 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micro-scale blood plasma separation: from acoustophoresis to egg-beaters
Maïwenn
Kersaudy-Kerhoas
a and
Elodie
Sollier
b
aInstitute of Biological Chemistry, Biophysics and Bioengineering, Heriot-Watt University, Edinburgh Campus, Edinburgh, EH14 4AS, United Kingdom. E-mail: m.kersaudy-kerhoas@hw.ac.uk
bDepartment of Bioengineering, University of California Los Angeles, 420 Westwood Plaza, 5121 Engineering V, P.O. Box 951600, Los Angeles, CA 90095, USA. E-mail: elodie.sollier@gmail.com
First published on 4th July 2013
Abstract
Plasma is a rich mine of various biomarkers including proteins, metabolites and circulating nucleic acids. The diagnostic and therapeutic potential of these analytes has been quite recently uncovered, and the number of plasma biomarkers will still be growing in the coming years. A significant part of the blood plasma preparation is still handled manually, off-chip, via centrifugation or filtration. These batch methods have variable waiting times, and are often performed under non-reproducible conditions that may impair the collection of analytes of interest, with variable degradation. The development of miniaturised modules capable of automated and reproducible blood plasma separation would aid in the translation of lab-on-a-chip devices to the clinical market. Here we propose a systematic review of major plasma analytes and target applications, alongside existing solutions for micro-scale blood plasma extraction, focusing on the approaches that have been biologically validated for specific applications.
 Maïwenn Kersaudy-Kerhoas | Maïwenn Kersaudy-Kerhoas is a Royal Academy of Engineering Research Fellow in the Institute of Biological Chemistry, Biophysics and Bioengineering (IB3) at Heriot-Watt University, Edinburgh, Scotland. She received a PhD in Engineering and Physical Sciences from Heriot-Watt University in 2010. Her PhD work focused on the design, manufacturing and testing of microfluidic devices for blood plasma sample preparation. Following a post-doctoral position in an industrial programme on microfluidics for synthetic biology applications, her current research aims at applying microfluidic concepts to develop affordable, non-invasive, prenatal testing tools for use in clinics and hospitals. |
 Elodie Sollier | After receiving a Physical Engineering Degree from Grenoble Institute of Technology, Elodie Sollier obtained a PhD in Physics for Life Science from CEA LETI Minatec, at Grenoble. Her PhD was followed by post-doctoral research at the Department of Bioengineering, University of California, Los Angeles with Prof. Di Carlo. Her expertise is the development of microfluidic devices for biological applications, focusing especially on blood preparation and analysis. She has been investigating different microfabrication approaches for transitioning from laboratory research to commercialization. Elodie is now VP, Research and Development for Vortex BioSciences, focusing on the development of microfluidic devices for cancer diagnostics. |
1. Introduction
Despite all the recent advances made in the lab-on-a-chip field, sample preparation remains a major technical hurdle in the path of large scale commercialization and public access to point-of-care (POC) biomedical devices. With very few exceptions, such as the pregnancy test from a urine sample or glucose measurement from blood, sample preparation comes with any diagnostic or analysis, and has been branded “the weak link in microfluidics-based biodetection”.1 Mariella proposes several reasons for the lack of interest in on-chip sample preparation. The first reason is economical, as the emerging microfluidic field has not triggered enough real commercial possibilities for sample preparation to be developed. Moreover, microfluidic technologies do not yet compete technically and cost-effectively with existing techniques; macroscale centrifugation remains just as simple, efficient and cheap. Another reason, more social, is the “inertia” of the current techniques and the lack of will for trained staff to change their traditional protocols. Last but not least, the integration of sample preparation is a challenging technical problem in itself. Sometimes referred to as the world-to-chip issue,2 the issue even has a dedicated yearly conference “Integrating Sample Preparation” which has been running for 6 years. The integration of sample preparation is complicated by various factors including the complexity of sample matrices and the use of disparate material between sample preparation and detection units, which may lead to leakage problems. These difficulties have deterred engineers and researchers from developing adapted sample preparation solutions in the first place, and the focus has been shifted towards other functionalities, such as detection. A lack of generic automated and integrated solutions for sample preparation means that most processes are still performed in macroscale tubes on the bench, potentially introducing human errors.Several reviews have targeted the challenges of sample preparation and its integration, including blood samples in particular.1,3–5 The complexity of the blood matrix is generally pointed out as the major hurdle towards on-chip blood preparation. Although very smart and precise tools have been developed, the main focus seemed to be on the demonstration of functionalities other than sample preparation, notably on the analysis and transduction of a chemical or physical detection into a readable electronic signal. Indeed, most of the prototypes developed have been mainly focused on analytical sensitivity and how it compares to conventional techniques. Although a few commercial examples with integrated sample preparation are available, such as (i) Cholestech measuring aspartate and alanine aminotransferase for hepatic diagnostic, (ii) CoaguChek monitoring prothrombin time during anticoagulation therapy, or (iii) the BioSite chip for the detection of three proteins linked to stroke pathologies, blood sample preparation has been more rarely considered and fully integrated on-chip. It is worth mentioning that these examples detect the presence of plasma analytes at high concentration, with the capacity for capillary-scale volumes for facile detection. Besides these specific examples, a significant part of the blood preparation is still handled manually, off-chip, with variable waiting time, and under non-reproducible conditions that may impair the collection of analytes of interest. The lack of suitable devices to perform such preparation at the microscale is the bottleneck towards complete integration of blood analysis. Furthermore, the development of miniaturised modules capable of automated and reproducible blood separation would certainly aid in the translation of blood-related lab-on-a-chip devices to the clinical market.
Despite being overlooked in many studies and labs-on-chips, the separation of plasma from blood is a requirement in many medical diagnostic procedures. Plasma is a straw-colored liquid and accounts for around 55% of total blood volume. It must not be confused with serum, which is the supernatant before anticoagulants have been added to the blood (Table 1). Most of the examples detailed in this review consider plasma, unless otherwise specified. Plasma is an aqueous substance, made out of 95% water, and is host to a myriad of analytes including proteins, metabolites, circulating nucleic acids (CNAs) and other organisms. The efficient detection of these analytes requires plasma to be completely free of cells, thereby reducing the pollution of contaminants, which might otherwise interfere with the accuracy of analyses. The diagnostic and prognostic potential of plasma analytes was recently uncovered6,7 and has fuelled the research into miniaturised blood plasma separation (Fig. 1). For example, CNAs have been found to be of great utility for the diagnosis or prognosis of multiple cancers,8 malaria9 and stroke.10 CNAs are also used for the monitoring of pregnancies and transplantations.7 Similarly, various proteins have been accepted as biomarkers for the diagnosis of several cancers.11 The LOC community has recognised that the lack of convenient solutions for blood plasma separation does not match the possible breadth of applications. Thus, there has been a steady growth in the number of publications about blood plasma separation by microfluidics from 2005 onwards and an exponential growth in the number of citations of the subject (Fig. 1). New ways to diagnose and treat a patient are emerging and may lead to further opportunities for microscale blood plasma separation. For example, blood plasma separation is particularly relevant in the field of theranostics, or companion diagnostics, the new driver for bed-side diagnosis and treatment. Theranostics is defined as the combination of the diagnostic and therapeutic fields, whereby a disease could be screened daily, and is expected to reach a market worth $19.3 billion by 2023.12 Therefore it becomes necessary to get blood samples from patients one or more times each day. Microfluidics is particularly helpful in that respect, as it permits the analysis of a few microlitres of biological sample and avoids painful blood extraction of several dozens of millilitres.
| Term | Definition |
|---|---|
| Dilution ratio | Ratio of the unit volumes of blood over the total volume of sample after dilution. In most cases, diluent is Phosphate Buffered Saline (PBS). |
| Erythrocytes - RBCs | Most common type of blood cell, accounting for 98% of blood cells, at a concentration of 5 × 106 per μL of blood. Responsible for the transport of oxygen in blood. Erythrocytes are 7–8 μm large and have a discoid shape. Also known as red blood cells (RBCs). |
| Extraction rate | Flow rate at the plasma outlet in an experimental microfluidic set-up. |
| Hematocrit - Hct | Volume percentage of RBCs in blood. This value depends on the age and health of the patient. Approximated at 40–50% for healthy patients. |
| Leukocytes - WBCs | Account for 1% of the total blood cells, at a concentration of 4–10 × 103 per μL of blood, with sizes varying from 7 to 30 μm. Leukocytes play a major role in the immune response. Also known as white blood cells (WBCs). |
| Plasma | Liquid phase of the blood. Supernatant collected after centrifugation of a blood sample in which anticoagulant was added. Plasma contains fibrinogen and other clotting proteins. |
| Purity | Purity relates to the number of RBCs remaining in the plasma extracted. In this review it is defined as a percentage by: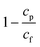 Where cp is the number of RBCs in the plasma fraction and cf is the number of RBCs in the feed (inlet) fraction; e.g.: 100% purity means no cells were detected in the plasma fraction. Where cp is the number of RBCs in the plasma fraction and cf is the number of RBCs in the feed (inlet) fraction; e.g.: 100% purity means no cells were detected in the plasma fraction. |
| Serum | Supernatant collected after centrifugation of a blood sample in which no anticoagulant was added. |
| Thrombocytes - PLTs | Account for 1% of the total blood cells, at a concentration of 150–400 × 103 per μL of blood. 2–4 μm in size. Small cells responsible for clot formation in blood and the coagulation process. Also known as platelets (PLTs). |
| Yield | The percentage of extracted plasma volume over the total volume of blood injected. |
 | ||
| Fig. 1 Graphs representing (top) the number of citations per year and (bottom) the number of publications between 1993 and 2012. These results were compiled using Thomson Reuters citation database on the “Web of Science” website using the key words “blood”, “plasma”, “separation” and “microfluidic”. While these results do not reflect exactly the number of publications in the field of microscale blood plasma separation, it gives a clear indication of the growing interest generated by the field. | ||
Recent reviews have already assessed microscale methods for continuous particle separation or sample preparation issues.1,5,13,14 In this article, we focus mainly on microscale blood plasma extraction solutions. This paper does not intend to give a historical review of the subject, but rather provides a critical review of the recent advances in the field as well as their applications. To achieve this purpose, the second part of this review assesses the analytes and biomarkers of interest in plasma and associated pathologies. The third part is focused on the challenges specific to blood plasma separation. The fourth part and core of the review is dedicated to the technological solutions currently available, which are divided into three formats: the chip format, the CD format, and the paper format. In this section, we will compare the different techniques and devices based on their performances such as purity, yield, and capacity to be integrated with analytical modules. In this purpose, a lexicon of the principal technical terms used in this review is available in Table 1. Subsequently, we will also discuss how these devices have been exploited for various applications and conclude by highlighting the possible future directions for blood plasma separation on-chip.
2. Plasma analytes and diagnostic applications
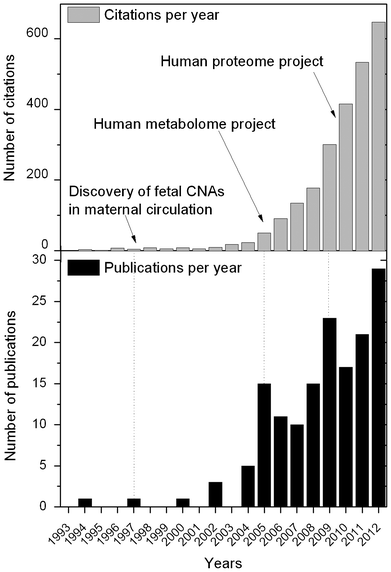
Plasma, a water-based biological buffer, constitutes more than 50% of the blood volume in most mammals including humans. Plasma can be rich with indicators of various diseases, which is why separating plasma from blood is of clinical importance. Plasma hosts many components, including but not limited to proteins, molecules, microorganisms or circulating nucleic acids, whose relative concentrations are indicated in Fig. 2. In this section we will detail a few of these useful biomarkers and their associated diagnostic applications. | ||
| Fig. 2 Blood composition. Plasma is host to a myriad of components, most of which have diagnostic and therapeutic potential. Nucleic acid levels are from ref. 18 and metabolite concentrations from ref. 6. | ||
2.1. Circulating nucleic acids
Circulating Nucleic Acids, or CNAs, are nucleic acids floating freely in human blood and other body fluids. CNAs include fragmented DNA and RNA, as well as microRNA. Although their origin is still obscure, they can be differentiated between cell-free (cf) nucleic acids and cell-associated (ca) nucleic acids.15 At the time of their discovery in 1948 by Mandel and Metais, they were not directly associated with pathologies.16 It was only 30 years later, in 1977, that CNAs were associated with a pathology in lung cancer patients.17 More recently, elevated levels of CNAs have been linked to various pathologies and emerged as potential biomarkers.18 While in healthy individuals, cfDNA concentration in blood ranges between 0 and 100 ng mL−1 with a mean value of 30 ng mL−1,19 cancer patients have higher levels of cfDNA usually around 180 ng mL−1. The release mechanisms of CNAs is thought to originate mostly from cell apoptosis.8 To support this hypothesis, CNA levels were demonstrated to drop by up to 90% after radiotherapy in several types of cancer, while more moderate drops (below 63%) have been reported in other cancers.20 Thus, CNAs may be used as a theranostic tool when assessing treatment effectiveness in patients affected by malignant tumors.Elevated levels of cell-free DNA have also been detected in patients suffering from malaria and are suspected of playing a role in malaria-induced inflammation.21 The monitoring of CNA levels could be used for the diagnosis and prognosis of this disease, along with a better clinical management in affected zones.9 Donor CNAs have been found in the plasma of hosts after organ transplantation and are described as a naturally occurring microchimerism (i.e. the presence in a small quantity of foreign genomic information in a person). Therefore they may also be used as rejection markers for the monitoring of graft versus host disease after transplantation.22
Another application of CNAs lies in Non-Invasive Diagnosis or Testing (NIPD or NIPT). Traditionally, methods to sample fetal material for prenatal diagnosis, like amniocentesis or chorionic villus sampling (CVS), are invasive with associated risks including bleeding, leakage, and foetal miscarriage. Amniocentesis leads to miscarriage in approximately 1% of cases and CVS in around 1–2% of cases.23,24 These percentages are quite significant as around 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 amniocenteses are conducted in the US each year, while 20000 amniocenteses and 5200 CVS occur every year in the UK alone. These procedures can also have undesirable collateral effects, such as infection to the mother or the foetus. Since their discovery in 1997,25 numerous studies have demonstrated the existence of cell free fetal nucleic acids (cffNAs) in maternal circulation. This flow of genetic information provides a unique opportunity for the development of techniques for NIPT. cffNA is routinely used today to diagnose gender-linked conditions26 or fetal rhesus D status and has also been demonstrated to be a potential indicator for pregnancy associated diseases such as pre-eclampsia and preterm labour.
000 amniocenteses are conducted in the US each year, while 20000 amniocenteses and 5200 CVS occur every year in the UK alone. These procedures can also have undesirable collateral effects, such as infection to the mother or the foetus. Since their discovery in 1997,25 numerous studies have demonstrated the existence of cell free fetal nucleic acids (cffNAs) in maternal circulation. This flow of genetic information provides a unique opportunity for the development of techniques for NIPT. cffNA is routinely used today to diagnose gender-linked conditions26 or fetal rhesus D status and has also been demonstrated to be a potential indicator for pregnancy associated diseases such as pre-eclampsia and preterm labour.
