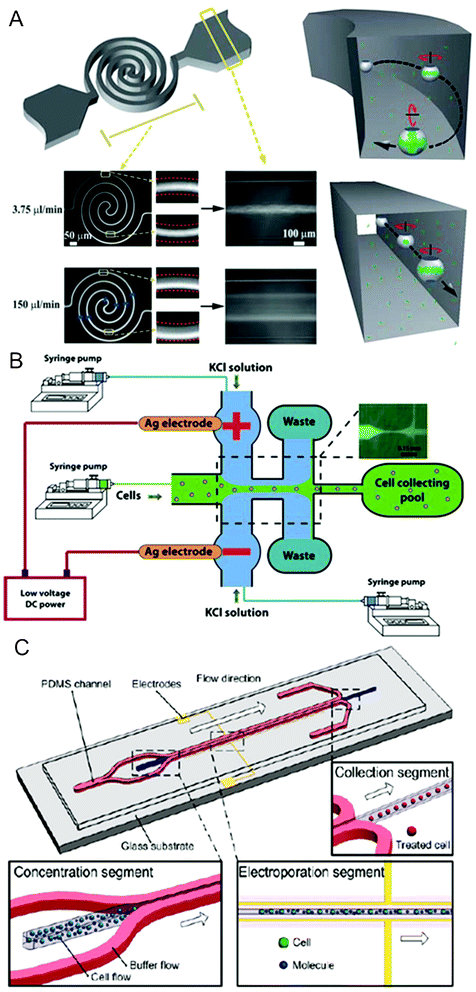 | ||
| Fig. 3 Hydrodynamics-enhanced electroporation. (A) Left: schematics of a flow-through electroporation chip featuring a spiral-shaped electroporation channel and two wide channels, and overlay fluorescent images of SYTO 16 stained cell migration at different flow rates on the chip. Right: proposed models of cell electroporation occurred in spiral (top) and straight (bottom) channels.26 Reproduced with permission from ref. 26. Copyright 2010 The Royal Society of Chemistry. (B) Schematics and fluorescent image of an electroporation chip based on hydrodynamic focusing under low dc voltage.157 Reproduced with permission from ref. 157. Copyright 2010 Springer. (C) Schematics of an electroporation chip that separated the cell suspension from the electrodes by hydrodynamic focusing.158 Reproduced with permission from ref. 158. Copyright 2011 American Chemical Society. | ||
By constructing a spiral-shaped electroporation channel, Wang et al.26 enabled the system to generate permeabilization over the whole cell membrane surface, thereby substantially enhancing the electroporation efficiency without compromising the cell viability (Fig. 3A). This was a significant extension to flow-through electroporation technology. This favorable flow condition (i.e. Dean flow) overcame the major limitation determined by both the physics and the practice of common electroporation during which electropermeabilization of the membrane mostly occurs at the poles of a cell where the surface normal is aligned with the field direction according to eqn (1). When entrained in Dean flow, cells were simultaneously involved in the flow along the channel path and the vortices in the secondary transverse direction. Such complex motions exposed a much larger fraction of the cell surface to the electroporation field.
Hydrodynamic focusing is a widely used technique that allows a central flow to be sandwiched by sheath flows. The technique is effective for creating a narrow stream that exchanges materials with the sheath flows only by diffusion under laminar flow conditions. Using the most common three-inlet configuration, Zhu et al.157 developed a electroporation strategy based on hydrodynamic focusing of cell suspension by highly conductive KCl sheath flows (Fig. 3B). When a constant dc voltage was established across the two side channels through Ag wire electrodes, the electric field was concentrated in the hydrodynamically focused cell flow region due to its much lower conductivity than that of KCl solution. Careful modulation of the central and side flow rates could precisely control the width of the focused stream containing cells, thereby varying the applied voltage to cells. A low voltage of <3 V was able to generate a sufficiently high field intensity to achieve successful electroporation. By placing the electrodes in side inlets, the system shielded the cell suspension from the metal electrodes with the sheath flows, and thus eliminated many adverse effects such as heating shock, localized pH variation and bubble generation. Alternatively a pair of surface Au electrodes were placed beneath the sheath flows (Fig. 3C).158 Hydrodynamic focusing was also used in our work to facilitate single cell electroporation. Bao et al.131 controlled the passage of cells suspended in the central stream in a single-line fashion to ensure uniform electric treatment. By avoiding the non-uniformity in the cell velocity caused by the parabolic profile of the flow field, each individual cell in this case was exposed to the identical treatment in terms of field intensity and duration.
Another effective method to improve the flow profile across the channel for electroporation was the introduction of two parallel stainless steel meshes perpendicular to the flow in the channel.159 In addition to serving as electrodes to generate a homogenous electric field, the mesh structures created a wealth of extremely thin channels which had a much higher hydrodynamic resistance than the main channel. The complex hydrodynamic effects produced a nearly uniform velocity distribution between the two meshes. By tuning up the electric pulse frequency, the flow rate and the volume of electroporation area, all cells experienced exactly one pulse under the same field intensity.
3.4. Compartmentalized electroporation
Confining cells into tiny reaction volumes offers a number of advantages for electroporation-based delivery including increased contact between the cell and the delivered molecule as well as the potential for single cell/molecule screening and analysis. Microfluidics provides an ideal platform for the spatiotemporally regulated electroporation processes in the form of either droplets57,160–162 or microwell arrays56,70,77,83,163–165 (Fig. 4).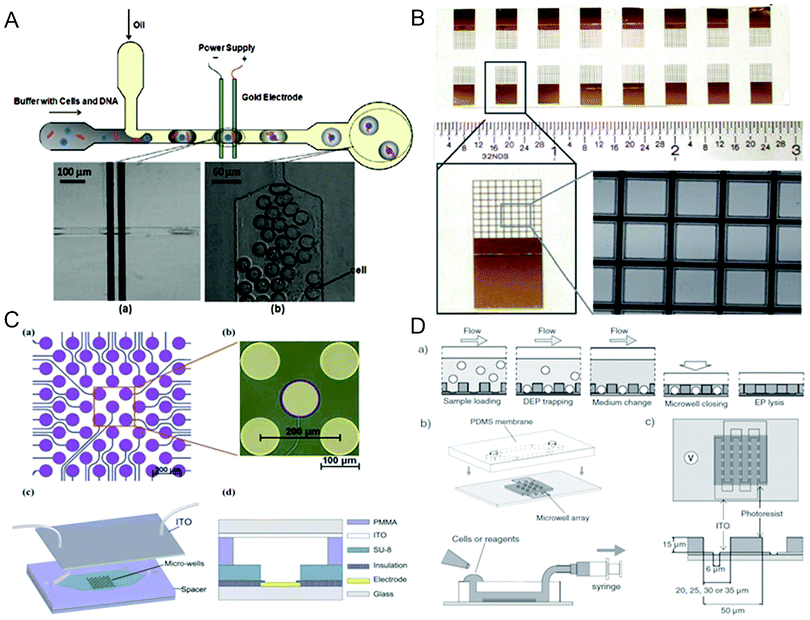 | ||
| Fig. 4 Compartmentalized electroporation. (A) Top: schematics of a droplet-based microfluidic electroporation chip. Bottom: images of (a) cell-containing droplet flowing through the two Au planar electrodes and (b) droplets after electroporation.57 Reproduced with permission from ref. 57. Copyright 2009 American Chemical Society. (B) 16 SU-8 nine-by-nine microwell arrays on an ITO glass.164 Reproduced with permission from ref. 164. Copyright 2012 The Royal Society of Chemistry. (C) Top: schematics and image of an individually addressable circular microelectrode array on a glass substrate. Bottom: schematics of the cell arraying-assisted electroporation chip featuring SU-8 photoresist microwell structures aligned with the microelectrode array and a plate ITO electrode on top.83 Reproduced with permission from ref. 83. Copyright 2011 The Royal Society of Chemistry. (D) Top: Schematics of the procedure for single-cell trapping and electroporation within a microwell array. Bottom: Schematics of the microfluidic system containing a PDMS membrane with access holes on the SU-8 photoresist microwell array (left) and the alignment of the microwell array with ITO interdigitated electrodes (right).70 Reproduced with permission from ref. 70. Copyright 2011 Wiley. | ||
Droplet microfluidics creates and manipulates monodispersed and subnanoliter aqueous droplets in an immiscible and inert carrier fluid (e.g. oil). These microscale droplets serve as microscale compartments which may encapsulate cells and other reagents (e.g. genes).166 The idea of performing electroporation within droplets was initially proposed by Luo et al.,160 in which an ac voltage was applied to parallel line microelectrodes in the downstream of droplet generation. Zhan et al.57 described a more advanced demonstration that was able to encapsulate single cells in picoliter aqueous droplets and then electroporate the encapsulated cells for gene delivery by applying a constant dc voltage on the microelectrode pair (Fig. 4A). The constant dc voltage ensured that every droplet experienced an electroporation field intensity that was higher than the electroporation threshold. Due to the non-conductivity of oil, cells only experienced a transient electric pulse when the conductive droplets passed across the electrodes. The shape and duration of the pulse was dependent on many factors including the velocity and dimensions of the droplet, the distance between the two electrodes, and the location of the cell inside the droplet. In a more recent work, Qu et al.161 increased the number of electrodes to 5 pairs to investigate the influence of the pulse number and the interval between pulses on electroporation. A serpentine channel was also used to improve the mixing of cells and reagents confined in discrete droplets. There was also a report of electroporation of cell-encapsulating microdroplets in the oil in a conventional cuvette.162 An oscillating radio frequency electric field was applied to the silicon oil phase, and a high voltage was required in this system (∼120 V) because of the high resistance of silicon oil.
