 Open Access Article
Open Access ArticleSample solution constraints on motor-driven diagnostic nanodevices†
Slobodanka
Korten‡
a,
Nuria
Albet-Torres‡
b,
Francesca
Paderi‡
c,
Lasse
ten Siethoff
b,
Stefan
Diez
ad,
Till
Korten
*a,
Geertruy
te Kronnie
*c and
Alf
Månsson
*b
aMax Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany. E-mail: skorten@mpi-cbg.de; korten@mpi-cbg.de
bLinnaeus University, School of Natural Sciences, SE-391 82 Kalmar, Sweden. E-mail: nuria.albet@gmail.com; alf.mansson@lnu.se
cUniversity of Padua, Department of Pediatrics “Salus Pueri”, SSD Clinical and Experimental Hematology, Padova, Italy. E-mail: truustekronnie@gmail.com; fran.paderi@gmail.com
dB CUBE - Center for Molecular Bioengineering, Technische Universität Dresden, Dresden, Germany. E-mail: diez@bcube-dresden.de
First published on 13th December 2012
Abstract
The last decade has seen appreciable advancements in efforts towards increased portability of lab-on-a-chip devices by substituting microfluidics with molecular motor-based transportation. As of now, first proof-of-principle devices have analyzed protein mixtures of low complexity, such as target protein molecules in buffer solutions optimized for molecular motor performance. However, in a diagnostic work-up, lab-on-a-chip devices need to be compatible with complex biological samples. While it has been shown that such samples do not interfere with crucial steps in molecular diagnostics (for example antibody-antigen recognition), their effect on molecular motors is unknown. This critical and long overlooked issue is addressed here. In particular, we studied the effects of blood, cell lysates and solutions containing genomic DNA extracts on actomyosin and kinesin–microtubule-based transport, the two biomolecular motor systems that are most promising for lab-on-a-chip applications. We found that motor function is well preserved at defined dilutions of most of the investigated biological samples and demonstrated a molecular motor-driven label-free blood type test. Our results support the feasibility of molecular-motor driven nanodevices for diagnostic point-of-care applications and also demonstrate important constraints imposed by sample composition and device design that apply both to kinesin–microtubule and actomyosin driven applications.
Introduction
Living cells work like autonomous miniaturized factories with the ability to sense their environment and adapt to it. These unique properties have inspired the development of novel nanoanalytical tools and engineering of nanoscale systems of increasing importance for human healthcare. Crucial in this regard are biosensors for molecular diagnostics, which combine specific biological recognition with microfluidics-based separation and specialized nanotechnology-based detection schemes.1–5Recently, transport by molecular motors of the cytoskeleton has been considered as an alternative to microfluidics.6–9 As one of a number of benefits (cf.6) of this approach, molecular motors efficiently transduce the chemical energy of ATP into mechanical work,10,11 without the requirement for external power supplies. This would enable portable diagnostics devices that are more heavily miniaturized, cheaper and more convenient to handle than conventional systems.
Among the best-studied motors are myosins moving along actin filaments (actomyosin systems) and kinesin moving along microtubules (kinesin–microtubule systems). Key studies have demonstrated that these motor systems can be used for transporting, sorting and assembling materials on a chip.12–22 In such applications, motors are adsorbed to flat or micro-/nano-structured surfaces, propelling cytoskeletal filaments that act as shuttles which carry cargoes.9,12,23,24 The two motor systems have distinct advantages8 that make them suitable for different purposes. For instance, actomyosin has a ten-fold higher speed, whereas microtubule–kinesin based systems are better at transporting cargoes such as cells, viruses, etc. These developments have recently evolved into proof-of-principle devices18,22 for the detection of biomarkers by adapting principles that are already widely used in diagnostics. In such devices, antibodies or oligonucleotides are immobilized on the cytoskeletal filaments for the binding of specific biomarkers (such as proteins or DNA sequences) (reviewed in8).
In addition to advantages over microfluidics driven separation, the actual detection of biomarkers may be achieved in unique ways using motor driven devices. One possibility is by observing changes in molecular-motor function,17,25e.g. inhibition of transport or markedly reduced filament velocity upon analyte binding. Another alternative, exemplified below, is by the actual observation of co-localization and co-transportation of cytoskeletal filaments and biomarkers.19,20,22 This method has the advantage that it distinguishes specific antibody–antigen binding from non-specific surface binding, thereby increasing the sensitivity and specificity compared to conventional methods of detection, e.g. enzyme-linked immunosorbent assays with antibodies immobilized on surfaces. A more advanced version of the concept with co-transportation of cytoskeletal filaments and biomarkers involves the initial transportation of the analyte to a nanoscale detector area.18,22,26 This achieves signal amplification by concentration and simultaneously exploits the inherent advantages of nanoscale sensors without suffering from slow diffusion limited transport.26
Up to now, molecular motor-based lab-on-a-chip devices have only been tested with buffer solutions optimized for in vitro function of the motors. However, a diagnostic device needs to be compatible with body fluids (such as blood and cell lysates), as well as buffer solutions essential for biomarker detection. Unlike the compatibility of most crucial steps in molecular diagnostics (for example antibody–antigen binding), it is not known whether molecular motors retain working abilities in body fluid solutions. Therefore, as a critical prerequisite for the development of motor-driven lab-on-a-chip devices for diagnostics we systematically studied the effect of a wide range of biological samples on both, the actomyosin and the kinesin–microtubule motor systems. Our extensive studies show full preservation of the function of both motor systems at defined dilutions of the biological sample solutions. Thus, this work helps to make well-founded decisions for the application of either motor system based on its biophysical properties and on its compatibility with the desired sample solution, laying the path for the development of novel motor driven diagnostic lab-on-a-chip devices in the near future.