Furthermore, CNAs have been put forward as biomarkers for auto-immune diseases, such as rheumatoid arthritis or systemic sclerosis, where the high levels of CNAs correlate with the intensity of the disease.7 In other diseases which lead to rapid cell death, such as trauma, sepsis, or cardio-vascular accidents, elevated CNA levels have been reported and may be used as prognostic markers to judge the efficacy of a treatment and the severity of the injury.7
In this section, we have cited a few of the possible applications of CNAs found in blood plasma. A recent and exhaustive review of the subject can be found in ref. 7. The excitement around CNAs largely originates from the possibility they offer to bypass invasive procedures in the diagnosis of life-threatening tumors and genetic conditions for prenatal testing. Analysis of CNAs first requires the separation of plasma or serum from whole blood. Another advantage of CNAs is their rapid clearance from the body, which minimises carry-over contamination. Studies have shown that fetal DNA found in maternal circulation is cleared out 20 min after the birth,27 which means that CNA detection is not susceptible to false-positives which would result from earlier pregnancies.
2.2. Proteins
Proteins play a predominant part in most biological processes. Studying proteins may give clues about the health of cells as evidenced in oncological studies.28 Sampling blood is a less invasive approach to detect and monitor cancer without having to subject patients to clinical interventions such as biopsies. Instead of studying the primary tumor, cancer can now be characterized at the molecular level not only by DNA copy number analysis or patterns in gene expressions, but also by changes in serum proteins. Cancer-associated proteins may be used not only to detect cancer tumors at an early stage of development but also to monitor its progression and response to therapy.29 In a relatively recent catalogue, 22% of 1261 proteins proposed as cancer biomarkers were traced in plasma.11 Among the most common protein cancer biomarkers, Prostate-Specific Antigen (PSA) detection has been widely used as a screening test for prostate cancer, both in clinical settings30 and as an application for lab-on-a-chip devices.31,32As another protein example, a group of plasma proteins called transaminases, including aspartate aminotransferase (AST) and alanine aminotransferase (ALT), are routinely used to study the hepatic (or liver) functions. The detection of ALT and AST in serum has been demonstrated on-chip in several formats.33 Although no plasma or serum extraction steps were integrated in this example, a more recent example from Whitesides' group includes plasma filtration and transaminase analysis in a paper format.34 An exhaustive review of AST/ALT detection techniques can be found in ref. 35. C-Reactive protein or CRP is another plasma protein widely used as a non-specific marker for the acute phase during an inflammation.36 While normal CRP levels in healthy individuals are reported to be less than 10 mg L−1, these can rise up to 350–400 mg L−1 in a number of pathologies, including appendicitis and pelvic inflammatory disease. As a non-specific marker, CRP is not recommended for diagnostic purposes but may be used to indicate failure of an antibiotic treatment.36,37 However, because of the large number of methods available for its rapid and precise quantification, CRP detection is still an appealing technique for clinicians and has consequently been largely applied in lab-on-a-chip devices.38–40 Aota et al. proposed for example a passive plasma separation device and validated it with microELISA analysis of CRP levels.40 A blood filtering membrane was also proposed in conjunction with a protein capture strategy to detect CRP levels.41 However, the device was loaded directly with human serum instead of whole blood.
Proteins can also be used to monitor pregnancy, as pregnancy-related proteins are present in the plasma of expectant mothers. Human Chorionic Gonadotrophin (hCG) protein is a hormone produced during pregnancy. hCG levels are undetectable in non-pregnant women but double every two days during early pregnancy. A blood hCG test is required to confirm whether a woman is pregnant, but hCG levels are also monitored for prenatal diagnosis.42 A number of other markers including Pregnancy-Associated Plasma Protein A (PAPPA)43 and pregnancy specific β1-glycoprotein (SP1)44 can be assessed, in the latter case for the detection of Trisomy 18 or Edwards Syndrome, a serious chromosomal abnormality.
Finally, sepsis, or inflammatory response, is often associated with elevated levels of proteins.45 Plasma proteins are also used to continuously monitor inflammatory response during surgeries, such as cardiopulmonary surgery.46 In this case, the lengthy separation time (during which the operation may last several hours) is a challenge that will require specific approaches with non-clogging systems, as demonstrated in a microfluidic format in ref. 46.
Protein analysis is widely used in clinical settings to diagnose various diseases or monitor treatment effectiveness in patients. As some of these proteins are available in high concentrations in blood, most of the applications presented in this section are compatible with plasma or serum extraction on chip or paper formats dealing with capillary volumes.
2.3. Metabolites
The Human Serum Metabolome project was set-up recently to develop a database of metabolites found in biological fluids, including serum or plasma.6 Metabolites are a group of small molecules including amino-acids, sugars and fats. Five of the most abundant organic metabolites found in serum are glucose (5 mM), total cholesterol (5 mM), melanin (5 mM), urea (4 mM), and adenosine triphosphate ATP (3 mM) (out of more than 4000 catalogued in ref. 6). Analysis of multiple metabolites in the bloodstream can be used to create a patient metabolic fingerprint or signature that could help to assess health status at any given time.Glucose, or blood sugar, measurement is widely used to monitor medical conditions including diabetes. Serum glucose levels are favored over whole blood glucose levels as the variation of hematocrit may influence the results.47 The reason behind this variability lies in the higher dissolution rate of glucose in serum. However, most POC devices still measure glucose levels in whole blood, which has cast a doubt over their reliability47,48 and which might necessitate restriction of use of POC devices with patients having a normal hematocrit level.49 Glucose measurements have been widely developed in LOC microfluidic formats,50–52 as well as LOC paper format.53 However, more devices should integrate a blood plasma extraction step, for the reasons detailed above. The detection of cholesterol, the second most abundant metabolite in blood, has also been demonstrated in various chip formats. The majority of them focus on electrochemical detection and few integrate a blood plasma extraction step.54,55 In ref. 56 however, Sun et al. proposed a technique using a disposable length of tubing to extract 60% of plasma from 1 μL of blood and characterised the extracted plasma with a cholesterol colorimetric test. ATP, the cellular energy carrier, has also been detected on-chip but without sample preparation and sometimes in urine samples rather than serum or plasma.57,58
Several hormones may be of interest for lab-on-a-chip detection, including thyrotropin (TSH) or thyroxine (T4), involved in the detection of diseases linked to thyroid function. A microchip using electrophoresis was developed to perform T4 detection on serum.59 Testosterone and estradiol, respectively the male and female sex hormones, may be used to diagnose ageing-related issues.60 While some hormones were detected in a lab-on-a-chip format,61 few of these analyses were done directly on blood. One example introduced the detection of progesterone from bovine serum, but did not integrate sample treatment in the device.62
Blood glucose and other metabolites are in concentrations high enough (4–8 mM for blood glucose in healthy adult) to be compatible with capillary volumes on the scale of a finger-prick blood droplet. These metabolic applications have been quite extensively demonstrated on-chip, however most examples failed to include blood plasma extraction, which renders such devices reliant on conventional centrifugation.
2.4. Viruses
Viruses are ubiquitous and can be transmitted via various routes, such as droplet transmission, direct or sexual contact, or even contaminated food and water, and may consequently cause worldwide outbreaks.63 According to the Bill & Melinda Gates Foundation, enteric viruses, dengue and yellow fever, HIV, viruses causing influenza, and pneumonia are considered the leading causes of childhood deaths, especially in the developing world.64,65 These viruses are now priority areas of focus66 for the global health community to tackle the lack of effective, fast, and cheap diagnostics and enable earlier detection of infections and prevention of pandemics. Viruses are also an established cause of cancer; for example, the human papilloma virus is known to cause cancers of the cervix, skin, anus, and penis.Viral diagnostics involve various kinds of biological samples, such as urine and saliva,67 but blood is the sample of choice for the detection of viral pathogens like HIV or Epstein–Barr virus among others.68,69 In blood, viral particles (20–400 nm) are surrounded by larger bacteria (300 nm to 10 μm) and cells (10 to 100 μm), which makes their detection a challenge in terms of sensitivity and specificity. Importantly, the viral concentration in samples is usually low, with only 102–106 HIV virions per mL of plasma.70 In effect, to avoid blood cell contamination, many laboratories process blood samples off-chip to directly detect viruses like HIV or dengue from plasma or serum.64,70,71
Schulze et al.72 and Cheng et al.73 have presented many recent integrated devices for molecular viral detection (antigenic and genomic), especially sample-to-answer systems. (i) Miniaturized lateral-flow systems have been developed for viral antigen detection, such as the Binax NOW Flu assay and Directigen Flu assay from BD for influenza A detection.74 These techniques suffer from a lack of sensitivity (from 53% to 59% compared to 80% for macro-scale methods)75 but are fast, simple, and automated. As an example of magnetic bead-based sandwich assays, the Architect by Abbott enables the high-throughput and automated detection of viral antigens from serum or plasma with a sensitivity of 99%.76 (ii) For detection of viral genomes, only few devices integrate all steps, including viral sample preparation, RT-PCR, target capturing, and detection on a single device, such as the droplet device proposed by Pipper et al. for avian influenza H5N1 detection on throat swab samples,77 or the fully integrated device by Cho et al.78 utilizing a CD platform for Hepatitis B detection. The protocol, which involves plasma separation from whole blood, mixing magnetic beads conjugated with target specific antibodies, washing and DNA extraction, was finished within 12 min. Only one initial manual step of loading 100 μL of whole blood was required, and real-time PCR results were as good as those from conventional bench-top protocols. However, the use of cartridges or devices pre-loaded with chemical reagents brings forth the issue of denaturation and shelf-life during transport and storage. For these reasons, a recent trend for integration of viral diagnostics involves exploiting direct virion detection on-chip, using for instance SPR imaging,79 mechanical cantilevers,80 or electrical conductance methods.81 These approaches are faster, cheaper, still sensitive, and specific, but operate reliably only with pure viral samples. Consequently, some techniques have also been integrated on chip for plasma extraction and viral particle concentration.69,70,82,83
The next step towards complete integration of on-chip viral diagnostics will be a unique system which combines plasma extraction and virus concentration with direct viral detection. Among others, the on-chip detection from blood and determination of viral load for HIV, that infects millions of people worldwide, would have a wide-reaching impact on healthcare. As an illustration of these future directions, Wang et al. developed a disposable, pump-free, size exclusion-based microfilter for both plasma and HIV virion extraction from 40 μL blood in resource-constrained settings.84 Seven HIV-infected patient samples were processed with recovery efficiencies as high as 82.5%. Due to the adjustability of the pore size in their approach, such microchips could target other viruses, such as influenza.
2.5. Bacteria, fungi and micro-organisms
Bacteria, fungi and various microorganisms potentially lead to infections and are detectable in plasma. With the rise of public and political attention on nosocomial (or hospital-acquired) infections (an average 8.7% of patients are affected through the world), the rapid and direct detection of microbial contamination and microorganisms in the bloodstream has become a pressing matter over the last 10 years.85 Among the bacteria detection techniques that have been developed on-chip, Staphylococcus aureus was used as a model in the development of a miniaturized diagnostic magnetic resonance (DMR) system.86 In this article, bacteria were detected directly in spiked human serum samples. While bacteria and fungi may be found in plasma or serum, it is often directly enriched from whole blood especially in microfluidic platforms.87,88 However, more recently, bacterial detection has also made use of PCR techniques for the molecular detection of bacterial DNA.89 Several groups have demonstrated the use of microfluidic platforms for detecting Staphylococcus aureus, Mycoplasma pneumonia, and Mycobacterium tuberculosis via a PCR amplification.90,91Candida albicans, a morbid type of fungus found in the blood and the fourth source of nosocomial infection in the US, has also been detected within microfluidic platforms via PCR.92 In the case of PCR amplification of bacteria, fungi, and microorganisms, the preliminary extraction of plasma from blood is likely to increase the sensitivity of the detection technique since it minimizes the presence of background DNA from other cells. However one article reports an improved amplification in blood rather than in serum.922.6. Plasma viscometry
Physical characteristics of the plasma itself can be used for diagnostic purposes and require plasma extraction from blood. Plasma Viscosity (PV) is considered a routine laboratory measurement and ranges in healthy adults around 1.40–1.75 mPa s.93,94 PV values between 1.75 and 2 mPa s suggest chronic disorders like infections, malignancy, vascular diseases, and auto-immune responses such as rheumatic diseases.93,94 Between 2 and 3 mPa s, the PV indicates a myeloma and is actually used as a diagnostic tool in more than 90% of myeloma cases. Additionally, PV is used to detect haematological cancers and cardiovascular diseases in conjunction with other methods.95In this section, we have highlighted the presence and diagnostic value of various analytes in plasma. The design of the plasma extraction device will ultimately depend on the analyte of interest with a specific application. The following section introduces some of the challenges encountered for these different diagnostic applications.