A microscale well format has also been used to implement electroporation in compartmentalized space.56,70,77,83,163–165 Inspired by conventional multiwell plates, microwell arrays provide further miniaturized and separated reaction chambers with increased well density by the state-of-the-art microfabrication technology.167 Jain et al. created microwell arrays shaped in circles (500 μm in diameter)163 and squares (400 μm × 400 μm)164 on glass substrates coated with a transparent conductor ITO using insulative laser-cut coverlays or biocompatible SU-8 photoresists, respectively (Fig. 4B). In the system, a metal (stainless steel or Al) piece placed on top of the microwell array as well as stainless steel or deposited Au on the bottom of the array were used to provide a uniform electric field to the entire chip. Alternatively, both cathodes and anodes could be deposited on a glass substrate.56 Rod-shaped single cardiomycytes were positioned within arrays of rectangular PDMS microwells aligned perpendicular to parallel Au line electrodes for electroporation.56 Huang et al.77 designed an annular interdigital Au microelectrode array on glass to increase the effective electroporation area and field uniformity in a circular well (Fig. 1C). The array of round-shaped microelectrodes was successfully aligned with a commercial silicone cell culture chamber with matching well dimensions. A more sophisticated device with individually addressable capability, reported by Xu et al., was constructed by aligning high-density microwells (100 μm in diameter) fabricated out of SU-8 photoresist with circular microelectrode arrays on glass substrates (Fig. 4C).83 Combined with the ITO electrodes on top, the chip enabled selective positioning of cells in the microwells through DEP force and subsequent electroporation in a spatially controlled fashion.
The microwell format was extremely suitable for single cell electroporation.70,165 By reducing the size of microwells to 20–35 μm, thousands of single cells could be trapped by DEP with high efficiencies (up to 95%) and high speed (less than 3 min), followed by efficient electroporation (Fig. 4D).70 This was performed on a SU-8 photoresist microwell array aligned with intedigitated ITO microelectrodes patterned on glass. The distance between the electrodes (6 μm) was smaller than the cell diameter (>10 μm) so that each cell was placed on the edge of the electrodes and treated by a local high-intensity electric field. The array was tightly enclosed after cell seeding by placing a PDMS membrane on top to confine cells and intracellular contents within defined wells. In a separate paper, single-cell DEP trapping and electroporation were demonstrated in an array of 10 μm microwell using voltage as low as 1 V, where the SU-8 photoresist microwell structure was sandwiched between the planar Au electrode and ITO electrode.165
3.5. Miscellaneous techniques
Miscellaneous electroporation methods have been developed on chips based on microvalves,120 salt bridges168 and light.169 Pneumatically actuated elastomeric valves made of PDMS are insulative, and thus the closure of these valves within microchannels could physically separate the ionic buffer and interrupt the circuit.170 Based on this feature, the valves were embedded in a microchip to facilitate electroporation under a constant dc voltage.120 Electric pulses of milliseconds could be generated by operating the valve in a close-open-close sequence. Both suspended and adherent cells were successfully electroporated under these microvalve-generated pulses. The promising technique not only eliminated the complex and expensive pulse generator, but also had the potential to be incorporated into large-scale integrated microfluidic systems based on microvalves. To focus an electric field into a specific area, Kim et al.168 proposed an interesting electroporation platform by introducing a pair of highly conductive polyelectrolytic gel plugs into the microchannel. The chip had a fluidic channel for cell flowing and two channels filled with hypertonic solution on the two sides. The gel plugs located at the interface of the two kinds of channels and acted as salt bridges because their ionic conductivity was similar to the hypertonic solution but 10 times higher than that of cell suspension. When a dc voltage was exerted though the external Ag/AgCl electrodes in hypertonic solution, the conductivity difference led to a focused electric field in the main channel that covered flowing cells. Valley et al.169 reported an optofluidics-based microsystem for parallel single cell electroporation and manipulation. The chip was composed of two layers of ITO-coated glass substrates and a SU-8 defined fluidic channel layer in between. A photosensitive film was coated on the bottom glass layer to create virtual electrodes when an ac bias was applied between two ITO layers. When a patterned light was applied, a localized electric field was exerted on the illuminated cells arrayed using optoelectronic tweezers due to the substantial drop of the photoconductive layer's resistance. By changing the applied electrical bias, the light could induce either DEP or electroporation of cells.4. The applications of microfluidic electroporation
The advances in microfluidic electroporation technologies have led to a variety of achievements. In addition to understanding and analyzing the electroporation process itself including pore formation and resealing dynamics,94,99,101,118,126,138–140,147,148 microfluidic electroporation has been mainly applied to cellular analysis and delivery. Based on the direction of movement for the materials across the cell membrane, we divided the applications of microelectroporation into two general categories: (1) Analysis of intracellular contents and inactivation; and (2) delivery of exogenous molecules.4.1. Analysis of intracellular contents and inactivation
An important application of electroporation in the context of analytical chemistry is to serve as a pretreatment for analysis of intracellular molecules. Electroporation provides a simple physical method for disruption of the cell membrane barrier and release of intracellular molecules. Many biomolecules other than nucleic acids (e.g.proteins and other metabolites) cannot be amplified. Thus highly sensitive approaches are required for their detection and quantification. Electroporation does not require chemical/biological reagents which may dilute the analytes or interfere with the downstream analysis. Furthermore, electroporation is very easy for integration with detection and analysis methods such as laser-induced fluorescence (LIF) and electrophoresis, especially on a microfluidic platform. Such features are important for works that require ultrahigh sensitivity, such as single cell analysis.Early practice of electroporation for cell lysis was often performed in combination with capillary electrophoresis (CE), inside a capillary tube and in the context of single cell analysis.171–174 More recent works based on microfluidics mainly focused on releasing low-molecular-weight dyes and metabolites from cells by fast electrical lysis and performing chemical analysis in a high-throughput manner. Such goals could be achieved by coupling electroporation with microchip-based CE and various sensitive detection methods such as LIF and electrochemical assays on an integrated platform.110–115,119 These microchips were typically composed of two channels intersecting with each other: cell flow channel and separation channel, and an electrical field established across the separation channel. Individual cells were transported, either hydrodynamically or electrically, to the junction of the two channels where cells were electrically lysed, and the lysate was electrokinetically injected into the separation channel for electrophoretic separation and LIF detection. Ramsey and colleagues reported an early example of a microfluidic chemical cytometer where fluorescent dyes in the cytoplasm of human T cell lymphoblast-like Jurkat cells could be completely released within <33 ms and single cells were analyzed at throughputs as high as 7–12 cells min−1.110,111 Wang and Lu further improved the throughput to 75–85 cells min−1 by designing a narrow lysis section to generate a much higher field intensity for electrical lysis than that for electrophoresis.119 Fang’s group developed a protocol in which each step could be modulated by means of simply changing the electrical potentials at the four reservoirs.113–115 Although the analysis rates were relatively low (15–25 cells h−1), they were able to efficiently detect a trace level of cellular component glutathione, a tripeptide, in human erythrocytes113 or simultaneously analyze glutathione and reactive oxygen species from a single cell lysate.115 While less common than LIF, electrochemical detection was also used in the detection of ascorbic acid from wheat callus cells.114 More recently, Ramsey’s group integrated electroporation with microchip-CE and electrospray ionization mass spectrometry (ESI-MS) to detect hemoglobinprotein from human erythrocytes.112 Although a similar crossed-channel design was utilized, the working principle was very different from small-molecule analysis. Cells were continuously infused into a channel and lysed when they entered into the junction where the depth was 100 times shallower than other channel segments to focus the electrical intensity. Upon lysis, the intracellular contents were electrophoretically separated along the cell infuse channel towards an electrospray corner where ionization occurred. The other channel filled with electrolyte was used to provide the voltage to cells and rapidly exchange buffer at the lysis region. Compared with LIF detection, the MS-based technique offered more chemical information for analyte identification.