Materials and methods
Preparation of proteins for the motility assays
Myosin and actin were purified from rabbit fast skeletal muscle.27,28 After purification, myosin was chymotryptically cleaved to yield the heavy meromyosin (HMM) motor fragment.29 After preparation, the proteins were frozen in liquid nitrogen and stored at −80 °C. Actin was fluorescently labeled using tetramethylrhodamine-isothiocyanate-phalloidin (TRITC-phalloidin) or Alexa Fluor 488® phalloidin (APh), both from Molecular Probes (Invitrogen, Eugene, OR, USA).Full length kinesin-1 from drosophila was expressed in bacteria and purified as previously described.30 This protein will be referred to simply as kinesin from now on. Tubulin was isolated from porcine brain and subsequently labeled with rhodamine as previously described.31 Microtubules were polymerized from 5 μl rhodamine labeled tubulin in BRB80 buffer (80 mM PIPES/KOH, pH 6.8, 1 mM EGTA, 1 mM MgCl2; if not specifically mentioned, all chemicals were purchased from Sigma) with 4 mM MgCl2, 1 mM Mg-GTP and 5% DMSO at 37 °C for 60 min. Afterwards, microtubules were stabilized and diluted 100-fold in BRB80 containing 10 μM taxol at room temperature.
In vitro motility assays
For actomyosin in vitro motility assays, glass cover-slips were silanized with trimethylchlorosilane (TMCS) and mounted in a flow cell.32 Unless indicated otherwise, standard incubation steps were used as follows: HMM (120 μg ml−1 [∼300 nM]) in buffer A (1 mM MgCl2, 10 mM MOPS pH 7.4, 0.1 mM EGTA, 1 mM dithiothreitol (DTT) and 50 mM KCl), bovine serum albumin (BSA; 1 mg ml−1 in buffer A), non-fluorescent blocking actin (1 μM in buffer A; to block rigor heads). This was followed by an MgATP-containing solution (buffer A supplemented with 1 mM MgATP and 25 instead of 50 mM KCl) and a wash in buffer A. Next, TRITC-phalloidin labeled actin filaments where added (2–20 nM in buffer A) and, finally, imaging of actin filaments was performed in an assay solution (aMC130; buffer A supplemented with final concentrations of 1 mM MgATP, 115 mM KCl and 10 mM DTT, 3 mg ml−1 glucose, 20 units/ml glucose oxidase, 870 units/ml catalase and 0.6% methylcellulose). Separate flow cells using the above standard motility assay conditions were studied at each experimental occasion to obtain control values for the gliding speed and the fraction of motile filaments. The effects of body fluids etc. were investigated in separate flow cells with modified incubation steps, as described in detail below. Temperature in the actomyosin motility assays varied between 22 and 30 °C in different experiments, but was constant to ±0.5 °C during a given experiment. Actin filaments were imaged using an inverted epifluorescence microscope (Nikon Eclipse TE300) and recorded using a CCD camera.Kinesin–microtubule gliding assays were performed as described by Nitzsche et al.33 Briefly, flow cells were constructed from two clean glass cover-slips (Menzel, 18 × 18 mm2 and 22 × 22 mm2 separated by strips of Nescofilm (Roth). Flow cells were perfused with casein-containing solution (0.5 mg ml−1) in BRB80 and left to adsorb for 5 min. Next, 15 μl of kinesin solution (2 nM full length kinesin unless specified otherwise), was perfused into the flow cells and incubated for another 5 min. A considerably lower motor concentration was used than for HMM, since kinesin is a processive motor with the capability to transport microtubules at maximum speed in spite of very low surface densities. Thereafter, motility solution (1 mM ATP, 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase, 10 mM DTT, 10 μM taxol in BRB80) containing rhodamine labeled taxol-stabilized microtubules was applied. Microtubules were left to bind to kinesin for 5 min and unbound microtubules were washed out using motility solution without microtubules. As a control for each experiment, fluorescence time-lapse movies for microtubule speed measurements were recorded directly after the wash-out step (referred to as BRB80 control from now on). After imaging, assay buffer was perfused into the flow cells and microtubule gliding speeds were measured every 10 min for the time course of 1 h. In order to reduce photobleaching and to power motility, the above motility buffers were supplemented with: 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase, 10 mM DTT and 1 mM ATP. Fluorescence time-lapse movies were recorded in these buffers and microtubule gliding speeds were measured from the movies. Imaging was performed using an Axiovert 200 M inverted optical microscope (Zeiss) equipped with a back-illuminated CCD camera (MicroMax 512 BFT, Roper Scientific) in conjunction with a Metamorph imaging system (Universal Imaging Corp.).
Blood and blood components
Whole blood, serum and plasma were drawn from volunteers who consented to the procedures under a protocol approved by the Linnaeus University and the Max-Planck-Institute of Molecular Cell Biology and Genetics.For actomyosin experiments, whole blood was collected in Vacutainer™ plastic tubes with EDTA (final concentration 5 mM) (Becton, Dickinson and Co., Plymouth, UK). In the case of plasma collection, cells were removed by centrifugation immediately after sampling (3000 g for 20 min at room temperature; 20 °C). Serum was obtained after drawing blood into a Vacutainer™ glass tube. After 1 h at room temperature the sample was centrifuged as for the collection of plasma to remove blood cells and solidified fibrin. All body fluids were used after dilution in assay buffer or wash buffer. The actomyosin experiments were performed on different occasions and with different batches of HMM and glass surfaces causing some differences in gliding speeds. Therefore, speeds are given relative to control values during each given experiment to ensure comparability of the data. In accordance with the demonstrated lack of reversibility (see below; Fig. 1), similar effects on actomyosin motility were observed whether blood samples were added to the flow cell prior to, or together with, the assay solution. These different cases are therefore pooled in the analysis.