3. Challenges
Miniaturization and integration of blood plasma extraction offer many advantages. First of all, it allows the separation of very low-volume samples, from a few microlitres to several millilitres in a relatively short time, from a few seconds to several minutes. The scale reduction and lower dead volumes limit the amount of sample processed to the amount necessary and sufficient for the analysis. Hence, these systems are more portable, i.e. transportable to the patient bedside or in the physician’s office, and several functions can be integrated in one device. Automated microfluidic systems should also be easier to use without the need for special training, dedicated labs, or cumbersome equipment. Secondly, the miniaturization of dimensions gives access to additional physical phenomena, which may enhance the separability of some biological components compared to macro-scale physics. Finally, prices are theoretically lower compared to their lab-scale counterparts, although this varies greatly depending on the chosen technology, the throughput, and the level of integration, as the cost expected for a paper device will be different from a glass or silicon device. However, designers of microsystems for blood plasma separation also face several choices and challenges, which are detailed below.3.1. Sample volume
Miniaturization should not necessarily imply the drastic reduction of sample volume; despite having a more sensitive detection in smaller systems, the volume should remain statistically representative of the analyte targeted. For such reason, two types of volumes are usually considered, also corresponding to two distinct methods of blood collection. (i) Capillary samples refer to sample volumes in the microliter range, typically a drop of blood collected from the fingertip (∼10 μL). These volumes are especially relevant when the target is highly concentrated, i.e. present in statistically high quantity for such a small volume of blood. Several commercial systems have demonstrated the use of a blood drop for the analysis of cardiac biomarker proteins such as the BioSite chip,96 for glucose dosage such as in the FreeStyle Lite system,97 or for the identification of blood type.98 In all these examples, the concentration of analytes is high enough to be detected in a drop of blood with high sensitivity. In the lab-on-a-chip community, a lot of the research has been focused on handling these microvolumes. However, the lack of “killer applications” has more recently led to a reconsideration of this focus. (ii) In most other applications, such as the amplification of rare circulating DNA from plasma, analytes are present below a meaningful quantity in a blood drop, and/or detection methods are not sensitive enough. In these cases, volumes in the range of millilitres are more relevant, and are collected by venous puncture (1–50 mL), but few commercially viable solutions have been proposed for these samples.3.2. Sample preparation time
Studies have shown that the quality of analytes in plasma and serum decreases after a prolonged contact with blood cells. Plasma and serum are more stable following rapid separation after blood collection.99 The issues with extended contact with blood cells may be multiple, including cell degradation leading to the release of unwanted genomic DNA, haemoglobin, or protein degradation. For example, in the case of non-invasive prenatal diagnostics, the total level of cfNA increases with time after blood collection as cell apoptosis occurs. Consequently the ratio of fetal CNAs versus maternal CNAs is adversely affected. Plasma extraction is therefore desirable as soon as possible after blood collection, and in a reproducible manner as the variation in preparation and consequent degradation time is a major cause of variability in analysis results. Microfluidic devices which enable fast and reproducible plasma extraction, ideally coupled with analyte stabilization in situ, would significantly improve analysis reproducibility and sensitivity.3.3. Hematocrit level
Hematocrit, the volume percentage of RBCs in blood, often directly impacts the performance of the separation device. Hematocrit levels in humans range from 29% in one-year old infants or patients presenting specific pathologies up to 68% in newborn babies, with 38 to 54% for healthy adults. Depending on the targeted applications, some devices may have to perform similarly in this large range. Some examples presented in this review show severe drops in separation performance with higher hematocrit levels, and require blood dilution before the actual separation to circumvent this performance drop.3.4. Clogging
One of the greatest difficulties faced by the engineer creating a plasma extraction system is the extremely high proportion of cells in blood, which makes blood very prone to cause clogging issues in microchannels.14,100 Added to the relative volume of cells, the intrinsic property of blood to coagulate and the platelets to aggregate add to the challenge of blood manipulation. Clogging is typically an issue for applications with long separation time in continuous systems. Some teams propose perfusion cycles to tackle this issue and periodically wash the filters,101 but this approach remains unusual and difficult to integrate in a long-term device. The most common solution to avoid such clogging is to dilute the blood with an appropriate stabilizing buffer. However, this approach introduces not only an additional mixing step, but also a change in the composition of the material. Furthermore, it is performed at the expense of the target analyte concentration downstream.3.5. Mechanical stress
Under relatively high pressure in microfluidic structures, several biological entities are at risk of rupturing, such as RBCs, or starting an adverse activation, such as platelets. In vivo, the formation of platelet aggregates is triggered by a range of parameters; however, in vitro, platelet aggregates are mainly due to a local zone of shear acceleration. A small aggregate first forms off the vertical wall on the channel and is gradually expanded by the accumulation of platelets in a tail made out of fibrin clots and restructured membrane tethers.102 Yoshimura et al. noticed that, despite a large amount of heparin in blood, fibrin clots could form in a microchannel and hinder the normal flow.103 In parallel, hemolysis describes the rupture of RBCs under mechanical stress, caused by excessive pressure or shear stress (>1500 dynes cm−2),104 or chemical stress such as osmotic pressure. The main drawback of hemolysis is the release of hemoglobin in plasma, which may contaminate or hinder subsequent reactions such as PCR or protein analysis, while RBC ghosts (membranes) may disturb the flow patterns and separation mechanism. Therefore, microsystem design for blood plasma extraction should, as much as possible, avoid the creation of local zones of high shear stress. The minimisation of local high shear zones may be possible before manufacturing, in the development phase, with the use of Computational Fluid Dynamics (CFD) software.3.6. Non-specific absorption
Non-specific absorption of biological material on surfaces such as silicone, plastics or even silicon and glass, is a common issue in miniaturisation of blood plasma extraction due to the important surface-to-volume ratio. The lower the quantity of analytes, the more important the issue. As assessed by Wong and Ho,105 common solutions to eliminate the problem include the preliminary saturation of surfaces with proteins, for instance with Bovine Serum Albumin (BSA),106 or with a specific surface treatment to render the surface of the PDMS channel less hydrophobic and less favorable for biological materials to anchor. Most common surface treatments include PEG, different types of silanization, or oxygen plasma treatment.107,1083.7. Contamination
Contamination from one blood sample to the other is generally averted by using single-use devices. However, this attribute must be determined in the design phase as it impacts the manufacturing technique associated with the device, the packaging, and instrument used for injection, and ultimately on its total cost.1093.8. Biocompatibility
Biocompatibility is an ambiguous concept and a vastly complex subject. Here, the challenge is to find the right material adapted to a specific application, which minimises the risk of non-specific absorption, hemolysis, and platelet activation. Though essential, this point seems too rarely emphasized in the literature, and may be a source of failure in the translation of research to market.109–1113.9. Product life cycle
The life cycle of microfluidic chips is rarely considered. As most blood plasma separation devices will not be reusable due to contamination issues between samples, they are usually thrown away. This may result in a vast amount of non-degradable plastics being thrown away each year. Few research groups have addressed this issue, but some examples of “green” microfluidic chips, made out of poly(lactic acid) (PLA) or natural-based materials, such as zein, have been demonstrated in the literature as early as 2004, though none of them have been demonstrated in the field of blood plasma separation.112–114 These studies have shown the possibility to manufacture microfluidic structures out of biodegradable polymers. However, and regrettably, the shelf life of the devices was not investigated. Shelf-life of biodegradable polymer chips might be significantly lower than conventional plastic ones, and should be considered during technology commercialisation efforts.3.10. Characterisation techniques
Optimum on-chip blood plasma separation techniques should have a large throughput, a high plasma yield and purity, and minimum cellular damage in order to prevent contamination of the plasma with cellular DNA and haemoglobin. Although not a direct engineering challenge, characterisation techniques should be taken into account during the testing phase of blood plasma separation microdevices. Performance in terms of separation efficiency and purity have been assessed in the literature using different devices, including hemocytometers, automated cytometers, and flow cytometers, which all provide different metrics and precisions. These differences have to be considered with caution as they make comparisons of existing approaches and performances quite challenging. Furthermore, blood plasma separation devices may be further validated using common biological techniques as previously described by the authors,115 such as (i) spectrophotometry for red cell lysis and evaluation of hemoglobin release (Cripps Method116), (ii) absorbance measurements at 280 nm to estimate the total amount of proteins and potential protein loss through the device, (iii) capillary electrophoresis, such as Bioanalyser Agilent, to confirm the presence of major proteins, (iv) enzymatic assays for evaluation of protein denaturation or (v) by performing PCR on DNA present in the plasma extracted.117 Such biological characterisation is important to demonstrate the utility of the device in a real-life clinical setting and should be considered cautiously depending on the application targeted.4. Formats and solutions for miniaturised blood plasma extraction
Various solutions are available for plasma extraction at the macro-scale depending on the desired sampling volume: blood transfusion volume (500 mL), analytic venous sample (1–50 mL), or blood droplet from a finger-prick (∼10 μL). Plasma extraction from a large volume of blood for transfusion purposes (a procedure named plasmapheresis) specifically requires continuous separation, with high-throughput and efficiency over time. Such separation is performed using centrifugation and rotation chambers, coupled with single-use plastic consumables. Transfusion equipment is large (over 1 m3), heavy (over 10 kg), expensive, and necessitates complex fluid handling, but efficiently extracts and cleans plasma before re-injection in the donor or in the patient. A review of the macro-scale techniques existing for plasmapheresis has been presented by Stegmayr.118 Filtering membranes are also commercially available and commonly used in clinical settings for dialysis, transfusion or therapeutic plasmapheresis. For venous sampling and most blood analyses performed in tubes, the conventional procedure used to separate plasma from blood cells includes centrifugation (5 min at 1500g), followed by physical separation with a pipette. This technique is simple, commonly and daily performed by clinicians, researchers, and technicians all over the world, but is still manually intensive at a macro scale, with a multi-step process that is prone to human error. Analytical centrifugation can be high throughput and fully automated, but necessitates relatively expensive equipment. Filtration may also be used for such volumes, usually directly at the point of extraction, i.e. with a filter adjustable on a syringe. Therefore, whatever the blood volume processed, the two conventional mechanisms—centrifugation or filtration—exploited for plasma separation at the macro-scale remain ubiquitous.On the contrary, a variety of solutions are available for miniaturised blood plasma extraction, which has currently been performed in three main formats (Fig. 3): (i) the microfluidic chip format, featuring fluid flowing through microchannels and based on classical microfabrication techniques, (ii) the CD format, based on a radial geometry and exploiting the laser CD developments, and (iii) the paper format, based on capillary flow migration on a cheap and biodegradable resource. Within the microfluidic chip format, separation techniques are generally classified in two distinct categories; the passive methods, based on flow properties in the microscale, as opposed to the active methods, that require an additional external field. Active and passive microfluidic chip approaches are assessed in Parts 1 and 2 of this section, while the CD and paper alternatives are detailed in Parts 3 and 4 respectively.
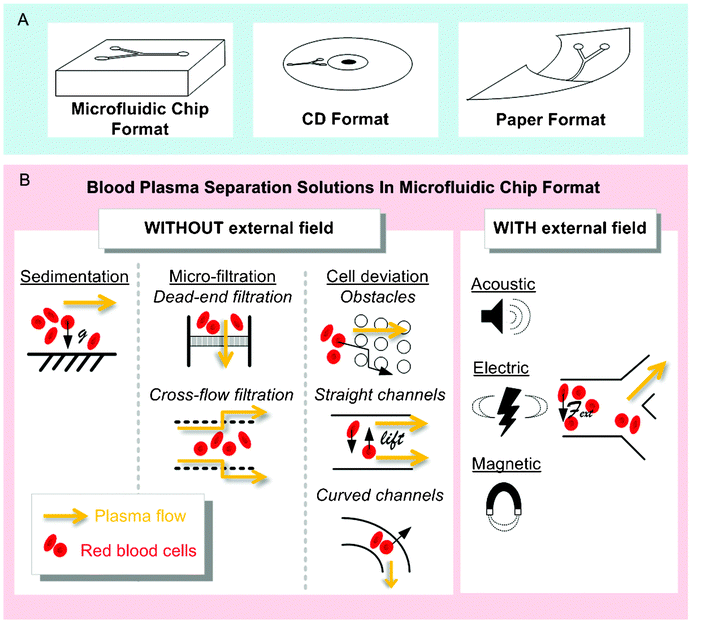 | ||
| Fig. 3 Technological solutions for micro-scale plasma separation. (A) Three different formats are currently used for micro-scale blood plasma separation. (B) A wide range of solutions have been developed for blood plasma separation in microfluidic chip format. Two categories make up the top-level classification of blood plasma separation techniques in microfluidic chip formats; techniques without external field (i.e. where separation occurs with hydrodynamic forces and built-in geometrical features) and techniques where an external field is used to force the separation of plasma from blood cells (Acoustic, Electric or Magnetic field). | ||
4.1. Microfluidic chip format and passive separation
Passive separation techniques, i.e. separation techniques without external fields, refer to those methods utilising only physical forces within the flow to induce plasma extraction from cell components. It is opposed to the sorting methods which apply an external force field, such as acoustic, electrical or magnetic field. To tackle the potential problems of such outer force field requirements in microdevices, researchers proposed some techniques typically coined as “passive” or “fluidic-only”. In these separation methods, particles are sorted exclusively by their mechanical properties, such as size, shape, and deformability, using only the geometries of microchannels or microstructures within the microchannels, the flow itself, and inherent hydrodynamic forces. Passive techniques were also the bases behind the first attempts of separation miniaturization, as researchers logically tried to first reproduce in smaller scales some principles that were well known and validated in the macroscale.119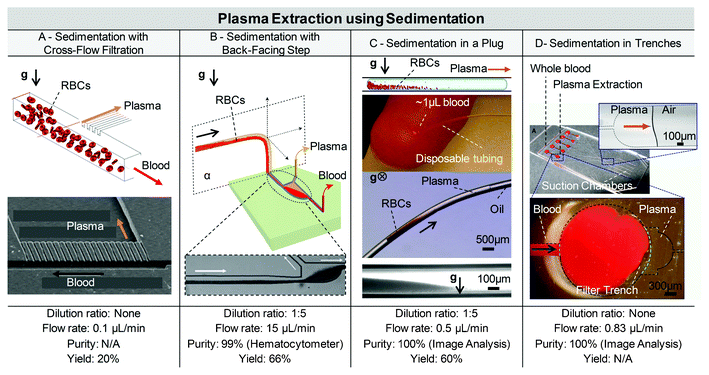 | ||
| Fig. 4 Plasma extraction using sedimentation. (A) Sedimentation and filtration, with 20% plasma collected in upper branch channels. Adapted from ref. 128. (B) Sedimentation is advantageously exploited with a back-facing-step, for 4 h of continuous blood perfusion at 15 μL min−1. Adapted with permission from ref. 129. Copyright 2012 American Chemical Society. (C) Sedimentation in a blood droplet blocked by oil plugs, with plasma extraction combined with cholesterol colorimetric assay. Adapted with permission from ref. 56. (D) Sedimentation of red cells in trenches for capillary extraction of plasma, combined with ELISA assay to measure biotin levels. Adapted with permission from ref. 127. | ||
Sedimentation advantageously reduces clogging issues inherent to microfiltration, as RBCs sediment away from the entrance of filter parts, enabling the free flow of plasma through the filter. As an illustration, Tachi et al. combined sedimentation and cross filtration featuring two parallel main channels connected via 1 μm deep shallow trenches.122 1:6 diluted blood samples are injected at 1 μL min−1 into the blood inlet, while a buffer is injected in the second channel at the same rate. RBCs gradually sink down the 20 mm long channel while plasma flows freely within the second channel. Although few platelets passed in the plasma collection channel, no blood cells were detected. Moreover, the haemoglobin concentration was measured to be less than 1 mg dL−1, which is not likely to influence further molecular analysis.
A sedimentation approach with a backward facing step for continuous plasma separation was described by Zhang et al.123 The key strategy in this design is to keep the natural sedimentation of blood cells unperturbed in a glass capillary channel, while the separation of plasma from the cell phase is facilitated by the addition of a constriction and a back-facing step (Fig. 4B). The purity reached 99% at 15 μL min−1, but decreased at higher flow rates and for 8% Hct (∼1:5 dilution). No clogging was observed during 4 h of continuous perfusion.
A capillary-driven system taking advantage of RBC sedimentation rate has been recently demonstrated by Wu et al.124 The system comprised two deep-seated reservoirs connected by a series of parallel channels sitting above the reservoirs and featuring filter pores and chambers, respectively 6–10 μm and 50–100 μm high. On one end, the reservoir is filled with blood, where RBCs start sinking toward the bottom. The top part of the reservoir is sucked by capillarity into the parallel channels, where the passage of the remaining RBCs is hampered by the combination of pores and chambers. After a number of pores, no RBCs remain in the plasma channel (by visual observation) while the plasma keeps flowing towards the outlet reservoir. However these results are demonstrated with a diluted blood sample (Hct between 0.2% and 20%), since clogging issues leading to the device failure were reported for whole blood and 1:2 blood dilution.
Recently, Sun et al. also proposed a novel sedimentation strategy coupled with a droplet generation system.56 In this new approach, a finger-prick blood droplet is sucked by capillarity into an oil-filled piece of tubing and blocked by a second plug of oil. Blood samples need to be at least 1:5 diluted before separation can occur, within 120–240 s at 0.5 μL min−1, with higher flow rates leading to a weaker separation effect (Fig. 4C). While sedimentation was demonstrated as the principal effect responsible for blood cell separation, the kinematics of the plug may also play a role. This plug-based plasma separation offers an opportunity to incorporate low shear rate (1 s−1) blood plasma separation with droplet-based technologies. A colorimetric assay for the detection of cholesterol was elegantly integrated within the same device and demonstrated the biological validity of the plasma extracted.
Sedimentation is sometimes used in combination with weirs or trenches to optimise and speed up the plasma extraction process.125–127 Dimov et al. used a trench in a capillary-driven system to extract plasma from a 5 μL droplet of blood, and integrated an ELISA assay in their Stand-alone Integrated Microfluidic Blood Analysis System (SIMBAS) to detect biotin levels.127 While blood cells fall and are trapped in a 2 mm deep trench, the plasma is extracted by capillary force into upper channels, further coated with antibodies for the ELISA assay in situ (Fig. 4D). In this study, 100% separation purity was obtained through image analysis for flow rates lower than 50 mL h−1, even with artificially high hematocrit levels (74%). Biotin was detected on spiked blood samples in concentrations as low as 1.5 pM.