Electroporation could be used as a preparative tool during extraction and purification of intracellular molecules. Techniques other than electrophoresis were also used for analysis of intracellular molecules released by electroporation.58,67–70,79,80,104,123,124 An integrated device was reported on a F1-ATPase rotational assay at the single molecule level.80 In the work, genetically engineered E. coli expressing 6× His-tag fusion F1-ATPase was continuously loaded into the chip and initially lysed by interdigitated saw-tooth planar microelectrodes in a chamber. The motor protein was subsequently extracted from cell lysate by specific binding onto the surface of a serpentine channel coated with nickel-nitrilotriacetic acid. Following sequential introduction of various buffers, the ATP-driven F1-ATPase rotation could be visualized and quantitatively measured. In a separate work, antibody functionalized microbeads packed within microchannel were also used to capture β-actin from electrical lysate via immunobinding with high specificity and sensitivity.79 We detected the autofluorescence of the total lysate from bacterial cells following electroporation using LIF and were able to quantify the number of bacterial cells without tedious labeling procedures.123,124 Since the leakage of intracellular ions such as Na+ and K+ into the surrounding solution can dramatically boost its conductivity, electroporation was introduced as an approach for sensitive bacterial enumeration based on dielectrophoretic impedance measurement after electropermeabilization.67–69 Impedance analysis following electrical lysis was also applied to discriminate live and dead yeast cells in a continuous mode.104 Other than the continuous flow mode, confining single cells within an array of microwells offered an alternative approach for massively parallel assays. Meanwhile, the detection sensitivity could be substantially improved, as the structure physically limited the diffusion and dilution of cellular materials. Kim et al.70 described an integrated device consisting of a dense array (3600) of tightly enclosed microwells having similar dimensions to cells (Fig. 4D). Enzymatic activity of intracellular β-galactosidase was measured after a large population of single cells were efficiently trapped by DEP (trapping efficiency up to 95%) and then lysed under a uniform electrical field. For the vast majority of the works involving electroporation for intracellular molecule release and analysis, electroporation conditions used were harsh and led to cell death. Recently, we demonstrated that it was possible to extract tiny amounts of intracellular proteins from cells without killing the cells when fine-tuned electroporation conditions were used.58 This discovery paved the way to minimally invasive cellular analysis and continuous monitoring of live cells.
Other than small molecules, metabolites and proteins, genetic materials could also be extracted from cells by electroporation.59,72–76,82 A number of microsystems combined electroporation with DEP to concentrate and lyse target cells, such as bacterial cells,74–76,82 microalga cells,73 in order to release DNA or RNA. The nucleic acid samples were often recovered from the chips and further analyzed by off-chip molecular biology techniques such as real-time PCR73,75,76 and hybridization.82 To incorporate DNA purification into the systems, a bead-based method was often used to selectively capture DNA after electrical lysis. Ramadan et al.72 constructed a continuous flow microchip to extract DNA from human white blood cells and murine clonal MN9D cells. While cells and silica beads were simultaneously loaded into the chamber, the cells were trapped and lysed by an interdigitated castellated planar microelectrode array, and the beads were also dielectrophoretically trapped to selectively bind the released DNA. More recently, we developed an integrated microchip that allowed cell tapping, electrical lysis, as well as DNA extraction and concentration.59 ChargeSwitch magnetic beads were stacked in the microchannel to form a matrix-like structure for both physical cell trapping and DNA binding based on pH alternation. The results revealed that electroporation could produce similar or higher amounts of PCR-grade genomic DNA from both mammalian and bacterial cells, compared to the commonly-used chemical lysis methods.
We have established over the years that the release of intracellular molecules by electroporation has strong dependence on the molecular size and the subcellular localization.122,125,127,128 Not surprisingly, large molecules are harder to release than small molecules125 and the nuclear fraction is harder to release than the cytosolic fraction for the same molecule.128 Using a transcription factor NF-κB which resides in the cytosol or nucleus in different cell states as a proof-of-principle, we showed that selective release of the cytosolic fraction of NF-κB was possible by tuning the electroporation parameters.128 This single-step procedure avoided laborious operations in traditional subcellular fractionation protocols. Based on the concept, we developed a novel flow cytometry tool referred to as “electroporative flow cytometry” which combined electroporation with flow cytometry for rapid analysis (∼200 cells s−1) of subcellular localization of intracellular proteins.122,127 We demonstrated that by using electroporative flow cytometry, we were able to detect protein translocations (i.e. change in the protein subcellular localization) both from cytosol to plasma membrane122 and from cytosol to nucleus.127 In both cases, we detected the fluorescence of the intracellular protein (tagged by a fluorescent protein marker) after electroporation-based release and then correlated the residual fluorescence intensity of the single cell with the original protein localization (i.e. before electroporation treatment) and quantitatively characterized the percent localization between two subcellular compartments for the protein at the single cell level. In the case of cytosol to plasma membrane translocation, the interaction between the molecule (i.e.tyrosine kinase Syk122) and the plasma membrane prevented the membrane fraction of the protein from being released by electroporation. Thus if there was a larger presence of the protein at the plasma membrane, there would be more residual fluorescence emitted by the cell after electroporation. Similarly, NF-κB localizes between the nucleus and cytosol, a larger percentage of NF-κB in the nucleus would leave higher residual fluorescence intensity after electroporative release for a cell.127 With electroporative flow cytometry, we were able to rapidly determine the protein localization information of single cells with a high speed and without imaging.
Unrelated to protein translocation and electroporation-based release, we also used combined cell electroporation and flow cytometric examination for study of cell biomechanics.126,131 We found that the expansion in the cell size during electroporation had strong correlation with the cell deformability.126 Thus we used this as a quantitative parameter to characterize cell deformability. In the follow-up work, we recorded the lysis kinetics of single erythrocytes and found that such lysis kinetics were characteristic of the defects in the cytoskeleton (due to mutated or missing proteins).131 We found that the method could efficiently discriminate red blood cell subpopulations with protein mutations and serve as a rapid tool for characterizing the deficiency involved in the cytoskeletal protein network at the single cell level.