 | ||
| Fig. 1 Effects of blood samples on cytoskeletal filaments and molecular motor function. (A) Actin filament and (B) Microtubule speeds after application of the given dilutions of whole blood (red squares, solid line), plasma (black circles, dashed line) and serum (green diamonds, dotted line) in the given buffers. Nonlinear regression (solid thin blue line) and 95% confidence interval (CI; thin blue dotted lines) of the pooled serum and plasma data gives average IC50 values of 0.68% (95% CI: 0.42%–1.12%) for actomyosin motility and 0.48% (95% CI: 0.33%–0.70%) for kinesin–microtubule motility (see also Supplementary Fig. 1–3 for further analysis, ESI†). (C) Time dependence of microtubule speeds in 1% whole blood. (D) Microtubule speeds in 1% whole blood diluted in assay buffers with the given pH and ionic strengths, measured directly after injection (1% blood) and 30 min after injection (after 30 min). As an additional control, the microtubule speeds were measured in the respective assay buffers without blood (assay buffer). These assay buffer controls were used for the determination of statistical difference. Speeds in this panel are the mean ± standard error of the mean of the speed of 16 microtubules. (E) Actin filament and (F) microtubule speeds measured before the injection of the blood component, in the presence of plasma, or serum, and after washing with the blood free control buffer. Data of serum and plasma (E) or of serum, plasma and whole blood (F) was pooled. The effect of the analyzed blood component was not reversible for either microtubule or actin motility as analyzed by repeated measures ANOVA (p < 0.05). Further, Bonferroni post-tests suggest significant differences (p < 0.05) from initial control values upon washing with control solution. For actin data, additional regression analysis found a significant linear trend (p < 0.05). (G) Fluorescence micrographs of actin filaments without blood (control), 1 min and 10 min after injection of 1% of serum. Upper panel: filament positions at the start of imaging. Lower panel: maximum projection of the filament positions during 7 s of imaging. In the lower panel, filament positions at the start of imaging are highlighted in green. (H) Fluorescence micrographs of antibody-cluster conjugated microtubules (red) and transmitted light micrographs of erythrocytes (green). Capillary blood samples of blood type A (top row) or blood type O (bottom row) were diluted to 1% (v/v) in BRB80 pH 7.5 IS 95. Clusters transported by molecular motors are indicated by white arrows. The paths travelled in the previous frames are indicated by white dashed lines. (A–F) Unless mentioned otherwise, speeds are given as mean ± standard error of the mean of several different flow cells. The number of different flow cells for each data point is given in parentheses. The speed in each cell was averaged from several representative filament paths. (B–F) Unless marked with n.s. (not significant) all data points are statistically significantly (p < 0.05) different from the respective controls. Data in A was analyzed separately (see ESI†). | ||
For kinesin–microtubule gliding assays, whole venous blood was collected in EDTA, Heparin or Serum Primavette® V (Kabe Labortechnik GmbH) and processed as described for the actomyosin experiments. The blood was then diluted to the given concentrations in BRB80, supplemented with 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase, 10 mM DTT and 1 mM ATP and injected into flow cells for in vitro motility assay experiments as described above. For the transport of blood type A erythrocytes, tubulin was conjugated with mouse anti human blood type A antibody (clone 9A, Abcam) using HyNic protein conjugation toolkit (SoluLink) according to the instructions of the manufacturer. This tubulin was mixed with rhodamine labeled tubulin in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and polymerized into microtubules. A gliding assay with these microtubules was then performed in the presence of blood (blood group A or O, respectively) diluted to 1% v/v in BRB80 (supplemented with 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase, 10 mM DTT and 1 mM ATP and 20 mM KCl and imaged after 10 min alternately using transmitted light microscopy to visualize the cells and fluorescence microscopy to visualize the microtubules.
:1 ratio and polymerized into microtubules. A gliding assay with these microtubules was then performed in the presence of blood (blood group A or O, respectively) diluted to 1% v/v in BRB80 (supplemented with 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase, 10 mM DTT and 1 mM ATP and 20 mM KCl and imaged after 10 min alternately using transmitted light microscopy to visualize the cells and fluorescence microscopy to visualize the microtubules.
Cellular proteins
For actomyosin experiments, cytosolic and nuclear protein fractions were obtained from the leukemia cell line MHH CALL4. A total of 107 cells (from DSMZ collection ACC 337) were lysed with minor modification as described.34 Briefly, cells were lysed on ice for 20 min in lysis buffer (10 mM HEPES pH 7.8, 15 mM KCl, 2 mM MgCl2, 1 mM EDTA). Cell lysates were then cleared by centrifugation and supernatants were collected and assayed for protein concentration using the Bradford assay. The protein concentrations were 0.9 and 1.5 g l−1 for the cytosolic fraction and 0.5 and 0.8 g l−1 for the nuclear fraction in two experiments.Cell lysates for the kinesin–microtubule experiments were prepared as follows: 107 HMEC-1 (human microvascular endothelial cell line-1) were resuspended in lysis buffer (with 1 mM EGTA instead of EDTA and 1× protease inhibitor cocktail). To release the nuclear extract, cell lysate was transferred to a Potter–Dounce homogenizer (10 strokes). All steps were performed on ice. Microtubule gliding assays were prepared as described above. Afterwards, cell lysates (supplemented with 20 mM D-glucose, 20 μg ml−1 glucose oxidase, 10 μg ml−1 catalase and 10 mM DTT) were injected into flow cells and microtubule gliding speeds were measured directly after injection and over a time period of 1 h.
DNA hybridization buffers
A formamide buffer for actomyosin motility assays was prepared as follows: 50% (v/v) deionized formamide was dissolved in a buffer containing, 0.01% (w/v) dextran sulfate, 150 mM NaCl and 14 mM trisodium citrate (pH 7.0). The formamide solution was further diluted to 10%, 33%, 66% and 90% (v/v) in Buffer B (10 mM MOPS pH 7.4, 0.1 mM EGTA, 3 mM NaN3). A dextran sulfate buffer (10% (w/v) dextran sulfate, 20% (w/v) glycerol and 150 mM NaCl was diluted to 10% (v/v) in Buffer B. In these diluted buffers, actin was preincubated overnight and then used for an actin motility assay (in the assay the buffers were diluted again tenfold in aMC130). The final concentrations of formamide were 5%, 15%, 30% and 45% during the preincubation of the actin filaments and 0.5%, 1.5%, 3% and 4.5% in the flow cells for gliding assays. The final concentrations of dextran sulfate were 1% during the preincubation and 0.1% in the gliding assay.For the kinesin–microtubule motility assays, formamide was diluted to final concentrations of 5%, 15% and 30% in BRB80. Dextran sulfate was dissolved at a concentration of 25% (w/v) in BRB80.