In all these examples, the separation throughput is necessarily low, as any increase in flow rate will result in the instability of the separation interface. In consequence, the sedimentation approach is not adapted for rapid separation of large volumes of blood, a major limitation to this technique. However, sedimentation has several specific advantages, which still makes it a powerful solution compatible with small volumes (finger-prick volume) and capillary pumping, and explains the recent publication of new devices. Sedimentation can easily be used without the need for blood dilution and offers a very effective solution in terms of separation efficiency. Also, coupling with filtering geometries can be further optimized to increase extraction yield.
Dead-end microfiltration. Membranes, pores, and packed beads have been created to allow the flow-through (or dead-end) filtration of plasma from blood for miniaturised applications.
The membrane often corresponds to the integration of a macroscale and commercial filtering membrane within a microsystem for a 2D-filtration.130 Thorslund et al. integrated a polymeric membrane with pore size of 0.40 μm between two PDMS slabs. To avoid hemolysis and RBC leakage, the blood samples had to be diluted down to 20% Hct with a sealed polypropylene (PP) device. Testosterone absorption tests showed that some protein fouling happening on the PDMS surface did not preclude its detection (91% recovery). In ref. 84, a filter membrane (with 2 μm pore size) cut from chromatographic paper is sandwiched in a reservoir between several layers made out of several laminated PMMA laser-cut layers. 40 μL of blood were injected, followed by a minimum of 200 μL of PBS buffer, at flow rates ranging from 100 to 500 μL min−1, forcing the cells against the filter and washing plasma through the collection chamber. Plasma purity reached 97.9% with pore sizes of 0.4 μm, but the collected volume was only 55 μL since the filter clogged very quickly. The collected volume reached 220 μL with pore sizes of 3 μm, but the purity dropped to 74.9%. The plasma quality was successfully assessed by using HIV recovery test, which showed up to 89.9% recovery for a viral load of 1000 copies per mL. Moorthy et al. presented a device with an integrated membrane, or porous plug, allowing the filtration of cells larger than 3 μm.131 The porous plug is a pre-polymer mixture containing a cross-linker for hardening the plug after photopolymerisation. After the pre-polymer mixture is drawn into a channel by capillary action and cured, incubation in water prior to the separation allows the plug to swell from its dried shrunken state. Rabbit blood was diluted 1:20 and incubated in hypotonic solution to make the cells swollen and less flexible, thus less likely to pass through the porous pores. After a certain volume, the filter is clogged by cells, and the cross-sectional area of the filter governs the total plasma yield. Here 20 μL of plasma could be extracted for an unknown volume of blood, at flow rates up to 10–20 μL min−1. The concentration of G-6-PDH enzymes in the plasma extracted on-chip was found to be equivalent to plasma extracted by centrifugation.
Silicon-based filter materials were used by Lee et al. to manufacture a planar filter with well-defined pore geometry.132 These filters, made out of a Si3N4 layer and a SiO2 stress-release layer, allowed for the separation of 1 μL plasma out of 5 μL blood. Although the authors reported visual observation of hemolysis at the filter outlet, an SDS-PAGE analysis did not reveal significant amount of haemoglobin (data not communicated). A small agarose gel block was attached to the back of the filter and reported to enhance the plasma flow through the filter via an increase of the capillary suction due to the porosity of the gel. Microscopic images of the gel do not show RBCs passing through the filter. The gel was melted down and analysed for the presence of plasma proteins, such as albumin. Clogging of the filter occurred after 5 min of use, and although the filter seems effective, it is not clear why such complexity would be necessary, unless the silicon filter was integrated to a silicon chip for a specific purpose such as refining the sensing of plasma analytes.
Bead-packed microchannels may be an easier way to incorporate effective blood plasma separation functions into capillary-driven blood analysis systems.133–136 In ref. 133, 10 to 20 μm silica beads were packed against a bump in the main channel of a Cyclic-Olefin Copolymer (COC) device. Protein-blocking solution was coated on the channel and bead walls in order to change the hydrophobicity of the channel surfaces. Some improvements include the use of two different bead sizes, with 100 μm-sized beads to block the inlet channel entrance and 10 μm ones to act as a filter, which leads to the separation of 350 nL of plasma from 5 μL of undiluted blood in less than 2 min.134 In another example, the plug was created by the evaporation of water in a bead slurry (beads ranging from 0.9 to 10 μm) injected at the entrance of the separation channel in a PDMS slab.135 10 μL of a whole sheep blood droplet was drawn by capillary action through the separation channel and bead slurry (Fig. 5A). 188 nL of plasma was extracted from 10 μL of whole blood in 10 min. In another article, plasma extracted on the same system from whole blood was validated through the detection of immunoglobulin (IgG), the main antibody responsible for fighting infection in body tissues.136 The detection occurred on antibody strips immobilised on the PDMS slab in the reaction zone located downstream from the deposited microbead-plug (DMBP). Here, the DMBP has a double function by also serving as a carrier for the labelled conjugates, which combines elegant plasma extraction with target analysis.
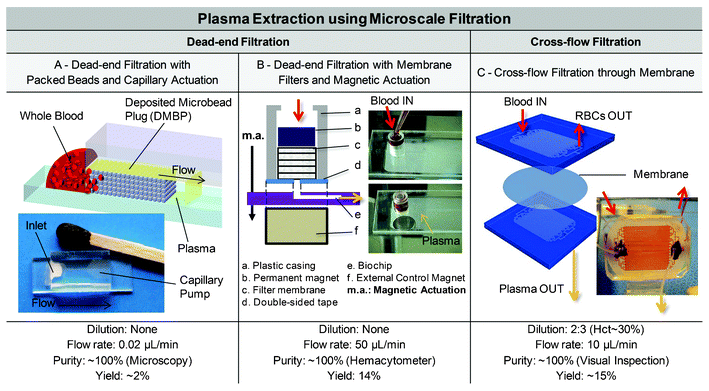 | ||
| Fig. 5 Examples of blood plasma separation using microscale filtration. (A) Dead-end filtration with packed beads (DMBP) and capillary-flow actuation.135 Red blood cells remain trapped within the bead plug while plasma can flow through the beads. The DMBP can be further used as a carrier for labelled conjugates, for bioassay on the plasma extracted. Adapted with kind permission from Springer Science and Business Media from ref. 136. (B) Dead-end filtration with membranes and magnetic actuation. A non-diluted blood sample is dropped into the filter unit. Magnetic attraction is applied to squeeze out only plasma while blood cells are filtered by the 6 membranes stacked together in the filter unit. Adapted from ref. 137. (C) Example of cross-flow membrane filtration. Left. The bottom compartment is a filtrate channel, where plasma flows across a semipermeable membrane from the top reservoir into the filtrate channels. The activated membrane and these 2 PDMS layers are bonded together to form a sandwich structure. Adapted from ref. 46. Right. This device is shown during blood perfusion for cardiopulmonary bypass procedures. Original photograph kindly provided by J. D. Zahn and K. Aran. | ||
While most examples require some pumping techniques or are self-actuated via capillary pumping, Chung et al. proposed an interesting magnetic actuation, where 3 to 8 filter membranes were sandwiched between a permanent magnet and a chip with a single channel in a plastic casing (Fig. 5B).137 After a few drops of blood have been pipetted on the membranes, (i) an external control magnet is brought underneath the chip, which (ii) attracts the permanent magnet located on the filter membranes, and (iii) squeezes the membranes to (iv) enable the extraction of 7 μL of plasma from 50 μL of whole undiluted blood in less than 1 min. The purity obtained was close to 100% (via hemacytometer counting). Although the system could be further optimized, it might be a practical actuation mechanism to replace or complement capillary actuation in some cases.
Cross-flow filtration. In cross-flow filtration, the fluid flows tangentially rather than through the filter as it does in membrane filtration. This type of filtration is widely used in several industrial applications, including those in chemical engineering and bioengineering, and has been rationally applied in microsystems for blood plasma separation and on-chip extraction purposes.
Crowley et al. proposed several planar microfilters138 based on cross-flow filtration and capillary actuation. The plasma can flow freely through pores (200 μm wide, 0.5 μm high, and 50 μm long) arranged longitudinally across the channel length. 14 to 45 nL of plasma were extracted from 5 μL of whole (40% Hct) or diluted (19% Hct) bovine blood, with an output plasma flow rate ranging between 35 and 175 μm s−1. The plasma was observed by microscopy and showed no trace of cell or cell debris. No further characterisation was performed on the extracted plasma. In ref. 100, a cross-flow design was proposed with a network of channels to counteract the effect of PDMS compliance, which can lead to channel deformation and thus create uneven device performance. Furthermore, the authors also used h-PDMS, which has a Young's modulus four times higher than PDMS. Interestingly, a back-pulsing strategy minimised the filter structure clogging and 10% of plasma was extracted from 20% Hct blood samples at 1 μL min−1, while hemolysis was measured via the Cripps method at 0.08%. Chen et al. developed cross-flow filtration devices using an array of posts as well as weir structures, and indicated the capability of retrieving plasma containing “only a few blood cells”, with no further characterisation.139 Sollier et al. proposed original cross-flow filtering geometries, featuring U-shapes and serpentine designs,140 but performing only 5% extraction from 1.4 μm deep filters and 1:10 diluted blood. In another cross-flow solution proposed by Aran et al. and dedicated to the continuous monitoring of inflammatory response during cardiopulmonary bypass procedures (cf. section 2.2), the device consists of two slabs of PDMS separated by a porous polycarbonate (PCTE) membrane with 100 to 200 nm pore sizes as pictured in Fig. 5C. While the 200 nm pore size initially provides a higher output flow rate, it also clogs faster.46 Heparin was used on the blood samples in order to avoid protein fouling on the membranes and reduce clogging. 15% of plasma was extracted from heparin-treated and slightly diluted blood (27–30% Hct) with high purity (100%, the characterisation technique not being specified) at 80 μL min−1. The device was connected to a cardiopulmonary bypass (CPB) circuit, and plasma was collected over a 4 h period with 80% of plasma proteins recovered.
As shown in these examples, dead-end and cross-flow filtrations are efficient but most of the time relatively short-term solutions for separating plasma from cell suspensions. A low flow rate is preferable, but it increases the risk of protein adhesion in the filters. The major drawback of such techniques is the risk of clogging, which limits their performance by the saturation time of the filter. The entrance to the filter pores is quickly clogged and eventually red blood cells might pass through the filter pores due to their deformability. Thus, these techniques are efficient but only for a certain time defined by saturation, for a given volume and a diluted sample, at low flow rates. Optimized filtration might call for more complex fabrication technologies, especially when small filter pores are required,132 but can also make use of simple chromatographic paper integrated in microfluidic channels.84,137 Most importantly, these examples differ fundamentally from the paper format devices described in Section 4.3, where whole assays are performed on paper, which acts as the filter as well as the support.
Deterministic deviation by obstacles. As emphasized with microfiltration approaches, microstructures can be arranged with intervals well adapted to the dimensions of the cells being filtered. Microstructure arrays can be also exploited to alter cell streamlines, as in Deterministic Lateral Displacement (DLD). The first DLD system was set up and tested by Huang et al.141 with micro-pillars of circular cross-section and arranged in rows within a microchannel. Each row of pillars is shifted from the other by a distance which partly sets the critical size cut-off. The asymmetric bifurcation of laminar flow around these obstacles leads particles to choose their paths deterministically on the basis of their size. A small particle will have a zig-zag displacement path, whereas a large particle will tend to flow straight. After several rows, the particles can be collected separately. In ref. 142, this method was successfully applied to blood samples, with one device (FD) demonstrating the fractionation of WBCs and RBCs and a second one (PD) showing plasma extraction. In the PD device, plasma was separated from RBCs, WBCs and PLTs in a cascade device with anti-clogging features, at an injection rate of 0.4 μL min−1. The flow of plasma was visually observed with the unbound PLT antibodies forming a uniform bright background in the device. However no biological validation was performed on the extracted plasma. An optimized version of the same device was later focused on the measurement of platelet size and morphology in platelet-rich-plasma (PRP) and included upstream a DLD array for the removal of RBCs and WBCs from whole blood.143 However, plasma itself was not characterised in this second study either.
In another example by Choi et al., a micro-pillar array is replaced by slanted obstacles for the efficient extraction of plasma.144,145 The combination of hydrophoretic separation with a vertical weir placed in the pathway of the lined up particles enabled size fractionation. The larger particles do not pass through the narrow gap but instead follow the obstacle to a larger gap at the other side of the channel. The yield of plasma in this device was close to 40% for 1:4 diluted rat blood at a flow rate of 4 μL min−1 (Fig. 6A).145
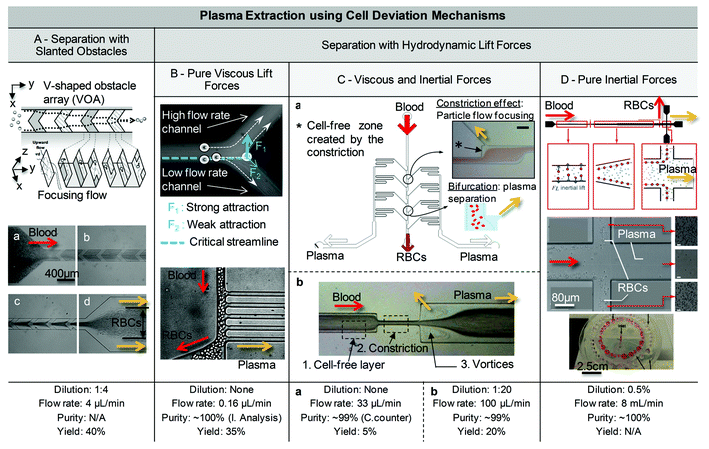 | ||
| Fig. 6 Examples of blood plasma separation using hydrodynamics and cell deviation mechanisms. (A) Separation with slanted obstacles. The generation of transverse flow due to the presence of slanted obstacles allows the concentration of red blood cells towards the center of the channel. Diluted rat blood was injected for demonstration in the ∼7 μm high channel Adapted from ref. 145. (B) Separation using a viscous lift force. Top: An illustration of the Zweifach–Fung effect at a bifurcation. As explained in ref. 149, the effect does not result, as previously thought, from an attraction towards the channel with the highest flow rate, but rather from the initial distribution of cells in the channel. Bottom: The use of several plasma channels placed in parallel and along the main channel allows the extraction of plasma. Adapted from ref. 154. (C) Top: Separation based on biomimetic effect, such as the Zweifach–Fung effect, but at a higher flow rate (up to 0.33 mL min−1). The scale bar is approximately 50 μm. Adapted from ref. 152. Bottom: Similarly, other devices can expand the initial cell-free layer to increase plasma extraction yield. Adapted from ref. 115. These designs are based on red blood cell lateral migration and the resulting cell-free layer locally expanded by geometric singularities such as the effect of a restriction, an enlargement of the channel or a cavity adjacent to the channel. (D) Separation using inertial lift forces. Top. Inertial lifts acting on large blood cells lead to their migration to equilibrium positions, located near the channel walls. RBCs can then be collected in side channels, while the plasma rich in bacteria is extracted in the middle outlet. This geometry can be advantageously parallelized, with 40 channels placed in a radial array, 1 inlet for injecting blood at 8 mL min−1, and 2 rings of outlets respectively for filtered blood and blood plasma collection. Adapted from ref. 106. Copyright © 2010 Wiley Periodicals, Inc. | ||
Laminar and deterministic deviation by microstructures is a powerful and robust separation mechanism, which also suffers from various drawbacks, including the necessity of extremely low flow rate, significant dilution of the sample, along with the precise and consequently expensive fabrication process. Last but not least, especially obvious for the DLD technique, the number of pillars employed and the narrow gaps between them bring a high risk of clogging, which explains why DLD has rarely been adapted exclusively for the separation of plasma.