Finally, in some works, electroporation or electric lysis was used to kill or remove undesired cells without involving analysis. Microfluidic chips allow flowing cells to be continuously lysed, thereby greatly enhancing the processing rate.45,117 By precisely controlling the temperature within the devices, flow-through electroporation using pulsed electric fields was used to inactivate bacteria (Lactobacillus plantarum) for food preservation.136,137 Based on the cell size difference, circulating tumor cells were selectively purged from blood by a flow-through electroporation technique with a clinical-scale throughput at the order of 1 mL min−1.39 Continuously and selectively dielectrophoretic concentrating leukemia cells while electrically lysing red blood cells was also demonstrated in a constriction microchannel.133
4.2. Delivery of exogenous molecules
Conventional cuvette-based electroporation has become a routine laboratory tool for delivering foreign molecules into living cells. Nevertheless, microfluidic electroporation offers a myriad of unique benefits for cellular delivery including capability for handling scarce cell samples, continuous flow mode, and potential for integration. A wide range of agents including dyes, plasmid DNA, oligonucleotides, short interfering RNA (siRNA), mRNA, proteins, polysaccharide, drugs, and nanoparticles have been delivered into cells or even embryos51,175 using the physical technique. Table S1 (in ESI†) summarizes the literature in the area and various works can be compared based on their designs and some quantitative performance metrics such as delivery efficiency and cell viability.The flow-through working mode of microfluidic electroporation substantially enhanced the processing rate of cell samples and also eliminated (or alleviated) many adverse effects associated with electroporation in static cuvettes, such as Joule heating, pH variation and bubble formation, thereby promoting the cell survival rate. In early flow-through electroporation work, consecutive electric pulses were applied to flowing cells for continuous delivery of genes,42 similar to the early flow-through electroporation work at benchscale.176 The improvement was achieved by modifying channel geometry116,121,129,168 and microelectrode architecture,71,90,108,109 optimizing the flow pattern,26,158,159 and introducing targeted nanoparticles91 as discussed above. For instance, we described a volume-scalable flow-through electroporation chip (Fig. 2A), where plasmid DNA could be efficiently delivered into mammalian cells with a high processing rate (up to ∼20 mL min−1) whiling maintaining high transfection efficiency (up to ∼80%) under optimized combination of channel geometry and electric parameters.129,130,132 Electrotransfer of mRNA into primary cells including T cells and dendritic cells has been recently demonstrated in high-throughput microfluidic electroporation systems.44,159 Wei et al.158 successfully loaded both plasmid DNA and siRNA into various cell lines in a laminar flow electroporation microchip (Fig. 3C). The system exhibited great performance with transfection efficiency as high as 70–90% and a gene silencing level up to 92%.
The other approach to improve the processing rate of electroporation was to perform massively parallel electroporation on patterned microelectrode arrays. Recently, a microarray-based electroporation system was created to be compatible with commercial multiwall plates and transfer genetic molecules into several difficult-to-transfect cell types such as MDCK, HUVEC, and DRG neurons with high transfection efficiency up to 30–90% (Fig. 1C).77 To test the electrochemotherapeutic effects on human breast cancer in parallel, Choi et al.81 miniaturized six clinical electroporators on a single chip to electrotransfer the drugbleomycin into tumor cells. Additionally, individually addressable high-density arrays of microelectrodes provided a high level of spatial and temporal control over the delivery of reagents into cells.84–86 Based on the technique, site-specific delivery enabled optimization of multiple electroporation parameters in a single experiment. Microfluidic devices with microwell structures and microelectrode arrays could physically separate cell populations, thereby restricting cell migration and contamination between neighboring cultures as well as facilitating the identification during image processing.83,163,164 Such schemes also allowed different molecules to be sequentially loaded.83 Alternatively, genetic materials could also be attached onto the electrode substrate followed by seeding cells on the surface for electroporation delivery, referred to as reverse transfection.49,50,164 The applied electric pulses facilitated desorption of nucleic acids from the substrate and translocation into the cells. Jain et al.164 applied non-contact inkjet technology to automatically and precisely introduce various nucleic acid molecules into the microwell array and this approach holds great promise for high-throughput genomic screening.
Another important advantage associated with microscale electroporation is its capability for single cell processing. Closely related to microfluidic approach, Orwar’s and Weber’s groups demonstrated the use of electrolyte-filled capillary for delivery of small molecules and biomolecules into single cells or subcellular regions.177,178 In a recent report, a variety of molecules including propidum iodide dyes, oligodexynucleotide, molecular beacon (mRNA), siRNA, plasmid DNA, and quantum dots were delivered into single cells using a nanochannel electroporation device.153 Precise amounts of these molecules could be transported into the cell by fine-tuning the number and duration of electrical pulses. Kurosawa et al.143 explored the dynamic responses of single myocytes by electrotransfer of three metabolic substrates for tricarboxylic acid metabolic cycle (glutamic acid, maleic acid, and succinic acid) into cells. Single cell manipulation may also be compatible with continuous-flow and array arrangements to facilitate cell population studies with improved throughput. A single cell could be flowed to a microhole, trapped by backside pressure, electroporated, and finally released.141,150 Using this procedure, Valero et al.150 transfected vector DNA encoding the EGFP-ERK1 fusion protein into single human mesenchymal stem cells (MSCs) and single mouse myoblastic C2C12 cells with efficiency as high as >75% without causing cell death. Nuclear translocation of the EGFP-ERK1 fusion protein in MSCs was observed upon external growth factor stimulation. Alternatively, cells could be continuously flowed through a microchannel with constriction segments whose spaces were only enough to accommodate one cell at any given moment.118,121,134 A chip with arrayed microholes allowed a nuclear transport factor importin β to be simultaneously transferred into 200 cells.144 Besides microhole arrays, single cells could be introduced to chips featuring high-density microelectrode arrays for cellular delivery. To achieve single-cell resolution, the gaps between adjacent electrodes should be close enough to allow only one cell to be confined between them. By replacing the planar electrodes,54 3D vertical sidewall electrodes were used to improve the spatial uniformity of electric treatment on single cells.93,95,96 Using this configuration, single neurons could be transfected with plasmid DNA, and in particular, local delivery of EGTA into a subcellular axon was also demonstrated.95 In an alternative method, the individual addressability was demonstrated on an array of planar circular microelectrodes with sizes similar to that of cells (15–50 μm).87,88 The array was constructed to deliver dyes, oligonucleotides, siRNA, plasmid DNA into adhere mammalian cells in a highly spatiotemporal controlled manner. In addition, single-cell electrotransfer was also performed in microfluidic droplets due to the high-throughput and tiny compartment volume offered by droplet microfluidics. Although the transfection efficiency was relatively low (∼11%), Zhan et al.57 for the first time delivered the plasmid DNA into mammalian cells encapsulated within microdroplets by electroporation. By optimizing electrode design and channel geometry, the genetic transformation of green microalgae cells with thick walls could be accomplished in microdroplets with 2–3 orders of magnitude higher efficiency than that performed in macroscale.161 This could be attributed to rapid mass and heat transfer in the device which facilitated the access of DNA to cells while avoiding adverse effects.