Genomic DNA
Genomic DNA (gDNA) was obtained from REH or CALL-4 cells (from DSMZ collection ACC 22 and ACC 337 respectively) by using Gentra Puregene blood kit (Qiagen) following manufacturer instructions. Effects of gDNA on actomyosin motility were tested by incubating the HMM-coated surface with gDNA directly before addition of the assay.Other buffers and Ca2+
Assay solutions based on different biological buffers were also tested on actomyosin. These buffers (pH 7.4) were similar to Buffer A, but MOPS was exchanged for 20 mM HEPES or 20 mM PIPES. Microtubule–kinesin motility was studied under the range of conditions given in Fig. 4. To test the effects of free calcium in experiments with actomyosin aMC130 was supplemented with 1 mM EGTA or 0.3 mM CaCl2 (giving a final concentration of 0.2 mM free Ca2+).Data analysis
To measure the actin filament speed, the positions in each frame were tracked using an automatic analysis program developed in MATLAB35 and only filaments with a coefficient of variation <0.2 for the frame-to-frame speeds during 10 frames were used to obtain the average gliding speeds. The fraction of motile filaments was determined using the multivariate statistical approach described previously.35 This fraction also included filaments with a coefficient of the variation of the frame-to-frame speed > 0.2. Therefore, fewer filaments were used to obtain gliding speeds than indicated by the fraction of motile filaments. All actin filament speed data and also data for the fraction of motile filaments are expressed as mean ± standard error of the mean, with n equal to the number of filaments if not otherwise stated. For each flow cell a mean speed value was estimated from ≥10 (unless stated otherwise) and generally several tens of randomly selected independent filament paths. The standard error of the mean for fractions was estimated using the expression for the standard error of the mean of a binomial distribution (SEM = where f is the observed fraction and n is the number of observations.
where f is the observed fraction and n is the number of observations.
The kinesin–microtubule gliding assays were analyzed as follows: The speed of 16 randomly picked microtubules was measured by evaluating the distance travelled over 20 s using the ImageJ image analysis software.
Kinesin-1 is a processive motor. Therefore, the microtubule speed is independent of the motor density over a wide range of densities.11,36 At constant temperature, microtubule gliding speeds are therefore largely inert to variations in motor and surface preparation allowing the use of absolute speed measurements using each filament path as an independent random observation. In experiments with actomyosin, 10% variability in speed may be expected between flow cells, even if other experimental conditions are kept constant.37 Therefore, and to accommodate differences in temperature between some replicated experiments (see above), actin speeds are generally given relative to controls measured with the same motor and surface preparation during the same experimental occasion. Note that this normalization has the inherent effect that the errors for the control used for normalization are always zero.
Unless mentioned otherwise, statistical analysis was performed by one-way analysis of variance (ANOVA). Repeated measures ANOVA were used when studying dependent data (e.g. reversibility). In cases when data was normalized to the control value, an independent control was also normalized in the same way (e.g.Fig. 3a) to serve as an independent group in the ANOVA or regression analysis. Bonferroni or Dunnet’s post tests were applied if all groups were compared to any other group, or to the control group, respectively. Linear regression, non-linear regression and statistical analyses were performed using Graph Pad Prism (v. 5.01, GraphPad Software, San Diego, CA, USA).
We noted, that filament speeds in blood-derived samples varied significantly more between flow cells (and experiments) than in experiments with samples that were not derived from blood. Therefore, we chose to consider the average speed of the filaments within one flow cell as one data point and give the average ± standard error of the mean of several flow cells for the analysis of blood samples (Fig. 1 and Supplementary Fig. 1–3†).
Results and discussion
Effects of blood samples on cytoskeletal filaments and molecular-motor function
Blood analysis is a major laboratory test for medical diagnostics. Therefore, we investigated whether the actomyosin and kinesin–microtubule systems – as potential components in novel devices – remain functional in the presence of blood. Fig. 1 shows that in vitro motility assays based on either motor system were adversely affected by increasing concentrations of whole blood, serum and plasma. Immediately after injection of the diluted blood samples, the filament gliding speeds decreased with increasing blood sample concentration (Fig. 1 A and B). Actomyosin gliding appeared to be faster in whole blood than in plasma and serum at intermediate dilutions. However, while regression analysis showed this effect to be significant (see Supplementary Fig. 1†), we caution over-interpretation of this finding due to the complexity of blood. From a practical point of view it is important that the inhibiting effect of whole blood is not greater than with plasma and serum. On the other hand, the kinesin–microtubule gliding speed was very similar for all blood derivatives. Because we did not detect significant differences between the effects of plasma and serum for either actomyosin or kinesin–microtubule motility (see Supplementary Fig. 1–3† for analysis of difference), we pooled this data to obtain estimates of the plasma/serum concentration at which the speed was reduced by 50% (IC50; see Supplementary Fig. 1†). Nonlinear regression analysis gave point estimates of IC50 = 0.68% (95% CI: [0.42, 1.12]%) for actin filament motility and IC50 = 0.48% (95% CI: [0.33, 0.70]%) for microtubule motility. These mean IC50-values correspond to plasma protein concentrations of ∼480 μg ml−1 for actomyosin and ∼340 μg ml−1 for microtubule–kinesin assuming a protein concentration in plasma of 70 g l−1. Interestingly, the IC50 values are not significantly different (overlapping 95% CI) for the two motor systems, suggesting similar constraints in the presence of blood samples. Another interesting observation (see below for further information) is that the IC50 values are very similar to that for the effects of cytosolic proteins on actomyosin motility.In a detection device, the motility assay needs to run for a certain amount of time to allow complex formation with the analyte and transport to the detection area. For the example of 1% whole blood and kinesin–microtubule gliding, Fig. 1 panel C (and Supplementary Movie 1†) shows that the gliding speed stays constant for the entire observation time of one hour (individual time points do not differ significantly; p > 0.05).