Hydrodynamic separation, starting from viscous lift forces. Hydrodynamic separation techniques are particularly relevant in the case of blood plasma separation, where the natural axial migration of blood cells in microchannels, sometimes referred to as the Fahraeus effect, or plasma skimming, can be exploited as the sorting mechanism. Briefly, the Reynolds number, a dimensionless number, gives a ratio of inertial forces to viscous forces.146 For Reynolds numbers lower than 1, viscous forces are expected to overcome inertial forces, yielding flows in microchannels that are expected to be laminar and devoid of turbulence. Such flow characteristics also occur in blood capillaries where, due to lift forces applied to cells in microchannels, deformable red cells undergo a lateral migration.147 Thus, a boundary-layer a few microns thick, named the cell-free layer, becomes depleted of red cells, making it possible to extract a clear plasma volume by branching off this cell-free layer into an appropriate side channel. Microcirculation effects also include the Zweifach–Fung bifurcation law. Fung et al. demonstrated that cells at a bifurcation have a tendency to travel to the highest flow rate daughter channel, provided that the flow rate ratio is at least 2.5
:1 and the dimensions of the cells are comparable to the channel diameter.148 This effect has often been described by the asymmetrical distribution of pressure and shear forces applied on the cell at the bifurcation, which might “pull” the cell to the channel with the highest flow. However, it was recently demonstrated that the basis of this effect is probably due to the initial repartition of particles in the channel, as the cell-free boundary layer itself is enough to allow the extraction of plasma though lateral bifurcations.149
These physical laws derived from microcirculation have been studied, used, and mimicked to design microstructures for continuous plasma separation.115,150–152 Faivre et al. first mentioned the use of this natural cell-free layer for plasma extraction on-chip, further demonstrating that a restriction or constriction of the microchannel increases this cell-free layer up to 1 cm downstream for specific geometries.150 During the continuous injection of 16% Hct blood at 200 μL h−1, 24% of the plasma originating from this expanded cell-free layer was extracted with two lateral collection channels. However, the blood was initially modified with washing steps, and no further estimation of the extraction purity nor biological validation were provided. By using a pure viscous lift force,153 Geislinger et al. separated platelets from RBCs from highly diluted samples (Hct = 0.1%) at low flow rates, ranging between 20 μL h−1 to 750 μL h−1. Citing the Zweifach–Fung law, Jaggi et al. presented a three-dimensional structure that took advantage of a high-aspect ratio to produce a better plasma yield.151 Yang et al. also used the Zweifach-Fung law in their design,154,155 where 100% pure plasma (purity checked through image analysis) from sheep blood was extracted with up to 25% yield and an extraction rate of 4 μL min−1 (Fig. 6B). A bar-coded protein detection system integrated in the plasma channels was coupled with this technique,156 and although the system was not tested with whole blood, the detection of hCG and a dozen protein cancer biomarkers was demonstrated on clinical serum.
Although all these examples draw their inspiration from long-standing microcirculation laws with low Re, the flow rates used in most of these extraction devices are relatively high. Inertial forces probably occur and combine with viscous forces, leading to an even stronger lift on the blood cells, which increases the efficiency and throughput of blood separation devices, as this is certainly the case in ref. 115, 117, 152 and 157. For instance, Kersaudy-Kerhoas et al. presented a system with a network of bifurcations allowing the separation of plasma from blood with up to 60% purity and 40% yield for diluted blood at relatively high flow rates (2–20 mL h−1) (Fig. 6C).117 Later on, after adding a constriction which enhances the cell-free plasma layer before each bifurcation, the purity was raised to 100% for whole blood at high flow rate (10 mL h−1), but at lower plasma yield (∼8%).152 The purity of the extracted plasma was successfully validated by the direct PCR amplification of a house-keeping gene (GAPDH) from CNAs. Furthermore, the CNA detection was validated for various dilution ratios and purity levels. Other original geometries have been proposed to locally amplify the initial cell-free layer. For instance, Sollier et al. showed that the clear plasma region could be expanded locally by certain geometric singularities, such as an abrupt enlargement of the channel or a cavity along the channel (Fig. 6C).115 These singularities create vortex flow patterns, which help by cleaning the plasma from contaminating red cells and increasing the extraction yield, providing a 3-fold improvement over the reference device simply extracting the initial cell-free layer. The extraction yield was around 18% for 1:20 diluted blood injected at 100 μL min−1. Additionally, extracted plasma was biologically validated, with no protein loss or denaturation, no hemolysis, and with excellent cell purity for red cells, platelets and white cells. Temperature was found to increase the cell-free layer by up to 250% in another device using relatively high flow rates (up to 12 mL h−1) to extract plasma through constrictions and bifurcations.157 A purity of 97% was obtained with an entrance flow rate of 200 μL min−1 and a temperature of 37 °C. A plasma yield of 3.5% was reported but no biological validation of the plasma was proposed.
These devices, mimicking blood microvascular networks, are especially attractive for continuous plasma extraction. Based on a purely passive fluidic principle, with no need for filters or external force fields, these devices are compatible with high flow rates. The designs are simple, with no restricting dimensions and do not require any complex fabrication technology. For all these reasons, such designs make it possible to integrate rapid plasma extraction in a lab-on-a-chip or could even serve as a stand-alone application. A new range of separation techniques has also recently arisen from the use of pure inertial hydrodynamic effects.
Moving on to inertial hydrodynamic separation within straight channels. Against widespread thinking that inertial forces have no action in microfluidic systems, Di Carlo and others have to be acknowledged for demystifying the area158 and demonstrating that inertial focusing can actually separate and concentrate microparticles and cells in microchannels63,158,159 at Reynolds numbers higher than 1. Briefly, inertial particle focusing is based on the superposition of two inertial lift forces, (i) a shear gradient lift force, due to the shear rate of a parabolic flow, that is directed towards the channel walls, and (ii) a wall effect lift force, due to reflection of a particle's wake, that is directed away from channels walls. Consequently, diluted suspensions of particles flowing in confined channels with finite inertia will migrate across streamlines to occupy dynamic equilibrium positions located approximately halfway between the channel centerline and walls. This phenomenon has been first emphasized by Segré and Silberberg in cylindrical channels, where particles focus to an annulus within the channel cross-section.160 In square or rectangular channels, inertial lift forces also focus particles, reducing these positions from an annulus to four precise locations, at a rate that depends on the particle and channel diameter. By changing the symmetry of the channel (i.e. for a very wide or very tall channel), single-point focusing can be achieved and has been successfully applied to blood cells for high-throughput cytometry.159 Subsequently, by splitting the outlet channel in correspondence to the streamlines, focused particles can be collected in a smaller volume and significantly concentrated.161
A device using such technology was introduced by Di Carlo et al. to remove plasma and pathogenic bacteria during transfusion (Fig. 6D).106 Each single channel consists of a focusing length followed by a gradual expansion and collection region. Blood cells align themselves near the walls, where they can be collected in side channels, while plasma is extracted in the middle outlet with bacteria. Separation experiments were conducted with whole human blood diluted to 0.5% v/v in PBS, spiked with E. coli and injected at 200 μL min−1. More than 80% of plasma and pathogenic bacteria were extracted from blood after two passes, with 82% of the RBCs being collected in cell outlets (i.e. 18% of the RBCs remaining in the plasma), and 5% hemolysis. The main interest of this work was the massive parallelization of the separation unit, which enables the continuous processing of large volumes of sample (30 mL) with extreme throughput (8 mL min−1 as total flow rate, with 400 million cells min−1). 40 single devices were placed in parallel, with one single inlet and 80 outlets, for a final footprint of 7 by 7 cm. In this design, the side outlets formed an inner ring for blood cell collection, while an outer ring of outlets was used for collecting blood plasma. Lee et al. proposed a series of expansions and contractions to vary channel aspect ratio and modify the amplitude of inertial lift forces.162 This device was applied to the continuous plasma extraction from whole blood, at relatively high-throughput (1.2 mL h−1 and 1 × 108 cells min−1) and high efficiency, with 62.2% extraction of the plasma in lower outlet. However, plasma collected off-chip was significantly diluted by PBS via co-flow (12 mL h−1) and the plasma purity was poor, with a RBC rejection ratio of only 60% (i.e. collected plasma still contains 40% of the RBCs) which was also visually confirmed by the outlet photographs included in the paper. Multiple serial processing steps would be interesting with such a device to improve plasma purity while maintaining high-throughput. More recently, Amini et al. have introduced an innovative strategy to program fluid motion using the inertial flow deformations induced by sequences of microstructures combined with inertial focusing for accurately controlling the motion of particles/cells.163 Using a specific program, the cross-stream translation of the fluid surrounding particles has been demonstrated with 72% of the liquid being extracted in one outlet, with only 1.4% of particle contamination. Such deterministic control of both particles and fluid motion represents a promising approach for continuous and fast plasma extraction from blood.
Hydrodynamic separation in curved channels. By using channels with curvature, an additional drag force arising from secondary flows – Dean vortices – balances these inertial lift forces and has been shown to enhance the speed of particle migration to more stable equilibrium positions.164 Consequently, the focusing of particles is faster than in straight channels. The use of curved channels originally came from attempts to master centrifugation in microsystems, not by using the centrifugal force induced by a rotating element but instead the one induced by flow in curved channels.14 Dean vortices, well described in the literature,165,166 consist of a pair of counter-rotating helical vortices located symmetrically with respect to the mid-plane of symmetry of the channel curve. Cells or particles flowing through a curved channel are then exposed to two lateral forces in addition with inertial lift forces. The centrifugal force makes the particles with higher density travel towards the outer channel wall, whereas Dean vortices tend to mix and disperse the beads due to the Dean drag force. Di Carlo and others have demonstrated the precise control of these forces for accurate particle focusing and its application to high-throughput and continuous blood separation.166–168 However, such a mechanism requires significant blood dilution, such as the 2% dilution considered in ref. 167. An original 180° bend device was also proposed in ref. 14, coupling inertial migration in a straight channel with Dean forces in the curve. The device was tested with 1
:20 diluted blood and plasma separation was visually observed, but no quantitative data was provided. Blattert et al. proposed a bend structure to increase the plasma free layer existing naturally in microchannels by imparting a centrifugal effect to force them on one side of the channel.169 Li et al. reported the extraction of 124 μL of plasma (purity not communicated) from 250 μL of slightly diluted blood at a flow rate of 10 μL min−1 in a spiral device combining cross-flow filtration, Dean vortices and centrifugal effects.170,171 More recently, Morikawa et al. introduced a spiral microchannel for blood injection at 5 mL min−1, separation of plasma at 90% (no indication of the protocol used), and use of the plasma/serum extracted for dry eye therapy.172
While viscous lift forces and biomimetic effects may be effective, these techniques are severely limited by slow flow rates. On the contrary, inertial effects at the microscale appear extremely promising in enhancing the natural cell-free layer and addressing the plasma extraction challenge, potentially enabling an approach which is (i) simple and requires no complex fabrication, (ii) low-cost so that it can be widely deployed, with (iii) high filtration efficiency resulting in good purity performance, (iv) high throughput, and finally, (v) able to operate on large volumes of sample. For all these reasons, inertial focusing could critically address the current plasma separation challenges from relatively large sample volumes (0.5 mL and above), but may be less useful for blood screening from small finger-prick volumes (∼10 μL).
4.2. Microfluidic chip format and active separation
The main application of FFA consists of cell focusing and continuous separation of particles from its liquid phase. Hence it is well adapted for plasma extraction. For instance, in the device proposed by Nilsson et al.,179 non-diluted blood is injected at 600 μL min−1 in an 8-parallel channels separator. Piezoceramic excitation is transverse so that cells align themselves in the channel center, whereas plasma remains in both lateral channels. Four devices are disposed in series for a final purity close to 99%. Protein quality of plasma extracted by acoustophoresis has been additionally validated by the authors with the detection of PSA biomarker at clinically relevant levels (Fig. 7A).31 Doria et al. recently presented a novel acoustic plasma separation method,180 including the integration of air–liquid cavity transducers. These new types of transducers have the advantage of creating microstreams within the flow patterns and swirling eddies where the RBCs become aggregated. The authors reported a plasma extraction rate of 10.6 nL s−1. The plasma was allegedly pure at 90% or more, although no characterisation technique was mentioned in the article.
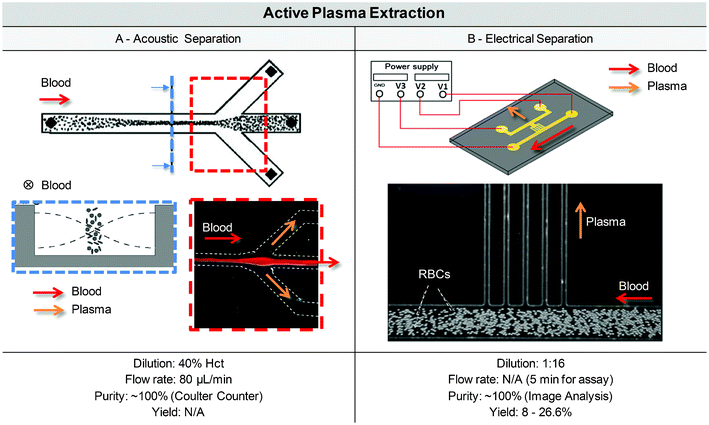 | ||
| Fig. 7 Active blood plasma separation techniques in microfluidic format. (A) Acoustic plasma extraction. As blood is injected within the microfluidic channel and through a standing wave field, red cells experience acoustic forces and align themselves in the channel centre, while plasma can be collected continuously and efficiently near the channel walls. Reprinted with permission from ref. 31. Copyright 2009 American Chemical Society. (B) Electrical plasma extraction. An electric field is applied across the main channel and generates the blood flow. A transverse field allows the plasma flow towards the bifurcation channels but is not strong enough to generate red cell motion. Adapted from ref. 183 with permission from IOP publishers. | ||
:9 diluted blood. A better yield was demonstrated, but at the expense of the purity. No biological characterisation of the extracted plasma was related.
Electro-osmotic flow was used by Jiang et al.183 to generate liquid flow, as well as to extract a fraction of blood plasma. Both of these functions were achieved by applying an electric field across a main channel, and a weaker transversal electric field across a series of bifurcations perpendicular to the main channel (Fig. 7B). The main electric field generates the blood flow across the main channel. At the same time, the transverse field allows the actuation of plasma flow in the bifurcation channels, but is not strong enough to pull the blood cells away from their paths. In the actual experiment, blood was diluted by 1:16 in PBS buffer, very few cells were seen entering the plasma channels, and the plasma yield was reported to be as high as 26%.
Although these techniques have to be optimized and fully characterized, they may have some potential for highly integrated plasma separation combined with in situ analysis. Indeed, the electric field could be used to actuate the flow and induce cell extraction from plasma as demonstrated in the latest example, but also to separate entities (CNAs, proteins) within the extracted plasma as well.