To promote the delivery efficiency on electroporation chips, several methods have been proposed to accelerate molecular uptake across the cell membrane. One strategy was pre-concentrating the molecules at the cell surface prior to electroporation to enhance the possibility of entry into cells. Besides loading DNA on micropatterned electrodes for reverse transfection,48,164electrophoresis could be incorporated to attract polyanionic DNA molecules to target regions.62,63,65 Lin et al.62 introduced an additional Au plate electrode on the top of a cell culture chamber with interdigitated planar microelectrodes. A low voltage pulse was initially applied between the plate electrode (cathode) and the interdigitated microelectrodes (anode) to induce DNA movement towards the adherent cells on the chip surface. Subsequently, electroporation was performed under a high voltage provided by only the interdigitated microelectrodes. To prevent the detrimental effects caused by plate electrodes such as bubble generation and solution contamination, they also fabricated the electrodes in a coplanar manner to provide a voltage for accumulating DNA.65 Adding the electrophoresis step significantly enhanced the transfection efficiency of plasmid DNA (from 16.62% to 35.89%) and Au-DNA(T21) nanoparticles. Besides electrophoresis, nanoscale physical barriers could also facilitate biomolecules bypassing the cell membrane. Two negatively-charged membranes with nanopores were used to sandwich the cells and push DNA molecules towards the cell surface.151,152 Protruded nanostraws closely underlying cultured cells further enhanced delivery efficiency because the hollow nanostructure not only concentrated the electric field at its interface with cell membranes to induce electroporation but also confined the biomolecule transport into cytosol through the electropores (Fig. 2D).155 In a different approach, superparamagnetic iron oxide nanoparticles (6 nm) were added together with plasmid DNA to improve the transfection.64 Magnetic complexes presumably formed via hydrogen bonding between DNA and the hydroxyl group of iron oxide, and then were condensed to the cell surface under strong magnetic fields. Finally, electrophoresis after electroporation was also used to actively drive genes deep into the cytoplasm and facilitate their eventual entry into the nucleus. As discussed above, electrophoretic force is able to facilitate DNA translocation into and inside the cytoplasm.31,32 We used the combination of high-intensity and low-intensity pulses by varying the width of the channel of flow-through microchips. After the high-intensity pulses in narrow segments generated pores in the membrane and enabled DNA binding to the cell membrane, low-intensity pulses in wide segments electrophoretically facilitated DNA translocation inward.121,129Electrophoresis for both pre-concentration and post-electroporation driving was combined to assist delivery of anionic calceindyes and oregon green dextran into single cells.149 The extremely high electric fields in nanochannel electroporation also created extremely strong electrophoresis to rapidly drive molecules into cells, as confirmed by the fact that the dye transport was not dominated by diffusion (Fig. 2E).153
5. Summary and outlook
The fusion of microfluidics with electroporation has created a myriad of versatile tools for analyzing intracellular contents and delivering foreign molecules into cells at the microscale. Generally speaking, microfluidic electroporation allows more precise control over the process and more flexible integration with other tools than their benchscale counterpart. Thus microfluidic electroporation provides unique advantages for single cell delivery and analysis, and integrated cell lysis and molecular analysis. Furthermore, microfluidic platforms provides unique opportunities for incorporating hydrodynamic effects into electroporation-based delivery processes and these effects often produce results that are otherwise not achievable. Most microfluidic electroporation devices are not designed for processing a large number of cells (with the exceptions of flow-through electroporation and some other devices with highly parallel operations).Future studies in this field will potentially focus on using electroporation to solve emerging and challenging cell biology problems. We will likely see major developments in several interesting areas. First, electroporation techniques will continue to play an important role in single cell studies. The works demonstrated so far have been largely proof-of-concept. The interface of electroporation with mass spectrometry and other techniques has been exciting and suggests the potential for generating real biochemical information on single cells. We expect that single cell studies will increasingly probe important molecular biological events involved in disease development with real clinical applications. Second, we will likely see more development in using electroporation for examining genetic materials in cells. Existent studies were mostly focused on peptides, proteins and small molecules. The extraction and examination of nucleic acids will open doors to genetic and epigenetic studies closely relevant to disease processes. Electroporation provides a physical approach that is clean and simple for this application. Third, gene delivery by highly precise and controlled electroporation will enable observation of cellular dynamics after genetic manipulation. We will probably see more interesting works in this direction. Fourth, delivery of nanoparticles by electroporation has been understudied and underdeveloped so far. We will potentially see an increase in the number of the papers in this area, given the lack of suitable physical approaches for nanoparticle internalization and the potential of electroporation for such applications. These exciting breakthroughs will benefit life science in broad realms including cell therapy, electrochemotherapy, clinical diagnosis, drug screening, large-scale functional screening of genome and epigenome, and cell biology studies.
Acknowledgements
We acknowledge financial support from Virginia Tech ICTAS NanoBio Trust and Center for Inflammation, NSF CBET 1016547, 0967069 and NIH NCI 1R21CA174577-01.References
- I. Hapala, Crit. Rev. Biotechnol., 1997, 17, 105–122 CrossRef CAS.
- E. Neumann, A. E. Sowers and C. A. Jordan, Electroporation and Electrofusion in Cell Biology, Plenum Press, New York, 1989 Search PubMed.
- T. Y. Tsong, Biophys. J., 1991, 60, 297–306 CrossRef CAS.
- D. C. Chang, B. M. Chassy, J. A. Saunders and A. E. Sowers, Guide to Electroporation and Electrofusion, Academic Press, Califonia, 1992 Search PubMed.
- H. Potter, Anal. Biochem., 1988, 174, 361–373 CrossRef CAS.
- P. J. Canatella, J. F. Karr, J. A. Petros and M. R. Prausnitz, Biophys. J., 2001, 80, 755–764 CrossRef CAS.
- M. B. Fox, D. C. Esveld, A. Valero, R. Luttge, H. C. Mastwijk, P. V. Bartels, A. van den Berg and R. M. Boom, Anal. Bioanal. Chem., 2006, 385, 474–485 CrossRef CAS.
- W. G. Lee, U. Demirci and A. Khademhosseini, Integr. Biol., 2009, 1, 242–251 RSC.
- S. Movahed and D. Li, Microfluid. Nanofluid., 2011, 10, 703–734 CrossRef CAS.
- S. Wang and L. J. Lee, Biomicrofluidics, 2013, 7, 11301 CrossRef.
- J. Olofsson, K. Nolkrantz, F. Ryttsen, B. A. Lambie, S. G. Weber and O. Orwar, Curr. Opin. Biotechnol., 2003, 14, 29–34 CrossRef CAS.
- M. Wang, O. Orwar, J. Olofsson and S. G. Weber, Anal. Bioanal. Chem., 2010, 397, 3235–3248 CrossRef CAS.
- J. C. Weaver and Y. A. Chizmadzhev, Bioelectrochem. Bioenerg., 1996, 41, 135–160 CrossRef CAS.
- S. Y. Ho and G. S. Mittal, Crit. Rev. Biotechnol., 1996, 16, 349–362 CrossRef CAS.
- C. Chen, S. W. Smye, M. P. Robinson and J. A. Evans, Med. Biol. Eng. Comput., 2006, 44, 5–14 CAS.
- J. Teissie, M. Golzio and M. P. Rols, Biochim. Biophys. Acta, Gen. Subj., 2005, 1724, 270–280 CrossRef CAS.
- J. M. Escoffre, T. Portet, L. Wasungu, J. Teissie, D. Dean and M. P. Rols, Mol. Biotechnol., 2009, 41, 286–295 CrossRef CAS.
- J. M. Crowley, Biophys. J., 1973, 13, 711–724 CrossRef CAS.
- H. G. Coster and U. Simmermann, J. Membr. Biol., 1975, 22, 73–90 CrossRef CAS.
- K. Kinosita Jr and T. Y. Tsong, Nature, 1977, 268, 438–441 CrossRef CAS.
- E. Neumann, M. Schaefer-Ridder, Y. Wang and P. H. Hofschneider, EMBO J., 1982, 1, 841–845 CAS.
- W. Krassowska and P. D. Filev, Biophys. J., 2007, 92, 404–417 CrossRef CAS.
- M. Golzio, J. Teissie and M. P. Rols, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1292–1297 CrossRef CAS.
- J. Teissie and M. P. Rols, Biophys. J., 1993, 65, 409–413 CrossRef CAS.
- M. Rebersek, C. Faurie, M. Kanduser, S. Corovic, J. Teissie, M. P. Rols and D. Miklavcic, BioMed. Eng. Online, 2007, 6, 25 CrossRef.
- J. Wang, Y. Zhan, V. M. Ugaz and C. Lu, Lab Chip, 2010, 10, 2057–2061 RSC.
- B. Gabriel and J. Teissie, Biophys. J., 1999, 76, 2158–2165 CrossRef CAS.
- M. P. Rols, C. Delteil, M. Golzio, P. Dumond, S. Cros and J. Teissie, Nat. Biotechnol., 1998, 16, 168–171 CrossRef CAS.
- A. Gothelf, L. M. Mir and J. Gehl, Cancer Treat. Rev., 2003, 29, 371–387 CrossRef CAS.
- F. Andre and L. M. Mir, Gene Ther., 2004, Suppl 11, S33–42 CrossRef.
- V. A. Klenchin, S. I. Sukharev, S. M. Serov, L. V. Chernomordik and A. Chizmadzhev Yu, Biophys. J., 1991, 60, 804–811 CrossRef CAS.
- S. I. Sukharev, V. A. Klenchin, S. M. Serov, L. V. Chernomordik and A. Chizmadzhev Yu, Biophys. J., 1992, 63, 1320–1327 CrossRef CAS.
- M. Kanduser, D. Miklavcic and M. Pavlin, Bioelectrochemistry, 2009, 74, 265–271 CrossRef CAS.
- T. Stroh, U. Erben, A. A. Kuhl, M. Zeitz and B. Siegmund, PLoS One, 2010, 5, e9488 Search PubMed.
- J. M. Escoffre, J. Teissie and M. P. Rols, J. Membr. Biol., 2010, 236, 61–74 CrossRef CAS.
- F. Vitzthum, G. Geiger, H. Bisswanger, B. Elkine, H. Brunner and J. Bernhagen, Nucleic Acids Res., 2000, 28, e37 CrossRef CAS.