Because a reduction in gliding speed could mean – in particular for the inherently slow kinesin–microtubule system – that the assay time of the device becomes too long to be practical, we performed a number of experiments (carried out only once) to increase microtubule gliding speeds in the presence of 1% whole blood. The most successful of these options was an increase of pH from 6.9 (the standard pH for BRB80) to 7.5 (close to the physiological pH of blood; Fig. 1 D). Under this condition, microtubule gliding speed in the presence of 1% whole blood was the same as in the BRB80 (pH 6.9) control and was less than 20% slower than the control without blood (pH 7.5). This favorable gliding speed was maintained during the observation time of 30 min. An increase of pH also increases the ionic strength (IS) of the buffer and thus reduces electrostatic interactions between biomolecules.38 To separate these two effects, we increased the IS while keeping the pH constant. This resulted in a small increase in gliding speed in the presence of 1% blood at pH 6.9 (Fig. 1 D; p < 0.05 between an ionic strength of 71 and 95). Other tested factors that did not show any significant effect were: kinesin density on the surface, subtilisin digestion of microtubules (which reduces the electrostatic charge of the microtubule surface39) and the presence of EDTA or heparin as anticoagulant in the blood (Supplementary Fig. 4†).
For both actomyosin and kinesin–microtubule motility, washing with control buffer after an initial 10 min incubation with diluted blood samples did not recover motility (p < 0.05; details in Fig. 1 E and F; see also Supplementary Movies 1 and 2†). In particular, no interaction of new filaments with the surface-bound motors was observed. Furthermore, the progressive slow-down was associated with a tendency for the formation of aggregates for both actin-filaments and microtubules (Fig. 1 G and Supplementary Movie 3†) as have been observed previously for streptavidin-cross-linked microtubules.40 In the case of HMM induced actin filament motility, the cross-linking caused the filaments to follow almost perfectly circular paths (Fig. 1 G). Both, motor inhibition and filament cross-linking occurred to a similar degree in whole blood, plasma and serum. Consequently, we hypothesize that the inhibition of both actomyosin and kinesin–microtubule motility is caused by the irreversible binding of blood serum components to both motors and filaments hampering their interaction and thus slowing down motility.
To demonstrate a practical diagnostics application, we conjugated tubulin with anti-blood type A antibodies. A gliding assay with these microtubules was then incubated with blood diluted to 1% in the optimized buffer condition we determined before (BRB80 pH 7.5, IS 95; see Fig. 1D). If the blood was blood type A, active transport of erythrocytes occurred. Erythrocyte transport was enhanced when tubulin and antibodies had clustered, presumably increasing the number of recognition sites on the gliding microtubules. In fact, 9 out of 11 observed clusters transported erythrocytes of blood type A (see Fig. 1 H top row and Supplementary Movie 4†). In a control sample from blood type O, none of the 27 observed clusters or any single microtubule transported erythrocytes, despite many direct contact events where erythrocytes were pushed out of the way (see Fig. 1 H bottom row and Supplementary Movie 5†). The test was performed with a sample volume of 30 nl whole blood obtained by capillary blood collection from a finger tip. Thus we established the ability of molecular motor-driven filaments to perform a label-free blood type analysis.
The results presented in this section indicate, that – quite surprisingly, considering the difference in biophysical properties – blood affects both motor systems in a very similar manner. Optimized conditions are present for both motor systems when blood samples are diluted about 200 fold before applying them directly to molecular motor-driven devices. This dilution can be partly compensated for by an increased sensitivity.26 Thus, if sensitivities in the fM range are desired, 200 fold dilution means that the effective sensitivity of the device needs to be in the 10 aM range. This appears to be an appreciable challenge. Therefore, further studies identifying the hindering blood components and designing ways to counteract these effects may be required.
Effects of lysed cell fractions on cytoskeletal filaments and molecular-motor function
Direct capturing of biomarkers from blood is only possible, if the target analyte is present in blood serum. Intracellular proteins, on the other hand, are only accessible after cell lysis. Ideally, lysis and transport would be performed within one device. Therefore we tested whether actomyosin and kinesin–microtubule motility works in cell lysates.Cytosolic proteins for actin gliding assays, prepared as described in Materials and Methods, led to concentration dependent reduction of actin gliding speed and fraction of motile filaments (Fig. 2 A and Supplementary Movie 6†) as suggested by linear regression analysis. Thus, both the speed and the fraction of motile filaments decreased significantly (p < 0.05) with increasing concentration (slopes of −16.5± 0.058% ml μg−1 (mean ± SEM) and −15.9 ± 2.6% ml μg−1, respectively). We also performed non-linear regression analysis (Supplementary Fig. 5)† to determine an IC50 value of 483 μg ml−1 (95% CI: 356 to 656 μg ml−1) for the reduction in actin filament speed. It is of interest to note the quantitative similarity between these values and the IC50 values for plasma and serum (when translated into plasma protein concentrations; see above). Inhibitory effects were also observed with nuclear proteins with a slightly lower IC50 value (Supplementary Fig. 5)†. After incubation in both cytosolic and nuclear proteins (at the highest concentrations tested), motility recovered to a large extent after the proteins were washed away by standard (protein-free) assay solution (Supplementary Fig. 5)†.