Magnetic separation of plasma from blood is theoretically possible, but slightly impractical due to the additional requirement of additives or membranes to render RBCs paramagnetic. It is more suited for niche applications where some cells might naturally be in a deoxygenated or paramagnetic state. This is the case, for example, for fetal nucleated red blood cells which are found to be naturally paramagnetic in maternal circulation,191 or malaria-infected red blood cells in the circulation of affected patients.192
4.3. Paper format
In the past 3 years (2009–2012) paper-based microfluidics has been the centre of a flurry of research activities.193,194 Paper has been widely used for lateral-flow assays, such as glucose and lipid measurements,195 coagulation measurements196 or pregnancy tests. But more recently, paper, a cost-effective, easily sourced, and readily biodegradable material, has been re-engineered to host a variety of integrated microfluidic functions, including plasma extraction from blood. The separation on paper-based format proposed in this section differs from the dead-end filtration section (cf. section 4.1.2) as in the latter, only the membrane is made of paper, not the entire system.Although paper is used in several devices to extract plasma from blood, few examples showcase paper as the only supporting format for the entire assay. In one example from Yang et al., chromatography paper was not only used as a substrate to agglutinate RBCs and extract plasma but also for glucose metering applications (Fig. 8A).53 As chromatography paper pores are large enough to let single RBCs flow through, an agglutination strategy was developed to form RBC aggregates and prevent them from flowing through the paper. On the other hand, plasma flows by capillary action away from the point where the finger-prick volume of blood has been deposited. The plasma extracted was reported to have a hematocrit value of 0.5 ± 0.2%, and was subsequently tested for glucose concentration with a colorimetric assay elegantly integrated in the paper structure (Fig. 8A). Glucose concentration was estimated to be within 10% of the concentration obtained with a spectrophotometer, demonstrating the validity of the device.
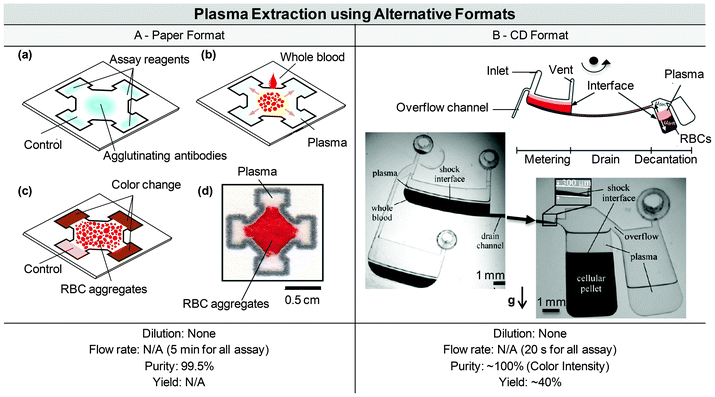 | ||
| Fig. 8 Blood plasma extraction on paper and CD format. (A) Design of a microfluidic paper-based analytical device (μPAD) with integrated plasma extraction. Adapted from ref. 53. Agglutinating antibodies and assay reagents are spotted on the μPAD. When a drop of whole blood is added on the device, RBCs react with agglutinating antibodies and remain in the central zone. Plasma migrates into the test zones and reacts with colorimetric assay reagents. (B) Plasma extraction on a CD format. Adapted from ref. 202. Whole blood is first centrifugally metered (5 μL), flows out of the metering chamber into the drain channel, where plasma and cellular constituents are separated along with centrifugation. Purified plasma is decanted into a separate reservoir while the cellular pellet is retained at the bottom of the decant chamber. | ||
Another example of blood plasma separation coupled with a direct assay on paper format was demonstrated by Songjaroen et al.197 The device was manufactured using a wax-dipping technique and featured a blood plasma separation membrane combined with patterned Whatman n°1 paper. The design comprises a separation chamber and two detection zones. 15 to 20 μL of blood with hematocrit varying between 24 to 55% are deposited in the separation chamber, and within 2 min, the plasma is extracted and enters by capillarity into the detection zones. This technique did not require an agglutination strategy as in ref. 53. Microscopy observation was used to check plasma purity, and no cells were found in the detection zone. Following the plasma separation, a bromocresol green colorimetric assay was used to detect the presence of albumin in the chip detection zones. The chip assay resulted in similar performance as the conventional method.
Whitesides' group proposed a design where the plasma is filtered vertically through the paper. The colorimetric detection of proteins related to liver functions (alkaline phosphatase (ALP) and AST) was integrated with plasma separation.34 For more convenience, the device was partially encapsulated in plastic, protecting the sample from excessive evaporation. The wax-printing technique employed allows hydrophobic barriers to be patterned in the paper and defines hydrophilic zones for the spotting of three independent assays. 15 μL of whole blood were applied on top of the filter, and the plasma was checked visually after 15 min. GX and GR filters from Pall were found to be suitable for this application. Despite discrepancies in the way the colorimetric assays were developed, the detection of ALP, AST and BSA was successfully demonstrated with whole blood samples.
While paper can be used conveniently to separate several microlitres of blood such as finger-prick volumes, it rapidly clogs and is unsuitable for larger volumes, limiting its use to the analysis of highly concentrated analytes, such as glucose53 or abundant proteins.197 Nevertheless, it is expected that researchers in the paper field will come up with several analysis techniques compatible with low plasma volume, which might in turn translate into a range of useful commercial lateral flow diagnostic kits.
4.4. CD format
Research on miniaturised CD-based labs-on-a-chip started in the 1970s, but really kicked-off in the early 2000s with the founding of Gyros and several research labs, including (but not limited to) Madou's and Ducree's. The CD-based format allows neat sample preparation as well as analysis, and therefore are good examples of lab-on-a-chip devices in terms of functional integration. An excellent exhaustive review of the subject is available.198,199 Taking full advantage of the centrifugation aspects, this format is obviously well-suited for blood plasma extraction and this has been demonstrated in several publications. Plasma extraction in a CD-based format relies on the centrifugal force forcing the heavy cellular content towards the external edge of the CD, while the plasma is siphoned out by various valving systems. The success of the plasma separation in CD format relies on these valving systems, which have included passive siphoning,200 hydrophobic, capillary,201,202 or ferrowax microvalves.203An early example of plasma extraction in a CD-format was demonstrated by Abaxis Inc and allowed plasma extraction from 40 μL of whole blood and subsequent glucose measurement in less than 10 min.201 The turbidity and amount of hemolysis were assessed photometrically, however this data was not communicated. Later, Haeberle et al. demonstrated the extraction of plasma from 5 μL of blood in less than 20 s.202 The plasma yield was 40%, with a residual hematocrit level of 0.11% (Fig. 8B). No biological validation of the plasma was presented at the time. A fully integrated immunoassay, including plasma extraction, was demonstrated by Lee et al.203 This system was able to retrieve 50 μL of plasma out of 150 μL of whole blood (33% yield) in less than a minute. It can be noticed that the color of the plasma extracted is pink, which is a sign of haemoglobin release. Whereas this was due to the use of old blood or to the characteristics of the separation device itself, it cannot be ascertained. Nevertheless, the subsequent concentration measurements of the antigen and the antibody of Hepatitis B virus were performed, and found to be within 14% of the conventional bench-top equivalent. The latest example of plasma extraction on a CD platform was demonstrated by Madou's group.200 Plasma separation from a 2 mL blood sample took 2.5 min and the authors claimed a plasma purity superior to 99.9%, although no mention of the characterisation technique was made. Looking at the pictures, the color of the plasma was pink, reflecting the possibility of hemolysis. Again, this could be due to the age of the blood, the parameters used for the centrifugation, or the device design. No associated biological assay was presented.
Protocols on the CD-based format are rapid, and the fabrication of CD cartridges is simple and cost-effective. Parallelization is enabled by having several similar chambers within one CD cartridge. However, the technology is limited by the use of numerous valves and the fact that the movement of fluid from one chamber to another relies only on the centrifugation speed. Despite these limitations, as noted by the authors of the “Centrifugal Microfluidics” review, it is surprising that no more commercial applications have been released.198
5. Future directions of microscale blood plasma separation
In the past five years (2008–2013), the LOC community has been sensitive to the challenges posed by blood sample preparation, and a high number of publications have demonstrated the clinical utility of this “sample prep” in a vast number of applications. This review has explored the diversified range of analytes found in plasma, which are attracting a growing interest for diagnostics and prognosis purposes, such as cancer biomarkers, viruses or circulating nucleic acids among others. As blood irrigates all our organs, the number of analytes of interest will not cease to rise, and we have yet to uncover diagnostic potential of even smaller plasma molecules, such as microRNA for example. MicroRNAs are short strands, less than 22 nucleotides, of non-coding RNA, which may play a key role in gene regulation and present therapeutic and diagnostic opportunities204 and, in particular, in a number of cancers.205–207In the field of plasma separation, three categories of sample volumes are prominent: large volume samples (over 500 mL) for transfusion, medium volume samples (0.5–50 mL) from venous collection for multi-parameter blood analyses, and small volume samples (∼10 μL) from finger-pricks for specific analyses. Due to the vast number of devices created and developed by LOC researchers and engineers, microfluidic solutions are capable of answering the needs of each of these situations. For instance, sedimentation strategies and capillary flow actuation lend themselves very well to the detection of highly concentrated analytes in capillary volumes of blood, such as glucose, some proteins and other metabolites, in which case slow flow rates do not hinder performance. For venous sample volumes, biomimetic hydrodynamic solutions at relatively high flow rates are especially well suited, as well as high-throughput active techniques like acoustophoresis, since despite needing more equipment, transducers are relatively easy to integrate. Some continuous solutions, such as inertial microfluidics that can operate at extremely high flow rates (mL–L min−1), may even be applicable to transfusion-scale volumes and plasmapheresis applications through the parallelization of several base units. Clearly, there is no single solution to solve all the challenges laid at the beginning of the review, but there are a myriad of systems which perform best for particular applications. Future solutions may lay in hybrid technologies, which integrate two or more of these technologies to achieve high throughput as well as high purity.
Generally speaking, the transposition of commonly used macro-scale technologies at the microscale, although tempting, has rarely been straightforward due to challenges such as clogging issues for microfiltration or the creation of Dean secondary flows in microchannels with curvature. Biomimetism works best at the microscale, and the techniques arising that exploit natural hemorheological phenomena, with some modifications, are producing some of the best results in microscale blood plasma separation.
Portability and ease of use are more than ever in the designers' mind, and sometimes lead to very unconventional innovations, with the example of the egg-beater developed by Wong et al. for hand-powered plasma extraction.208 By its simplicity and the familiarity of its actuation device, this example stands out in the blood plasma separation literature panorama (Fig. 9). In this example and several others over the past three years, ease of use, portability and cost-effectiveness come first, and these devices have been specifically designed for resource-poor settings. Paper devices and designs with capillary flow actuation also address the issue of equipment limitations relative to the operating environment, however they remain limited to concentrated analytes. On the other hand, portability may not be the only criteria of interest. Some devices solve other challenges, such as the collection of small volumes of plasma (less than a vacutainer tube) over a large period of time (>4 h), as with the cross-flow filtration technique applied to the cardiopulmonary bypass machinery.46
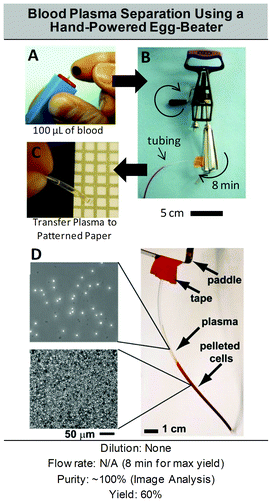 | ||
| Fig. 9 Plasma extraction using an egg-beater. (A) Approximately 100 μL of blood sample is collected in a polyethylene (PE) piece of tubing using a rubber bulb. Blood can also be drawn from a finger-prick. (B) The tubing is sealed at both ends and attached at the end of one paddle on the egg-beater. After 8 min of manual spinning at approximately 1200 rpm, the tubing is detached from the egg-beater and cut at the interface between plasma and supernatant. (C) The plasma is put in contact with the paper, which absorbs the plasma in the colorimetric cholesterol assay regions. (D) The purity was found to be close to 100% (50000 cells remaining per mL). Adapted from ref. 208. | ||
The importance of clinical validation was largely acknowledged by research communities in the past five years, and more and more articles demonstrate some sort of clinical validation, either through the demonstration of classical biological assays (such as ELISA or PCR), or in specific clinical settings through inter-disciplinary collaboration with clinicians. Furthermore, the failure of some lab-on-a-chip technologies may be attributed to the lack of clinician involvement, failing to leverage industrial interest, and subsequently having a lack of funds for commercialisation purposes. Clinicians and their staff are the end-users of most of these sample preparation systems and are thus best placed to define their need for new equipment, capable of reducing the cost of analysis or speeding or refining the acquisition of results. Furthermore, collaboration engineering is the best approach to raise awareness about real clinical needs and challenges, and consequently the key to developing potentially marketable technologies usable in clinical settings. Collaboration forces engineers to think innovatively by always adapting themselves to novel situations and should be the way forward for the LOC community.
Acknowledgements
The authors would like to thank Dr Holger Schulze (University of Edinburgh, Scotland), James Che (University of California, Los Angeles, USA) and Dr Rajan Kulkarni, M.D. (Division of Dermatology, UCLA Medical Center) for proofreading this manuscript and providing useful comments. MKK acknowledges the Royal Academy of Engineering for funding and Prof. Marc Desmulliez and Dr Helen Bridle for useful discussions. MKK would also like to thank Dr Deirdre Kavanagh for useful discussions on cffNAs. ES acknowledges the CEA and the Program for Health Technologies for funding and would like to thank Prof. Jean-Luc Achard, Dr Yves Fouillet, Dr Myriam Cubizolles, Dr Magalie Faivre, Dr Herve Rostaing and Prof. Dino Di Carlo for useful discussions on microfluidics and blood applications.References
- R. Mariella Jr, Biomed. Microdevices, 2008, 10, 777–784 CrossRef.
- J. H. Wang, Anal. Bioanal. Chem., 2005, 381, 809–811 CrossRef CAS.
- N. Pamme, Lab Chip, 2007, 7, 1644–1659 RSC.
- M. Toner and D. Irimia, Annu. Rev. Biomed. Eng., 2005, 7, 77–103 CrossRef CAS.
- S. Mukherjee, T. G. Kang, Y. Chen and S. Kim, Crit. Rev. Biomed. Eng., 2009, 37, 517–529 CrossRef.
- N. Psychogios, D. D. Hau, J. Peng, A. C. Guo, R. Mandal, S. Bouatra, I. Sinelnikov, R. Krishnamurthy, R. Eisner, B. Gautam, N. Young, J. Xia, C. Knox, E. Dong, P. Huang, Z. Hollander, T. L. Pedersen, S. R. Smith, F. Bamforth, R. Greiner, B. McManus, J. W. Newman, T. Goodfriend and D. S. Wishart, PLoS One, 2011, 6, e16957 CAS.
- J. Wagner, Biochemia Medica, 2012, 22, 24–38 CrossRef CAS.
- H. Schwarzenbach, D. S. B. Hoon and K. Pantel, Nat. Rev. Cancer, 2011, 11, 426–437 CrossRef CAS.
- B. S. Franklin, B. L. F. Vitorino, H. C. Coelho, A. Menezes-Neto, M. L. S. Santos, F. M. F. Campos, C. F. Brito, C. J. Fontes, M. V. Lacerda and L. H. Carvalho, PLoS One, 2011, 6, e19842 CAS.
- T. H. Rainer, L. K. S. Wong, W. Lam, E. Yuen, N. Y. L. Lam, C. Metreweli and Y. M. D. Lo, Clin. Chem., 2003, 49, 562–569 CAS.
- M. Polanski and N. L. Anderson, Biomarker insights, 2007, 1, 1–48 Search PubMed.
- J. P. G. Information, in PR Newswire, 2013 Search PubMed.
- M. Kersaudy-Kerhoas, R. Dhariwal and M. P. Y. Desmulliez, IET Nanobiotechnol., 2008, 2, 1–13 CrossRef CAS.