- V. Ganeva, B. Galutzov and J. Teissie, Anal. Biochem., 2003, 315, 77–84 CrossRef CAS.
- T. Oshima and M. Sato, Adv. Biochem. Eng. Biotechnol., 2004, 90, 113–133 CrossRef.
- N. Bao, T. T. Le, J. X. Cheng and C. Lu, Integr. Biol., 2010, 2, 113–120 RSC.
- H. M. Eppich, R. Foxall, K. Gaynor, D. Dombkowski, N. Miura, T. Cheng, S. Silva-Arrieta, R. H. Evans, J. A. Mangano, F. I. Preffer and D. T. Scadden, Nat. Biotechnol., 2000, 18, 882–887 CrossRef CAS.
- S. Jeyamkondan, D. S. Jayas and R. A. Holley, J. Food Prot., 1999, 62, 1088–1096 CAS.
- Y.-C. Lin, C.-M. Jen, M.-Y. Huang, C.-Y. Wu and X.-Z. Lin, Sens. Actuators, B, 2001, 79, 137–143 CrossRef CAS.
- X. Gong, X. Yi, K. Xiao, S. Li, R. Kodzius, J. Qin and W. Wen, Lab Chip, 2010, 10, 2622–2627 RSC.
- Y. Choi, C. Yuen, S. N. Maiti, S. Olivares, H. Gibbons, H. Huls, R. Raphael, T. C. Killian, D. J. Stark, D. A. Lee, H. Torikai, D. Monticello, S. S. Kelly, P. Kebriaei, R. E. Champlin, S. L. Biswal and L. J. Cooper, Biomed. Microdevices, 2010, 12, 855–863 CrossRef CAS.
- M. Shahini and J. T. Yeow, Nanotechnology, 2011, 22, 325705 CrossRef.
- M. Shahini and J. T. Yeow, Lab Chip, 2013, 13, 2585–2590 RSC.
- M. Shahini, F. van Wijngaarden and J. T. Yeow, Biomed. Microdevices, 2013 DOI:10.1007/s10544-013-9761-0.
- N. Miyano, Y. Inoue, Y. Teramura, K. Fujii, F. Tsumori, H. Iwata and H. Kotera, Lab Chip, 2008, 8, 1104–1109 RSC.
- F. Yamauchi, K. Kato and H. Iwata, Langmuir, 2005, 21, 8360–8367 CrossRef CAS.
- H. Fujimoto, K. Kato and H. Iwata, Anal. Bioanal. Chem., 2010, 397, 571–578 CrossRef CAS.
- K. S. Huang, Y. C. Lin, K. C. Su and H. Y. Chen, Biomed. Microdevices, 2007, 9, 761–768 CrossRef.
- N. Jokilaakso, E. Salm, A. Chen, L. Millet, C. D. Guevara, B. Dorvel, B. Reddy Jr, A. E. Karlstrom, Y. Chen, H. Ji, R. Sooryakumar and R. Bashir, Lab Chip, 2013, 13, 336–339 RSC.
- Y.-C. Lin and M.-Y. Huang, J. Micromech. Microeng., 2001, 11, 542–547 CrossRef CAS.
- H. He, D. C. Chang and Y. K. Lee, Bioelectrochemistry, 2006, 68, 89–97 CrossRef CAS.
- W. Cheng, N. Klauke, H. Sedgwick, G. L. Smith and J. M. Cooper, Lab Chip, 2006, 6, 1424–1431 RSC.
- W. Cheng, N. Klauke, G. Smith and J. M. Cooper, Electrophoresis, 2010, 31, 1405–1413 CrossRef CAS.
- Y. Zhan, J. Wang, N. Bao and C. Lu, Anal. Chem., 2009, 81, 2027–2031 CrossRef CAS.
- Y. Zhan, C. Sun, Z. Cao, N. Bao, J. Xing and C. Lu, Anal. Chem., 2012, 84, 8102–8105 CrossRef CAS.
- T. Geng, N. Bao, N. Sriranganathanw, L. Li and C. Lu, Anal. Chem., 2012, 84, 9632–9639 CAS.
- L. A. MacQueen, M. D. Buschmann and M. R. Wertheimer, Bioelectrochemistry, 2008, 72, 141–148 CrossRef CAS.
- Y.-C. Lin, M. Li, C.-S. Fan and L.-W. Wu, Sens. Actuators, A, 2003, 108, 12–19 CrossRef CAS.
- Y.-C. Lin, M. Li and C.-C. Wu, Lab Chip, 2004, 4, 104–108 RSC.
- C.-P. Jen, W.-M. Wu, M. Li and Y.-C. Lin, J. Microelectromech. Syst., 2004, 13, 947–955 CrossRef CAS.
- M. Li, Y.-C. Lin and K.-C. Su, in Progress on Advanced Manufacture for Micro/Nano Technology 2005, Pt 1 and 2, ed. W. Jywe, C. L. Chen, K. C. Fan, R. F. Fung, S. G. Hanson, W. H. Hsieh, C. L. Hsu, Y. M. Huang, Y. L. Hwang, G. Jager, Y. R. Jeng, W. Li, Y. S. Liao, C. C. Lin, Z. C. Lin, C. K. Sung and C. H. Tzeng, 2006, vol. 505–507, pp. 661–666 Search PubMed.
- K.-S. Huang, Y.-C. Lin, C.-C. Su and C.-S. Fang, Lab Chip, 2007, 7, 86–92 RSC.
- S.-C. Yang, K.-S. Huang, H.-Y. Chen and Y.-C. Lin, Sens. Actuators, B, 2008, 132, 551–557 CrossRef CAS.
- J. Suehiro, M. Shutou, T. Hatano and M. Hara, Sens. Actuators, B, 2003, 96, 144–151 CrossRef CAS.
- J. Suehiro, T. Hatano, M. Shutou and M. Hara, Sens. Actuators, B, 2005, 109, 209–215 CrossRef CAS.
- J. Suehiro, A. Ohtsubo, T. Hatano and M. Hara, Sens. Actuators, B, 2006, 119, 319–326 CrossRef CAS.
- S. H. Kim, T. Yamamoto, D. Fourmy and T. Fujii, Small, 2011, 7, 3239–3247 CrossRef CAS.
- A. Adamo, A. Arione, A. Sharei and K. F. Jensen, Anal. Chem., 2013, 85, 1637–1641 CrossRef CAS.
- Q. Ramadan, V. Samper, D. Poenar, Z. Liang, C. Yu and T. M. Lim, Sens. Actuators, B, 2006, 113, 944–955 CrossRef CAS.
- M. M. Bahi, M. N. Tsaloglou, M. Mowlem and H. Morgan, J. R. Soc. Interface, 2011, 8, 601–608 CrossRef CAS.
- C. de la Rosa and K. V. Kaler, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2006, 1, 4096–4099 Search PubMed.
- C. de la Rosa, R. Prakash, P. A. Tilley, J. D. Fox and K. V. Kaler, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2007, 2007, 6303–6306 Search PubMed.
- C. de la Rosa, P. A. Tilley, J. D. Fox and K. V. Kaler, IEEE Trans. Biomed. Eng., 2008, 55, 2426–2432 CrossRef.
- H. Huang, Z. Wei, Y. Huang, D. Zhao, L. Zheng, T. Cai, M. Wu, W. Wang, X. Ding, Z. Zhou, Q. Du, Z. Li and Z. Liang, Lab Chip, 2011, 11, 163–172 RSC.
- S.-W. Lee and Y.-C. Tai, Sens. Actuators, A, 1999, 73, 74–79 CrossRef CAS.
- H. Sedgwick, F. Caron, P. B. Monaghan, W. Kolch and J. M. Cooper, J. R. Soc. Interface, 2008, 5(Suppl_2), S123–130 CrossRef CAS.
- T. Nakayama, M. Namura, K. V. Tabata, H. Noji and R. Yokokawa, Lab Chip, 2009, 9, 3567–3573 RSC.
- Y. S. Choi, H. B. Kim, G. S. Kwon and J. K. Park, Biomed. Microdevices, 2009, 11, 151–159 CrossRef.