 | ||
| Fig. 2 Effects of lysed cell fractions on cytoskeletal filaments and molecular motor function. (A) Actin filament speed and fraction of motile actin filaments at different concentrations of cytosolic proteins. Nonlinear regression (solid thin blue line) and 95% confidence interval (CI; thin blue dotted lines) of the actin speed data gives average IC50 values of 483 μg ml−1 (95% CI: 356 μg ml−1–656 μg ml−1). Both nonlinear and additional linear regression analysis found a significant trend (p < 0.05). See also Supplementary Fig. 5† for further analysis. (B) Microtubule speeds measured after treatment with cell lysates obtained by lysing 1000–10000 cells μl−1. Unless marked with n.s. (not significant), all data points in this panel are statistically significantly (p < 0.05) different from the respective controls. (A, B) Recovery from treatment with cell lysates was measured after washing with the respective control buffers. Speeds are given as mean ± standard error of the mean of the number of actin filaments given in brackets next to the data points (A), or 16 randomly selected microtubules (B), respectively. | ||
For kinesin–microtubule motility, whole cell lysates with a variety of cell numbers were tested. Microtubule gliding speeds in these lysates were measured directly after injection and every 10 min for a time course of 1 h. Fig. 2 B shows that a lysate prepared with 1000 cells μl−1 had only a slight influence on the microtubule gliding speed. When increasing the cell number in the lysates, the gliding speed decreased considerably and some microtubule bundling could be observed. A lysate prepared with 10000 cells μl−1 caused strong microtubule bundling and after 20 min, it was no longer possible to reliably determine microtubule gliding speeds because microtubules were coiled into spools and circles (Supplementary Movie 7†). It is important to note that, just like for actomyosin motility, the slow-down and the bundling could be partly recovered by washing the flow cell with standard motility solution (Fig. 2 B and Supplementary Movie 7†).
The results presented in this section show that both actomyosin and kinesin–microtubule motility work in the presence of cytosolic as well as nuclear proteins. Again, the effects on both motor systems were similar: Bundling of the filaments and reduced speed as well as recovery after washing away the proteins was observed in both cases. This suggests that, in contrast to the experiments with blood, the slow-down of gliding motility is caused by the reversible binding of cellular proteins to the filaments. This is quite plausible because many such proteins that bind specifically and reversibly to actin filaments and microtubules are known and some of these have been shown to hinder motility.41
Compatibility between motor/filament function and detection of nucleic acid biomarkers
The detection of a specific sequence in a sample of double-stranded genomic DNA can be facilitated using oligonucleotide probes. Recognition of the target sequence requires that DNA is allowed to hybridize with these probes. In this process, the double-stranded DNA needs to be separated into single strands. Usually this is achieved by heating the sample to 90 °C and then cooling to the melting temperature of the oligonucleotide for specific annealing. However, none of the proteins required for molecular transport (microtubules, kinesin, actin filaments and myosin) are stable at these high temperatures. To achieve compatibility with actomyosin and kinesin–microtubule motility, alternative ways of separating double stranded DNA were tested: (i) chemical treatment with e.g. 45% formamide, is known to significantly reduce the DNA melting temperature and (ii) dextran sulfate, often used to prevent re-annealing after strand separation.In order to test the effect of formamide on actomyosin motility, actin filaments (200 nM) were preincubated overnight at 37 °C in standard buffer and up to 30% formamide (66% hybridization buffer with formamide). Thereafter, the solution was diluted ten times in aMC130 solution and added to a HMM coated surface (i.e. up to 3% formamide in the motility assay). Preincubation with up to 15% formamide (i.e. up to 1.5% formamide in the motility assay) did not hamper the actin gliding speed and fraction of motile filaments (Fig. 3 A). Notably, a fraction of the filaments preincubated in 15% formamide moved collectively together along narrow paths rather than randomly on the surface (Supplementary Movie 8†). This could be due to the self-organization phenomena observed previously.42 Similar phenomena were not observed after preincubation with lower dilutions of the hybridization buffer. After preincubation in ≥30% formamide, no filaments were observed, neither in solution nor attached to HMM on the surface, indicating that the filaments were disassembled under these conditions. However, incubation with new actin filaments largely restored motility (speed 75% of control and similar fraction of motile filaments as in control solution) suggesting that the 3% formamide in the motility assay solution did not irreversibly affect HMM. This was confirmed by direct incubation of the motility assay flow cell with formamide in assay solution (without preincubation of the actin filaments). In this case (Fig. 3 B) the motility was irreversibly blocked by 50% formamide, but was negligibly affected by 5% formamide.
 | ||
| Fig. 3 Effects of formamide and dextran sulfate on cytoskeletal filaments and molecular motor function. (A) Actin filament speeds after overnight preincubation of actin in 0–33% (v/v) formamide. In the motility assay, the formamide/dextran containing buffers were diluted again 10 times to give 0–3.3% formamide (B) Actin filament speed in the presence of 0.5–50% formamide (without preincubation of the filaments). (C) Microtubule gliding speed in the presence of 5–30% formamide or 25% dextran sulfate in the motility assay. (A–C) Speeds are given as mean ± standard error of the mean of the number of filaments given in brackets next to the data points. Unless marked with n.s. (not significant) or n.d. (not determined because no filaments could be observed) all data points are statistically significantly (p < 0.05) different from the respective controls. | ||
With kinesin–microtubule motility assays, the hybridization conditions were applied to the whole motility assay. The effects of formamide and dextran sulfate were similar to those observed with actomyosin: In the presence of less than 15% formamide, microtubule gliding could be observed (Fig. 3 C). However, at 15% formamide, microtubules started to depolymerize (Supplementary Movie 9†) and at concentrations ≥30% formamide no microtubules could be observed and gliding could not be recovered afterwards. Difficulties with the motility assay were also encountered with dextran sulfate. Microtubule gliding speeds in 25% dextran sulfate could not be determined because the microtubules did not glide in a directional manner, but rather moved back and forth along the surface, possibly because of microtubule entanglement in the dense dextran sulfate network. Again, gliding could not be recovered after washing with fresh motility solution.
A further pre-condition for the application of molecular motors and cytoskeletal filaments in the diagnostic detection of specific oligonucleotides from a pool of patient's gDNA specimens is that genomic DNA itself does not affect in vitro motility. In view of the discouraging experiments with hybridization conditions, the latter was investigated only for actomyosin. In particular, we were concerned about the effects on motor function due to electrostatic interactions between the positively charged actin binding sites on HMM and the negatively charged DNA. We therefore preincubated HMM surfaces with gDNA at concentrations of 100–1000 μg ml−1 before adding the motility assay solution (Supplementary Fig. 6†). Preincubation with 100 μg ml−1 gDNA had no apparent effect on motility. At 500 μg ml−1 gDNA motility was still observable but gliding speed and the fraction of motile filaments were reduced. After preincubation with 1000 μg ml−1 gDNA binding of actin filaments to HMM was irreversibly blocked. When gDNA was added to the assay solution, there was a slight reduction of the actin gliding speed and the fraction of motile filaments.