- E. Sollier, H. Rostaing, P. Pouteau, Y. Fouillet and J.-L. Achard, Sens. Actuators, B, 2009, 141, 617–624 CrossRef CAS.
- P. Pinzani, F. Salvianti, M. Pazzagli and C. Orlando, Methods, 2010, 50, 6–6 CrossRef.
- P. Mandel and P. Metais, C. R. Acad. Sci. Paris, 1948, 142, 241–243 CAS.
- S. A. Leon, B. Shapiro, D. M. Sklaroff and M. J. Yaros, Cancer Research, 1977, 37, 646–650 CAS.
- P. B. Gahan and R. Swaminathan, Ann. N. Y. Acad. Sci., 2008, 1137, 1–6 CrossRef CAS.
- P. Anker and M. Stroun, Medicina-Buenos Aires, 2000, 60, 699–702 CAS.
- S. A. Leon, B. Shapiro, D. M. Sklaroff and M. J. Yaros, Cancer Research, 1977, 37, 646–650 CAS.
- S. Gal, C. Fidler, S. U. E. Turner, Y. M. D. Lo, D. J. Roberts and J. S. Wainscoat, Ann. N. Y. Acad. Sci., 2001, 945, 234–238 CrossRef CAS.
- V. Garcia Moreira, B. Prieto Garcia, J. M. Baltar Martin, F. Ortega Suarez and F. V. Alvarez, Clin. Chem., 2009, 55, 1958–1966 Search PubMed.
- L. P. Shulman and S. Elias, Western Journal of Medicine, 1993, 159, 260–268 CAS.
- F. Mujezinovic and Z. Alfirevic, Obstet. Gynecol., 2007, 110, 687–694 CrossRef.
- Y. M. D. Lo, N. Corbetta, P. F. Chamberlain, V. Rai, I. L. Sargent, C. W. G. Redman and J. S. Wainscoat, Lancet, 1997, 350, 485–487 CrossRef CAS.
- K. M. Finning and L. S. Chitty, Semin. Fetal Neonat. Med., 2008, 13, 69–75 CrossRef.
- Y. M. D. Lo, J. Zhang, T. N. Leung, T. K. Lau, A. M. Z. Chang and N. M. Hjelm, Am. J. Hum. Genet., 1999, 64, 218–224 CrossRef CAS.
- C. L. Sawyers, Nature, 2008, 452, 548–552 CrossRef CAS.
- S. M. Hanash, S. J. Pitteri and V. M. Faca, Nature, 2008, 452, 571–579 CrossRef CAS.
- National Cancer Institute, Prostate-Specific Antigen (PSA) Test, http://www.cancer.gov/cancertopics/factsheet/detection/PSA, 2013 Search PubMed.
- A. Lenshof, A. Ahmad-Tajudin, K. Järås, A.-M. Swärd-Nilsson, L. Åberg, G. r. Marko-Varga, J. Malm, H. Lilja and T. Laurell, Anal. Chem., 2009, 81, 6030–6037 CrossRef CAS.
- Q. Zhu and D. Trau, Anal. Chim. Acta, 2012, 751, 146–154 CrossRef CAS.
- N. Ohgami, S. Upadhyay, A. Kabata, K. Morimoto, H. Kusakabe and H. Suzuki, Biosens. Bioelectron., 2007, 22, 1330–1336 CrossRef CAS.
- S. J. Vella, P. Beattie, R. Cademartiri, A. Laromaine, A. W. Martinez, S. T. Phillips, K. A. Mirica and G. M. Whitesides, Anal. Chem., 2012, 84, 2883–2891 CrossRef CAS.
- X.-J. Huang, Y.-K. Choi, H.-S. Im, O. Yarimaga, E. Yoon and H.-S. Kim, Sensors, 2006, 6, 756–782 CrossRef CAS.
- B. Clyne and J. S. Olshaker, J. Emerg. Med., 1999, 17, 1019–1025 CrossRef CAS.
- E. L. Tsalik, L. B. Jaggers, S. W. Glickman, R. J. Langley, J. C. van Velkinburgh, L. P. Park, V. G. Fowler, C. B. Cairns, S. F. Kingsmore and C. W. Woods, J. Emerg. Med., 2012, 43, 97–106 CrossRef.
- M. Wolf, D. Juncker, B. Michel, P. Hunziker and E. Delamarche, Biosens. Bioelectron., 2004, 19, 1193–1202 CrossRef CAS.
- S. Choi, S. Huang, J. Li and J. Chae, Lab Chip, 2011, 11, 3681–3688 RSC.
- A. Aota, S. Takahashi, K. Mawatari, Y. Tanaka, Y. Sugii and T. Kitamori, Anal. Sci., 2011, 27, 1173–1178 CrossRef CAS.
- L. Gervais and E. Delamarche, Lab Chip, 2009, 9, 3330–3337 RSC.
- P. Von Lode, J. Rainaho and K. Pettersson, Clin. Chem., 2004, 50, 1026–1035 CAS.
- P. Bischof, S. Duberg, W. Herrmann and P. C. Sizonenko, British Journal of Obstetrics and Gynaecology, 1981, 88, 973–975 CrossRef CAS.
- P. C. Ho and W. R. Jones, American Journal of Obstetrics and Gynecology, 1980, 138, 253–256 CAS.
- H. Freund, S. Atamian, J. Holroyde and J. E. Fischer, Ann. Surg., 1979, 190, 571–576 CrossRef CAS.
- K. Aran, A. Fok, L. A. Sasso, N. Kamdar, Y. Guan, Q. Sun, A. Undar and J. D. Zahn, Lab Chip, 2011, 11, 2858–2868 RSC.
- S. Kanji, J. Buffie, B. Hutton, P. S. Bunting, A. Singh, K. McDonald, D. Fergusson, L. A. McIntyre and P. C. Hebert, Crit. Care Med., 2005, 33, 2778–2785 CrossRef.
- R. A. Sidebottom, P. R. Williams and K. S. Kanarek, Clin. Chem., 1982, 28, 190–192 CAS.
- A. Cook, D. Laughlin, M. Moore, D. North, K. Wilkins, G. Wong, A. Wallace-Scroggs and L. Halvorsen, American Journal of Critical Care, 2009, 18, 65–71 CrossRef.
- C.-J. Huang, Y.-H. Chen, C.-H. Wang, T.-C. Chou and G.-B. Lee, Sens. Actuators, B, 2007, 122, 461–468 CrossRef CAS.
- J. H. Pei, F. Tian and T. Thundat, Anal. Chem., 2004, 76, 292–297 CrossRef CAS.
- V. Srinivasan, V. K. Pamula and R. B. Fair, Anal. Chim. Acta, 2004, 507, 145–150 CrossRef CAS.
- X. Yang, O. Forouzan, T. P. Brown and S. S. Shevkoplyas, Lab Chip, 2012, 12, 274–280 RSC.
- A. Wisitsoraat, P. Sritongkham, C. Karuwan, D. Phokharatkul, T. Maturos and A. Tuantranont, Biosens. Bioelectron., 2010, 26, 1514–1520 CrossRef CAS.
- S. Aravamudhan, A. Kumar, S. Mohapatra and S. Bhansali, Biosens. Bioelectron., 2007, 22, 2289–2294 CrossRef CAS.
- M. Sun, Z. S. Khan and S. A. Vanapalli, Lab Chip, 2012, 12, 5225–5230 RSC.
- B. F. Liu, M. Ozaki, H. Hisamoto, Q. M. Luo, Y. Utsumi, T. Hattori and S. Terabe, Anal. Chem., 2005, 77, 573–578 CrossRef CAS.
- Y. Du, C. Chen, M. Zhou, S. Dong and E. Wang, Anal. Chem., 2011, 83, 1523–1529 CrossRef CAS.
- D. Schmalzing, L. B. Koutny, T. A. Taylor, W. Nashabeh and M. Fuchs, J. Chromatogr., Biomed. Appl., 1997, 697, 175–180 CrossRef CAS.
- R. A. M. Lopes, K. B. Neves, F. S. Carneiro and R. C. Tostes, Front. Physiol., 2012, 3, 89–89 CrossRef CAS.
- K. Ino, Y. Kitagawa, T. Watanabe, H. Shiku, M. Koide, T. Itayama, T. Yasukawa and T. Matsue, Electrophoresis, 2009, 30, 3406–3412 CrossRef CAS.
- F. J. Arévalo, G. A. Messina, P. G. Molina, M. A. Zón, J. Raba and H. Fernández, Talanta, 2010, 80, 1986–1992 CrossRef.
- E. Sollier, in Microfluidic Technologies for Human Health, World Scientific Publishing Company, 2012 Search PubMed.
- P. Y. Shu and J. H. Huang, Clinical and Diagnostic Laboratory Immunology, 2004, 11, 642–650 Search PubMed.
- J. L. Greenwald, G. R. Burstein, J. Pincus and B. Branson, Curr. Infect. Dis. Rep., 2006, 8, 125–131 CrossRef.
- S. T. Nichol, J. Arikawa and Y. Kawaoka, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12411–12412 CrossRef CAS.
- H. D. H. Testereci, A. Ertekin and T. Kahraman, Eastern J. Med., 1998, 3, 62–66 Search PubMed.
- A. Dehee, C. Asselot, T. Piolot, C. Jacomet, W. Rozenbaum, M. Vidaud, A. Garbarg-Chenon and J. C. Nicolas, J. Med. Virol., 2001, 65, 543–552 CrossRef CAS.
- Y.-G. Kim, S. Moon, D. R. Kuritzkes and U. Demirci, Biosens. Bioelectron., 2009, 25, 253–258 CrossRef CAS.
- G. D. Chen, C. J. Alberts, W. Rodriguez and M. Toner, Anal. Chem., 2010, 82, 723–728 CrossRef CAS.
- P. Dussart, B. Labeau, G. Lagathu, P. Louis, M. R. T. Nunes, S. G. Rodrigues, C. Storck-Herrmann, R. Cesaire, J. Morvan, M. Flamand and L. Baril, Clin. Vaccine Immunol., 2006, 13, 1185–1189 CrossRef CAS.
- H. Schulze, G. Giraud, J. Crain and T. T. Bachmann, J. Biophotonics, 2009, 2, 199–211 CrossRef CAS.
- X. Cheng, G. Chen and W. R. Rodriguez, Anal. Bioanal. Chem., 2009, 393, 487–501 CrossRef CAS.
- M. Smit, K. A. Beynon, D. R. Murdoch and L. C. Jennings, Diagn. Microbiol. Infect. Dis., 2007, 57, 67–70 CrossRef CAS.
- C. DeLima, J. Blair, D. E. Low, L. Burton, T. Mazzulli and S. J. Drews, Int. J. Infect. Dis., 2009, 13, e327–E328 CrossRef.
- T. P. Leary, R. A. Gutierrez, A. S. Muerhoff, L. G. Birkenmeyer, S. M. Desai and G. J. Dawson, J. Med. Virol., 2006, 78, 1436–1440 CrossRef CAS.
- J. Pipper, M. Inoue, L. F. P. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS.
- Y.-K. Cho, J.-G. Lee, J.-M. Park, B.-S. Lee, Y. Lee and C. Ko, Lab Chip, 2007, 7, 565–573 RSC.
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS.
- L. Johnson, A. T. K. Gupta, A. Ghafoor, D. Akin and R. Bashir, Sens. Actuators, B, 2006, 115, 189–197 CrossRef CAS.
- F. Patolsky, G. F. Zheng, O. Hayden, M. Lakadamyali, X. W. Zhuang and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14017–14022 CrossRef CAS.
- D. S. Reichmuth, S. K. Wang, L. M. Barrett, D. J. Throckmorton, W. Einfeld and A. K. Singh, Lab Chip, 2008, 8, 1319–1324 RSC.
- J. D. Uram, K. Ke, A. J. Hunt and M. Mayer, Small, 2006, 2, 967–972 CrossRef CAS.
- S. Wang, D. Sarenac, M. H. Chen, S.-H. Huang, F. F. Giguel, D. R. Kuritzkes and U. Demirci, International Journal of Nanomedicine, 2012, 7, 5019–5028 CAS.
- Prevention of hospital-acquired infections, World Health Organization, 2002 Search PubMed.
- H. Lee, E. Sun, D. Ham and R. Weissleder, Nat. Med., 2008, 14, 869–874 CrossRef CAS.
- Z. Wu, B. Willing, J. Bjerketorp, J. K. Jansson and K. Hjort, Lab Chip, 2009, 9, 1193–1199 RSC.
- H. W. Hou, H. Y. Gan, A. A. S. Bhagat, L. D. Li, C. T. Lim and J. Han, Biomicrofluidics, 2012, 6 Search PubMed.
- R. P. H. Peters, M. A. van Agtmael, S. A. Danner, P. H. M. Savelkoul and C. Vandenbroucke-Grauls, Lancet Infect. Dis., 2004, 4, 751–760 CrossRef CAS.
- C. Ke, H. Berney, A. Mathewson and M. M. Sheehan, Sens. Actuators, B, 2004, 102, 308–314 CrossRef CAS.
- Z. S. Hua, J. L. Rouse, A. E. Eckhardt, V. Srinivasan, V. K. Pamula, W. A. Schell, J. L. Benton, T. G. Mitchell and M. G. Pollack, Anal. Chem., 2010, 82, 2310–2316 CrossRef CAS.
- W. A. Schell, J. L. Benton, P. B. Smith, M. Poore, J. L. Rouse, D. J. Boles, M. D. Johnson, B. D. Alexander, V. K. Pamula, A. E. Eckhardt, M. G. Pollack, D. K. Benjamin, J. R. Perfect and T. G. Mitchell, Eur. J. Clin. Microbiol. Infect. Dis., 2012, 31, 2237–2245 CrossRef CAS.
- R. M. Hutchinson and R. D. Eastham, J. Clin. Pathol., 1977, 30, 345–349 CrossRef CAS.
- R. Rosencranz and S. A. Bogen, Am. J. Clin. Pathol., 2006, 125 Suppl, s78–s86 Search PubMed.
- R. K. Jain, Cancer Research, 1988, 48, 2641–2658 CAS.
- HTI Medical, Biosite, Triage MeterPro-Fluorescence Meter, http://htimedical.mybigcommerce.com/biosite-triage-meterpro-fluorescence-meter-triage-meter/, 2013 Search PubMed.
- Abbott Diabetes Care, FreeStyle Lite Overview, https://www.abbottdiabetescare.com/products/patient/fs-lite-overview.html, 2013 Search PubMed.
- D. S. Kim, S. H. Lee, C. H. Ahn, J. Y. Lee and T. H. Kwon, Lab Chip, 2006, 6, 794–802 RSC.
- B. L. Boyanton and K. E. Blick, Clin. Chem., 2002, 48, 2242–2247 CAS.
- V. VanDelinder and A. Groisman, Anal. Chem., 2006, 78, 3765–3771 CrossRef CAS.
- V. VanDelinder and A. Groisman, Anal. Chem., 2007, 79, 2023–2030 CrossRef CAS.
- W. S. Nesbitt, E. Westein, F. J. Tovar-Lopez, E. Tolouei, A. Mitchell, J. Fu, J. Carberry, A. Fouras and S. P. Jackson, Nat. Med., 2009, 15, 665–673 CrossRef CAS.
- Y. Yoshimura, Y. Hiramatsu, Y. Sato, S. Homma, Y. Enomoto, Y. Kikuchi and Y. Sakakibara, Ann. Thorac. Surg., 2003, 75, 7–7 Search PubMed.
- L. B. Leverett, E. C. Lynch, C. P. Alfrey and J. D. Hellums, Biophys. J., 1972, 12, 257 CrossRef CAS.
- I. Wong and C.-M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS.