- J. Cheng, E. L. Sheldon, L. Wu, A. Uribe, L. O. Gerrue, J. Carrino, M. J. Heller and J. P. O'Connell, Nat. Biotechnol., 1998, 16, 541–546 CrossRef CAS.
- Y. Xu, H. Yao, L. Wang, W. Xing and J. Cheng, Lab Chip, 2011, 11, 2417–2423 RSC.
- T. Jain and J. Muthuswamy, Biosens. Bioelectron., 2007, 22, 863–870 CrossRef CAS.
- T. Jain and J. Muthuswamy, Lab Chip, 2007, 7, 1004–1011 RSC.
- T. Jain and J. Muthuswamy, IEEE Trans. Biomed. Eng., 2008, 55, 827–832 CrossRef.
- S. Vassanelli, L. Bandiera, M. Borgo, G. Cellere, L. Santoni, C. Bersani, M. Salamon, M. Zaccolo, L. Lorenzelli, S. Girardi, M. Maschietto, M. Dal Maschio and A. Paccagnella, New Biotechnol., 2008, 25, 55–67 CrossRef CAS.
- L. Odorizzi, C. Ress, C. Collini, E. Morganti, L. Lorenzelli, N. Coppedè, A. B. Alabi, S. Iannotta, E. Cazzanelli, L. Vidalino and P. Macchi, Sens. Actuators, B, 2012, 170, 182–188 CrossRef CAS.
- H. Lu, M. A. Schmidt and K. F. Jensen, Lab Chip, 2005, 5, 23–29 RSC.
- S. Wang, X. Zhang, W. Wang and L. J. Lee, Anal. Chem., 2009, 81, 4414–4421 CrossRef CAS.
- S. Wang, X. Zhang, B. Yu, R. J. Lee and L. J. Lee, Biosens. Bioelectron., 2010, 26, 778–783 CrossRef CAS.
- C. Dalmay, J. Villemejane, V. Joubert, A. Silve, D. Arnaud-Cormos, O. Francais, L. M. Mir, P. Leveque and B. Le Pioufle, Biosens. Bioelectron., 2011, 26, 4649–4655 CrossRef CAS.
- H. He, D. C. Chang and Y.-K. Lee, Bioelectrochemistry, 2007, 70, 363–368 CrossRef CAS.
- H. He, D. C. Chang and Y.-K. Lee, Bioelectrochemistry, 2008, 72, 161–168 CrossRef CAS.
- W. C. Chang and D. W. Sretavan, Biosens. Bioelectron., 2009, 24, 3600–3607 CrossRef CAS.
- S. Homhuan, B. Zhang, F. S. Sheu, A. A. Bettiol and F. Watt, Biomed. Microdevices, 2012, 14, 533–540 CrossRef CAS.
- G. Tresset and C. Iliescu, Appl. Phys. Lett., 2007, 90, 173901 CrossRef.
- K. Y. Lu, A. M. Wo, Y. J. Lo, K. C. Chen, C. M. Lin and C. R. Yang, Biosens. Bioelectron., 2006, 22, 568–574 CrossRef CAS.
- C. Xie, Z. Lin, L. Hanson, Y. Cui and B. Cui, Nat. Nanotechnol., 2012, 7, 185–190 CrossRef CAS.
- N. Wilke, C. Hibert, J. O'Brien and A. Morrissey, Sens. Actuators, A, 2005, 123–124, 319–325 CrossRef CAS.
- P. J. Koester, C. Tautorat, H. Beikirch, J. Gimsa and W. Baumann, Biosens. Bioelectron., 2010, 26, 1731–1735 CrossRef CAS.
- D. Braeken, R. Huys, D. Jans, J. Loo, S. Severi, F. Vleugels, G. Borghs, G. Callewaert and C. Bartic, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2009, 2009, 2756–2759 Search PubMed.
- D. Braeken, R. Huys, J. Loo, C. Bartic, G. Borghs, G. Callewaert and W. Eberle, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2010, 2010, 6473–6476 Search PubMed.
- G. Mernier, W. Hasenkamp, N. Piacentini and P. Renaud, Sens. Actuators, B, 2012, 170, 2–6 CrossRef CAS.
- D. Shah, M. Steffen and L. Lilge, Biomicrofluidics, 2012, 6, 014111 CrossRef.
- N. Demierre, T. Braschler, P. Linderholm, U. Seger, H. van Lintel and P. Renaud, Lab Chip, 2007, 7, 355–365 RSC.
- H. Shafiee, J. L. Caldwell, M. B. Sano and R. V. Davalos, Biomed. Microdevices, 2009, 11, 997–1006 CrossRef CAS.
- Y. S. Shin, K. Cho, J. K. Kim, S. H. Lim, C. H. Park, K. B. Lee, Y. Park, C. Chung, D. C. Han and J. K. Chang, Anal. Chem., 2004, 76, 7045–7052 CrossRef CAS.
- J. A. Kim, K. Cho, Y. S. Shin, N. Jung, C. Chung and J. K. Chang, Biosens. Bioelectron., 2007, 22, 3273–3277 CrossRef CAS.
- M. A. McClain, C. T. Culbertson, S. C. Jacobson, N. L. Allbritton, C. E. Sims and J. M. Ramsey, Anal. Chem., 2003, 75, 5646–5655 CrossRef CAS.
- A. D. Hargis, J. P. Alarie and J. M. Ramsey, Electrophoresis, 2011, 32, 3172–3179 CrossRef CAS.
- J. S. Mellors, K. Jorabchi, L. M. Smith and J. M. Ramsey, Anal. Chem., 2010, 82, 967–973 CrossRef CAS.
- J. Gao, X. F. Yin and Z. L. Fang, Lab Chip, 2004, 4, 47–52 RSC.
- F. Xia, W. Jin, X. Yin and Z. Fang, J. Chromatogr., A, 2005, 1063, 227–233 CrossRef CAS.
- Y. Y. Ling, X. F. Yin and Z. L. Fang, Electrophoresis, 2005, 26, 4759–4766 CrossRef CAS.
- H. Y. Wang and C. Lu, Anal. Chem., 2006, 78, 5158–5164 CrossRef CAS.
- H. Y. Wang, A. K. Bhunia and C. Lu, Biosens. Bioelectron., 2006, 22, 582–588 CrossRef CAS.
- H. Y. Wang and C. Lu, Biotechnol. Bioeng., 2006, 95, 1116–1125 CrossRef CAS.
- H. Y. Wang and C. Lu, Chem. Commun., 2006, 3528–3530 RSC.
- J. Wang, M. J. Stine and C. Lu, Anal. Chem., 2007, 79, 9584–9587 CrossRef CAS.
- H. Y. Wang and C. Lu, Biotechnol. Bioeng., 2008, 100, 579–586 CrossRef CAS.
- J. Wang, N. Bao, L. L. Paris, H. Y. Wang, R. L. Geahlen and C. Lu, Anal. Chem., 2008, 80, 1087–1093 CrossRef CAS.
- N. Bao, B. Jagadeesan, A. K. Bhunia, Y. Yao and C. Lu, J. Chromatogr., A, 2008, 1181, 153–158 CrossRef CAS.
- N. Bao and C. Lu, Appl. Phys. Lett., 2008, 92, 214103 CrossRef.
- N. Bao, J. Wang and C. Lu, Electrophoresis, 2008, 29, 2939–2944 CAS.
- N. Bao, Y. Zhan and C. Lu, Anal. Chem., 2008, 80, 7714–7719 CrossRef CAS.
- J. Wang, B. Fei, Y. Zhan, R. L. Geahlen and C. Lu, Lab Chip, 2010, 10, 2911–2916 RSC.
- Y. Zhan, V. A. Martin, R. L. Geahlen and C. Lu, Lab Chip, 2010, 10, 2046–2048 RSC.
- T. Geng, Y. Zhan, H. Y. Wang, S. R. Witting, K. G. Cornetta and C. Lu, J. Control. Release, 2010, 144, 91–100 CrossRef CAS.
- T. Geng, Y. Zhan, J. Wang and C. Lu, Nat. Protoc., 2011, 6, 1192–1208 CrossRef CAS.
- N. Bao, G. C. Kodippili, K. M. Giger, V. M. Fowler, P. S. Low and C. Lu, Lab Chip, 2011, 11, 3053–3056 RSC.