The results presented in this section indicate that neither actomyosin nor kinesin–microtubule motility are compatible with DNA hybridization conditions. This is not unexpected since melting double-stranded DNA requires the disruption of many hydrogen bonds and the proteins involved in the motility assays studied here are stabilized by hydrogen bonds. On the other hand, if the nucleic acid that is to be detected is single-stranded (as is the case for mRNA), it is not necessary to melt the nucleic acids. Under these latter conditions it has previously been shown that molecular beacons, transported by molecular motors, can detect certain nucleotide sequences.19,43
Effects of different buffers, pH and Ca2+-ions on cytoskeletal filaments and molecular-motor function
In our laboratories, actomyosin and kinesin–microtubule gliding assays are commonly performed in defined MOPS- (e.g. aMC130) and PIPES-based buffers (e.g. BRB80), respectively. These buffers are widely used throughout the field but little is known about how the motility assays perform in other buffer solutions. However, for the application in a lab-on-a-chip device it is important to know the flexibility of the working conditions, because some specimens or analytical tests might require different buffers. Therefore we analyzed the impact of switching from conventional solutions for each motor system to a range of alternative buffers.Previously, it has been shown that in addition to the commonly used MOPS based buffers of ionic strengths from 20–160 mM,44 actomyosin motility also works without limitations in imidazole based buffers45 and phosphate buffered saline.46 The flexibility of the actomyosin motility assay concerning the buffering agent is also shown in Fig. 4 A. Although the mean speeds (but not the fraction of motile filaments) for all conditions tested were significantly different from the control value it is important to note that no reduction in actin gliding speed of practical relevance was observed. To corroborate this, was the main purpose of the experiment. The increased gliding speeds seen in HEPES and BRB80 (Fig. 4 A last two groups of bars) potentially deserve further investigation. However, this is outside the scope of the present study. In addition, extra Ca2+, expected in diagnostic samples affected neither gliding speed nor the fraction of motile filaments Fig. 4 B).
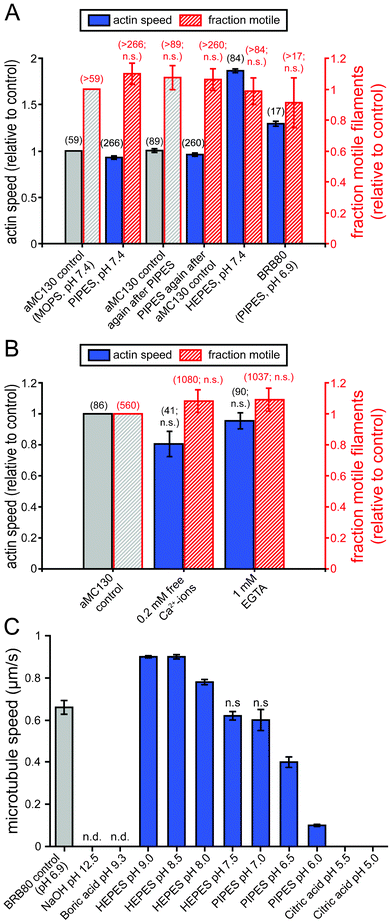 | ||
| Fig. 4 Effects of different buffers, pH and Ca2+-ions on cytoskeletal filaments and molecular motor function. (A) Actin filament speeds and fractions of motile actin filaments in various buffers. All velocity data points differ from the control value(s) (p < 0.05), although the effects were small except for the effect of HEPES. (B) The effect of EGTA and free Ca2+-ions on actomyosin gliding speed and fraction of motile filaments. (C) Microtubule speeds in various buffers at pH 5.0–12.5. (A–C) Speeds are given as mean ± standard error of the mean of 16 randomly selected microtubules (C) or the number of actin filaments given in brackets next to the data points (A, B). Unless marked with n.s. (not significant) or n.d. (not determined because no filaments could be observed) all data points are statistically significantly different (p < 0.05) from the respective controls. | ||
Chemical reactions required for the labeling of cytoskeletal filaments with antibodies or oligonucleotides often require increased or decreased pH. However, the effect of pH has previously only been studied for actomyosin47 and for a different kinesin motor than the one commonly used for nanotechnological applications.48 Therefore, in addition to testing the buffers, we also varied the pH ranges of these buffers for the kinesin–microtubule system. Fig. 4 C (and Supplementary Movie 9†) shows microtubule gliding speeds in common biological buffers for a range of pH values. From pH 6 up to pH 9, microtubule gliding speeds constantly increased. However, between pH 8 and 9, microtubule tips started to detach from the motor-coated surface after approximately 10 min and shorter microtubules detached completely, indicating a lower active kinesin density on the surface. On the other hand, microtubules alone were stable over a wider range from pH 5.5 up to pH 9.3. At pH 5.0 microtubules were observed to slowly depolymerize. At pH 12.5 (in 50 mM NaOH) tubulin clusters were observed and adding fresh microtubules in motility solution did not restore motility indicating that both microtubules and kinesin denatured rapidly. These results confirm once more the incompatibility with DNA melting conditions (see above). Thus, the functional pH range for kinesin–microtubule motility was pH 6 to 8 with an optimum at pH 7.5. In this pH range, kinesin–microtubule motility performed equally well with HEPES and PIPES-based buffers. The usable pH range for microtubules was pH 5.5 to 9.3.
The results presented in this section show that both actomyosin and kinesin–microtubule motility assays work in a wide range of buffering conditions. This supports the feasibility of replacing microfluidic transport by molecular-motor based transport in a variety of miniaturized biological assays.