- A. J. Mach and D. Di Carlo, Biotechnol. Bioeng., 2010, 107, 302–311 CrossRef CAS.
- N. J. Shirtcliffe, R. Toon and P. Roach, Methods Mol. Biol., 2013, 949, 241–268 CAS.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS.
- C. D. Chin, V. Linder and S. K. Sia, Lab Chip, 2012, 12, 2118–2134 RSC.
- G. M. Whitesides, Lab Chip, 2013, 13, 11–13 RSC.
- P. M. van Midwoud, A. Janse, M. T. Merema, G. M. Groothuis and E. Verpoorte, Anal. Chem., 2012, 84, 3938–3944 CrossRef.
- D. K. Armani and C. Liu, J. Micromech. Microeng., 2000, 10, 80–84 CrossRef CAS.
- K. R. King, C. C. J. Wang, M. R. Kaazempur-Mofrad, J. P. Vacanti and J. T. Borenstein, Adv. Mater., 2004, 16, 2007 CrossRef CAS.
- A. Hsiao, J. Luecha, J. Kokini and L. Liu, Green Microfluidics Made of Corn Proteins, Lab. Chip, 2011, 11, 3419–3425 RSC.
- E. Sollier, M. Cubizolles, Y. Fouillet and J.-L. Achard, Biomed. Microdevices, 2010, 12, 485–497 CrossRef.
- C. M. Cripps, J. Clin. Pathol., 1968, 21, 110–112 CrossRef CAS.
- M. Kersaudy-Kerhoas, R. Dhariwal, M. P. Y. Desmulliez and L. Jouvet, Microfluid. Nanofluid., 2010, 8, 105–114 CrossRef CAS.
- B. G. Stegmayr, Science, 2005, 32, 209–220 Search PubMed.
- T. Crowley, M. Hayes and V. Pizziconi, J. Assoc. Lab. Autom., 2003, 8, 78–80 CrossRef.
- M. L. Brigden, American Family Physician, 1999, 60, 1443–1450 CAS.
- S. Y. Yoon, S. Yang, Y. Moon and K. C. Kim, presented in part at MicroTas 2006, Tokyo, Japan, 2006.
- T. Tachi, N. Kaji, M. Tokeshi and Y. Baba, Anal. Chem., 2009, 81, 3194–3198 CrossRef CAS.
- X.-B. Zhang, Z.-Q. Wu, K. Wang, J. Zhu, J.-J. Xu, X.-H. Xia and H.-Y. Chen, Anal. Chem., 2012, 84, 3780–3786 CrossRef CAS.
- C. C. Wu, L. Z. Hong and C. T. Ou, Journal of Medical and Biological Engineering, 2012, 32, 163–168 CrossRef.
- C.-T. Huang, H.-H. Chang, P.-N. Li and C.-P. Jen, presented in part at the 5th IEEE International Conference on Nano/Micro Engineered and Molecular Systems, Xiamen, China, 2010.
- C.-T. Huang, P.-N. Li, C.-Y. Pai, T.-S. Leu and C.-P. Jen, Sep. Sci. Technol., 2010, 45, 42–49 CrossRef CAS.
- I. K. Dimov, L. Basabe-Desmonts, J. L. Garcia-Cordero, B. M. Ross, A. J. Ricco and L. P. Lee, Lab Chip, 2011, 11, 845–850 RSC.
- J. S. Yoon, J. T. Germaine and P. J. Culligan, Water Resour. Res., 2006, 42, W06417 CrossRef.
- X.-B. Zhang, Z.-Q. Wu, K. Wang, J. Zhu, J.-J. Xu, X.-H. Xia and H.-Y. Chen, Anal. Chem., 2012, 84, 3780–3786 CrossRef CAS.
- S. Thorslund, O. Klett, F. Nikolajeff, K. Markides and J. Bergquist, Biomed. Microdevices, 2006, 8, 73–79 CrossRef CAS.
- J. Moorthy and D. J. Beebe, Lab Chip, 2003, 3, 62–66 RSC.
- D. S. Lee, Y. H. Choi, Y. D. Han, H. C. Yoon, S. Shoji and M. Y. Jung, ETRI J., 2012, 34, 226–234 CrossRef.
- A. W. B. Joon, S. Shim, Se Hwan Lee and C. H. Ahn, An On-chip Whole Blood/Plasma Separator with Colloidal Silica Bead-Packed microchannel on coc polymer, San Diego, California, USA, 2008.
- J. S. Shim and C. H. Ahn, presented in part at the 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences Groningen, The Netherlands, 2010.
- C. Li, C. Liu, Z. Xu and J. Li, Biomed. Microdevices, 2012, 14, 565–572 CrossRef CAS.
- C. Li, C. Liu, Z. Xu and J. Li, Talanta, 2012, 97, 376–381 CrossRef CAS.
- K. H. Chung, Y. H. Choi, J.-H. Yang, C. W. Park, W.-J. Kim, C. S. Ah and G. Y. Sung, Lab Chip, 2012, 12, 3272–3276 RSC.
- T. A. Crowley and V. Pizziconi, Lab Chip, 2005, 5, 922–929 RSC.
- X. Chen, D. F. Cui, C. C. Liu and H. Li, Sens. Actuators, B, 2008, 130, 216–221 CrossRef CAS.
- E. Sollier, H. Rostaing, P. Pouteau, Y. Fouillet and J. L. Achard, Sens. Actuators, B, 2009, 141, 617–624 CrossRef CAS.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS.
- J. A. Davis, D. W. Inglis, K. J. Morton, D. A. Lawrence, L. R. Huang, S. Y. Chou, J. C. Sturm and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14779–14784 CrossRef CAS.
- D. W. Inglis, K. J. Morton, J. A. Davis, T. J. Zieziulewicz, D. A. Lawrence, R. H. Austin and J. C. Sturm, Lab Chip, 2008, 8, 925–931 RSC.
- S. Choi and J. K. Park, Lab Chip, 2007, 7, 890–897 RSC.
- S. Choi and J.-K. Park, presented in part at the microTAS 2007, Paris, 2007.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- G. B. Thurston, Biorheology, 1989, 26, 199–214 CAS.
- Y. C. Fung, Biomechanics: Mechanical Properties of Living Tissues, Springer, 1993 Search PubMed.
- V. Doyeux, T. Podgorski, S. Peponas, M. Ismail and G. Coupier, J. Fluid Mech., 2011, 674, 359–388 CrossRef CAS.
- M. Faivre, M. Abkarian, K. Bickraj and H. A. Stone, Biorheology, 2006, 43, 147–159 Search PubMed.
- R. D. Jaggi, R. Sandoz and C. S. Effenhauser, Microfluid. Nanofluid., 2007, 3, 47–53 CrossRef.
- M. Kersaudy-Kerhoas, D. M. Kavanagh, R. S. Dhariwal, C. J. Campbell and M. P. Y. Desmulliez, Lab Chip, 2010, 10, 1587–1595 RSC.
- T. M. Geislinger, B. Eggart, S. B. Ller, L. Schmid and T. Franke, Appl. Phys. Lett., 2012, 100, 183701 CrossRef.
- S. Yang, A. Undar and J. D. Zahn, Lab Chip, 2006, 6, 871–880 RSC.
- S. Yang, A. Undar and J. D. Zahn, ASAIO J., 2005, 51, 585–590 CrossRef.
- R. Fan, O. Vermesh, A. Srivastava, B. K. H. Yen, L. D. Qin, H. Ahmad, G. A. Kwong, C. C. Liu, J. Gould, L. Hood and J. R. Heath, Nat. Biotechnol., 2008, 26, 1373–1378 CrossRef CAS.
- A. I. Rodríguez-Villarreal, M. Arundell, M. Carmona and J. Samitier, Lab Chip, 2010, 10, 211–219 RSC.
- D. Di Carlo, Lab Chip, 2009, 9, 3038–3046 RSC.
- S. C. Hur, H. T. K. Tse and D. Di Carlo, Lab Chip, 2010, 10, 274–280 RSC.
- G. Segré and A. Silberberg, Nature, 1961, 189, 209–210 CrossRef.
- M. Masaeli, E. Sollier, H. Amini, W. Mao, K. Camacho, N. Doshi, S. Mitragotri, A. Alexeev and D. Di Carlo, Phys. Rev. X, 2012, 2, 031017 CrossRef CAS.
- M. G. Lee, S. Choi, H. J. Kim, H. K. Lim, J. H. Kim, N. Huh and J. K. Park, Appl. Phys. Lett., 2011, 98, 253702 CrossRef.
- H. Amini, E. Sollier, M. Masaeli, Y. Xie, B. Ganapathysubramanian, H. A. Stone and D. Di Carlo, Nat. Commun., 2013, 4, 1926 CrossRef.
- D. R. Gossett and D. Di Carlo, Anal. Chem., 2009, 81, 8459–8465 CrossRef CAS.
- A. P. Sudarsan and V. M. Ugaz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7228–7233 CrossRef CAS.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897 CrossRef CAS.
- D. Di Carlo, J. F. Edd, D. Irimia, R. G. Tompkins and M. Toner, Anal. Chem., 2008, 80, 2204–2211 CrossRef CAS.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Lab Chip, 2008, 8, 1906–1914 RSC.
- R. J. C. Blattert, I. Tahhan, A. Schoth and H. Reinecke, presented in part at MicroTAS 2006, 2006.
- D. X. W. Wang and Z. Li, presented in part at Micrototal Analysis Systems, Tokyo, Japan, 2006.
- Y. R. Ju, Z. X. Geng, L. Q. Zhang, W. Wan and Z. H. Li , presented in part at Transducers, Beijing, China, 2011.
- J. Morikawa, Plasma separation from human blood using spiral microchannels for dry eye treatment, Japan, 2012.
- F. Petersson, A. Nilsson, C. Holm, H. Jonsson and T. Laurell, Lab Chip, 2005, 5, 20–22 RSC.
- F. Petersson, L. Aberg, A.-M. Sward-Nilsson and T. Laurell, Anal. Chem., 2007, 79, 5117–5123 CrossRef CAS.
- A. Lenshof, C. Magnusson and T. Laurell, Lab Chip, 2012, 12, 1210–1223 RSC.
- M. Wiklund, Lab Chip, 2012, 12, 2018–2028 RSC.
- T. Laurell, F. Petersson and A. Nilsson, Chem. Soc. Rev., 2007, 36, 492–506 RSC.
- A. Lenshof and T. Laurell, Chem. Soc. Rev., 2010, 39, 1203–1217 RSC.
- A. Nilsson, F. Petersson and T. Laurell, presented in part at μTAS, Tokyo, Japan, 2006.
- M. P. A. Doria and A. P. Lee, presented in part at MicroTAS, Seattle, US, 2011.
- C. M. Das, F. Becker, S. Vernon, J. Noshari, C. Joyce and P. R. Gascoyne, Anal. Chem., 2005, 77, 2708–2719 CrossRef CAS.
- Y. Nakashima, S. Hata and T. Yasuda, Sens. Actuators, B, 2010, 145, 561–569 CrossRef CAS.
- H. Jiang, X. Weng, C. H. Chon, X. Wu and D. Li, Journal of Micromechanics and Microengineering, 2011, 21, 085019 CrossRef.
- S. A. Peyman, E. Y. Kwan, O. Margarson, A. Iles and N. Pamme, J. Chromatogr., A, 2009, 1216, 9055–9062 CrossRef CAS.
- A. I. Rodriguez-Villarreal, M. D. Tarn, L. A. Madden, J. B. Lutz, J. Greenman, J. Samitier and N. Pamme, Lab Chip, 2011, 11, 1240–1248 RSC.
- N. Pamme and C. Wilhelm, Lab Chip, 2006, 6, 974–980 RSC.
- K. H. Han and A. B. Frazier, Lab Chip, 2006, 6, 265–273 RSC.
- J. Jung and K.-H. Han, Appl. Phys. Lett., 2008, 93, 223902 CrossRef.
- C. W. Yung, J. Fiering, A. J. Mueller and D. E. Ingber, Lab Chip, 2009, 9, 1171–1177 RSC.
- M. Takayasu, D. R. Kelland and J. V. Minervini, IEEE Trans. Appl. Supercond., 2000, 10, 927–930 CrossRef.
- D. M. Kavanagh, M. Kersaudy-Kerhoas, R. S. Dhariwal and M. P. Y. Desmulliez, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1905–1911 CrossRef CAS.
- F. Paul, S. Roath, D. Melville, D. C. Warhurst and J. O. S. Osisanya, Lancet, 1981, 318, 70–71 CrossRef.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS.
- X. Li, D. R. Ballerini and W. Shen, Biomicrofluidics, 2012, 6, 011301 CrossRef.
- Alere, Alere Cholestech LDX® System, http://www.alere.com/us/en/product-details/cholestech-ldx-system.html, 2013 Search PubMed.
- CoaguChek, CoaguChek® systems are easy to use, http://www.coaguchek.com/uk/index.php?target=/en/patients/products Search PubMed.
- T. Songjaroen, W. Dungchai, O. Chailapakul, C. S. Henry and W. Laiwattanapaisal, Lab Chip, 2012, 12, 3392–3398 RSC.
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. S. Lee, J.-M. Park, J. Kim, H. Kim, M. Madou and Y.-K. Cho, Lab Chip, 2010, 10, 1758–1773 RSC.
- J. Steigert, M. Grumann, T. Brenner, K. Mittenbühler, T. Nann, J. Rühe, I. Moser, S. Haeberle, L. Riegger, J. Riegler, W. Bessler, R. Zengerle and J. Ducrée, J. Assoc. Lab. Autom., 2005, 10, 331–341 CrossRef CAS.
- M. Amasia and M. Madou, Bioanalysis, 2010, 2, 1701–1710 CrossRef CAS.
- C. T. Schembri, V. Ostoich, P. J. Lingane, T. L. Burd and S. N. Buhl, Clin. Chem., 1992, 38, 1665–1670 CAS.
- S. Haeberle, T. Brenner, R. Zengerle and J. Ducree, Lab Chip, 2006, 6, 776–781 RSC.
- B. S. Lee, J.-N. Lee, J.-M. Park, J.-G. Lee, S. Kim, Y.-K. Cho and C. Ko, Lab Chip, 2009, 9, 1548–1555 RSC.
- S. Gilad, E. Meiri, Y. Yogev, S. Benjamin, D. Lebanony, N. Yerushalmi, H. Benjamin, M. Kushnir, H. Cholakh, N. Melamed, Z. Bentwich, M. Hod, Y. Goren and A. Chajut, PLoS One, 2008, 3, e3148 Search PubMed.
- P. S. Mitchell, R. K. Parkin, E. M. Kroh, B. R. Fritz, S. K. Wyman, E. L. Pogosova-Agadjanyan, A. Peterson, J. Noteboom, K. C. O'Briant, A. Allen, D. W. Lin, N. Urban, C. W. Drescher, B. S. Knudsen, D. L. Stirewalt, R. Gentleman, R. L. Vessella, P. S. Nelson, D. B. Martin and M. Tewari, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10513–10518 CrossRef CAS.
- Z. S. K. Wang, B. Marzolf, P. Troisch, A. Brightman and Z. Hu, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4402–4407 CrossRef.
- D. J. Starkey, P. J. Lewis, V. Platt, K. J. Simpson, D. G. Craig and D. J. Antoine, et al. , Hepatology, 2011, 54, 1767–1776 CrossRef.
- A. P. Wong, M. Gupta, S. S. Shevkoplyas and G. M. Whitesides, Lab Chip, 2008, 8, 2032–2037 RSC.
| This journal is © The Royal Society of Chemistry 2013 |