- Y. Zhan, Z. Cao, N. Bao, J. Li, J. Wang, T. Geng, H. Lin and C. Lu, J. Control. Release, 2012, 160, 570–576 CrossRef CAS.
- C. Church, J. Zhu, G. Huang, T. R. Tzeng and X. Xuan, Biomicrofluidics, 2010, 4, 044101 CrossRef.
- R. Ziv, Y. Steinhardt, G. Pelled, D. Gazit and B. Rubinsky, Biomed. Microdevices, 2009, 11, 95–101 CrossRef.
- N. Klauke, G. Smith and J. M. Cooper, Anal. Chem., 2010, 82, 585–592 CrossRef CAS.
- M. Fox, E. Esveld, R. Luttge and R. Boom, Lab Chip, 2005, 5, 943–948 RSC.
- M. B. Fox, D. C. Esveld, H. Mastwijk and R. M. Boom, Innov. Food Sci. Emerg. Technol., 2008, 9, 101–108 CrossRef CAS.
- Y. Huang and B. Rubinsky, Biomed. Microdevices, 1999, 2, 145–150 CrossRef.
- Y. Huang and B. Rubinsky, Sens. Actuators, A, 2001, 89, 242–249 CrossRef CAS.
- Y. Huang, N. S. Sekhon, J. Borninski, N. Chen and B. Rubinsky, Sens. Actuators, A, 2003, 105, 31–39 CrossRef CAS.
- Y. Huang and B. Rubinsky, Sens. Actuators, A, 2003, 104, 205–212 CrossRef CAS.
- R. E. Diaz-Rivera and B. Rubinsky, Biomed. Microdevices, 2006, 8, 25–34 CrossRef.
- O. Kurosawa, H. Oana, S. Matsuoka, A. Noma, H. Kotera and M. Washizu, Meas. Sci. Technol., 2006, 17, 3127 CrossRef CAS.
- T. Suzuki, H. Yamamoto, M. Ohoka, A. Okonogi, H. Kabata, I. Kanno, M. Washizu and H. Kotera, Conf. Proc. IEEE Solid-State Sensors, Actuators and Microsystems, 2007, 2007, 687–690 Search PubMed.
- E. S. Lee, D. Robinson, J. L. Rognlien, C. K. Harnett, B. A. Simmons, C. R. Bowe Ellis and R. V. Davalos, Bioelectrochemistry, 2006, 69, 117–125 CrossRef CAS.
- D. B. Robinson, E. S. Lee, Z. Iqbal, J. L. Rognlien and R. V. Davalos, Sens. Actuators, B, 2007, 125, 337–342 CrossRef CAS.
- M. Khine, A. Lau, C. Ionescu-Zanetti, J. Seo and L. P. Lee, Lab Chip, 2005, 5, 38–43 RSC.
- M. Khine, C. Ionescu-Zanetti, A. Blatz, L. P. Wang and L. P. Lee, Lab Chip, 2007, 7, 457–462 RSC.
- C. Ionescu-Zanetti, A. Blatz and M. Khine, Biomed. Microdevices, 2008, 10, 113–116 CrossRef CAS.
- A. Valero, J. N. Post, J. W. van Nieuwkasteele, P. M. Ter Braak, W. Kruijer and A. van den Berg, Lab Chip, 2008, 8, 62–67 RSC.
- Z. Fei, S. Wang, Y. Xie, B. E. Henslee, C. G. Koh and L. J. Lee, Anal. Chem., 2007, 79, 5719–5722 CrossRef CAS.
- Z. Fei, X. Hu, H. W. Choi, S. Wang, D. Farson and L. J. Lee, Anal. Chem., 2010, 82, 353–358 CrossRef CAS.
- P. E. Boukany, A. Morss, W. C. Liao, B. Henslee, H. Jung, X. Zhang, B. Yu, X. Wang, Y. Wu, L. Li, K. Gao, X. Hu, X. Zhao, O. Hemminger, W. Lu, G. P. Lafyatis and L. J. Lee, Nat. Nanotechnol., 2011, 6, 747–754 CrossRef CAS.
- T. Ishibashi, K. Takoh, H. Kaji, T. Abe and M. Nishizawa, Sens. Actuators, B, 2007, 128, 5–11 CrossRef CAS.
- X. Xie, A. M. Xu, S. Leal-Ortiz, Y. Cao, C. C. Garner and N. A. Melosh, ACS Nano, 2013, 7, 4351–4358 CrossRef CAS.
- E. M. Nelson, V. Kurz, J. Shim, W. Timp and G. Timp, Analyst, 2012, 137, 3020–3027 RSC.
- T. Zhu, C. Luo, J. Huang, C. Xiong, Q. Ouyang and J. Fang, Biomed. Microdevices, 2010, 12, 35–40 CrossRef.
- Z. Wei, D. Zhao, X. Li, M. Wu, W. Wang, H. Huang, X. Wang, Q. Du, Z. Liang and Z. Li, Anal. Chem., 2011, 83, 5881–5887 CrossRef CAS.
- D. Selmeczi, T. S. Hansen, O. Met, I. M. Svane and N. B. Larsen, Biomed. Microdevices, 2011, 13, 383–392 CrossRef.
- C. Luo, X. Yang, Q. Fu, M. Sun, Q. Ouyang, Y. Chen and H. Ji, Electrophoresis, 2006, 27, 1977–1983 CrossRef CAS.
- B. Qu, Y. J. Eu, W. J. Jeong and D. P. Kim, Lab Chip, 2012, 12, 4483–4488 RSC.
- K. Xiao, M. Zhang, S. Chen, L. Wang, D. C. Chang and W. Wen, Electrophoresis, 2010, 31, 3175–3180 CrossRef CAS.
- T. Jain, R. McBride, S. Head and E. Saez, Lab Chip, 2009, 9, 3557–3566 RSC.
- T. Jain, A. Papas, A. Jadhav, R. McBride and E. Saez, Lab Chip, 2012, 12, 939–947 RSC.
- J. Wang, S.-C. Yang, C. Wang, Q. Wu and Z. Wang, Conf. Proc. IEEE Sensors, 2010, 2010, 2097–2100 Search PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. Huck, Angew. Chem. Int. Ed., 2010, 49, 5846–5868 CAS.
- S. Lindstrom and H. Andersson-Svahn, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 308–316 CrossRef.
- S. K. Kim, J. H. Kim, K. P. Kim and T. D. Chung, Anal. Chem., 2007, 79, 7761–7766 CrossRef CAS.
- J. K. Valley, S. Neale, H. Y. Hsu, A. T. Ohta, A. Jamshidi and M. C. Wu, Lab Chip, 2009, 9, 1714–1720 RSC.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- Q. Xue and E. S. Yeung, J. Chromatogr., Biomed. Appl., 1996, 677, 233–240 CrossRef.
- F. Han, Y. Wang, C. E. Sims, M. Bachman, R. Chang, G. P. Li and N. L. Allbritton, Anal. Chem., 2003, 75, 3688–3696 CrossRef CAS.
- L. A. Woods, P. U. Gandhi and A. G. Ewing, Anal. Chem., 2005, 77, 1819–1823 CrossRef CAS.
- H. Zhang and W. Jin, J. Chromatogr., A, 2006, 1104, 346–351 CrossRef CAS.
- T. Bansal, J. Lenhart, T. Kim, C. Duan and M. M. Maharbiz, Biomed. Microdevices, 2009, 11, 633–641 CrossRef CAS.
- L. H. Li, R. Shivakumar, S. Feller, C. Allen, J. M. Weiss, S. Dzekunov, V. Singh, J. Holaday, J. Fratantoni and L. N. Liu, Technol. Cancer Res. Treat., 2002, 1, 341–350 CAS.
- K. Nolkrantz, C. Farre, A. Brederlau, R. I. Karlsson, C. Brennan, P. S. Eriksson, S. G. Weber, M. Sandberg and O. Orwar, Anal. Chem., 2001, 73, 4469–4477 CrossRef CAS.
- M. Wang, O. Orwar and S. G. Weber, Anal. Chem., 2009, 81, 4060–4067 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50566a |
| This journal is © The Royal Society of Chemistry 2013 |