Comparison of the effects on actomyosin and kinesin–microtubule motility
Table 1 summarizes the effects of the different sample types studied here on actomyosin and kinesin–microtubule motility. Except for DNA hybridization conditions, all sample types were compatible at defined dilutions. Stable motility was observed for both motor systems in blood samples when they were diluted ∼100 fold. The detrimental effects of blood could not be reversed for either system. Cell lysates allowed stable motility up to 160 μg ml−1 for actomyosin and 5000 cells μl−1 for kinesin–microtubule motility, respectively. The effects of cell lysates on both motor systems were reversible. DNA hybridizing conditions caused irreversible inactivation of both motor systems. Finally, both motor systems were fully compatible with various buffer systems frequently used in diagnostic assays. Surprisingly, these effects were very similar for both actomyosin and kinesin–microtubule motility, despite the fact that both systems have very different biophysical properties like filament rigidity, gliding speed and motor processivity.| Sample types/conditions | Actomyosin | Kinesin–microtubule |
|---|---|---|
| Blood samples | compatible when diluted to 1%; effect non-reversible | compatible when diluted to 1% effect non-reversible |
| Cell lysates | compatible up to 160 μg ml−1; effect reversible | compatible up to 5000 cells μl−1; effect reversible |
| DNA hybridization conditions | not compatible | not compatible |
| PIPES, HEPES, MOPS buffers | fully compatible | fully compatible |
Conclusions
We studied how diagnostic samples and derived solutions such as blood, cell lysates, gDNA and DNA-hybridization buffers affect in vitro gliding motility assays. This information is critical for the development of diagnostics applications where, starting from patient's specimens, separation, concentration and detection of biomarkers rely on molecular motors and their associated cytoskeletal filaments. The present study identifies important constraints on how to develop clinically useful devices based on actomyosin and/or microtubule–kinesin motility, contributing to close a long-standing information gap in the field. The most severe effects on both systems were observed under DNA hybridization conditions necessary for the separation of double-stranded DNA into single strands, which is a prerequisite for the analysis of gDNA. These conditions appear to be incompatible with motor function. For blood samples, however, approximately 100-fold dilution preserves motor function and allowed us to demonstrate label-free blood-group typing. Specific dilution of cell lysates also guarantees motor activity. No appreciable difference of the performance of two motor systems in the different sample solution types was observed. Therefore, the choice to use either system in future diagnostic devices can be made independent of the sample type to be analyzed and can focus on the respective advantages of either system. For example actomyosin transport could be preferred for the fast transport of small analytes, while kinesin–microtubule transport performs better for large analytes, such as the transport of erythrocytes demonstrated here. Together with the recent demonstrations of convenient long-term storage of both actomyosin and kinesin–microtubule based motility systems,49,50 our results demonstrate the feasibility of molecular-motor driven lab-on-a-chip devices. We envision that molecular motors will help to develop devices that are smaller, cheaper and more convenient to handle than conventional systems, making them ideal for diagnostic point-of-care applications.Acknowledgements
We gratefully acknowledge help with blood sampling from P Nilsson and K Sandahl in the research group of K Nilsson-Ekdahl at the Linnaeus University. We also acknowledge M Lard for valuable comments on the manuscript. This work was funded by the European commission (FP7) under the contract MONAD (NMP4-SL-2009-228971), the European Research Council (ERC starting grant), the Swedish Research Council (Project # 621-2010-5146), The Carl Trygger Foundation, the Deutsche Forschungsgemeinschaft (DFG Heisenberg Programme), and the Faculty of Natural Sciences and Engineering at Linnaeus University as well as the Max-Planck-Society.Alf Månsson is a co-founder, co-owner and CEO of the start-up company ActoSense Biotech AB (Kalmar, Sweden) aiming to develop diagnostic devices based on the aggregation of cytoskeletal elements, particularly actin filaments, in solution. Moreover, A Månsson holds two Swedish patents in this field and application for one of these patents (about aggregation of actin filaments by analyte molecules) has also been filed in the US and Europe.
Notes and references
- M. L. Chabinyc, D. T. Chiu, J. C. McDonald, A. D. Stroock, J. F. Christian, A. M. Karger and G. M. Whitesides, Anal. Chem., 2001, 73, 4491–4498 CrossRef CAS.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- E. D. Goluch, J. M. Nam, D. G. Georganopoulou, T. N. Chiesl, K. A. Shaikh, K. S. Ryu, A. E. Barron, C. A. Mirkin and C. Liu, Lab Chip, 2006, 6, 1293–1299 RSC.
- H. D. Hill and C. A. Mirkin, Nat. Protoc., 2006, 1, 324–336 CrossRef CAS.
- E. D. Goluch, S. I. Stoeva, J. S. Lee, K. A. Shaikh, C. A. Mirkin and C. Liu, Biosens. Bioelectron., 2009, 24, 2397–2403 CrossRef CAS.
- A. Månsson, M. Sundberg, R. Bunk, M. Balaz, I. A. Nicholls, P. Omling, J. O. Tegenfeldt, S. Tågerud and L. Montelius, IEEE Trans. Adv. Packag., 2005, 28, 547–555 CrossRef.
- A. Agarwal and H. Hess, Prog. Polym. Sci., 2010, 35, 252–277 CrossRef CAS.
- T. Korten, A. Månsson and S. Diez, Curr. Opin. Biotechnol., 2010, 21, 477–488 CrossRef CAS.
- H. Takatsuki, H. Tanaka, K. M. Rice, M. B. Kolli, S. K. Nalabotu, K. Kohama, P. Famouri and E. R. Blough, Nanotechnology, 2011, 22, 245101 CrossRef.
- S. J. Kron and J. A. Spudich, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6272–6276 CrossRef CAS.
- J. Howard, A. J. Hudspeth and R. D. Vale, Nature, 1989, 342, 8 CrossRef.
- H. Hess, J. Clemmens, D. Qin, J. Howard and V. Vogel, Nano Lett., 2001, 1, 235–239 CrossRef CAS.
- T. B. Brown and W. O. Hancock, Nano Lett., 2002, 2, 1131–1135 CrossRef CAS.
- J. A. Jaber, P. B. Chase and J. B. Schlenoff, Nano Lett., 2003, 3, 1505–1509 CrossRef CAS.
- L. Ionov, M. Stamm and S. Diez, Nano Lett., 2005, 5, 1914 CrossRef.
- C. Z. Dinu, J. Opitz, W. Pompe, J. Howard, M. Mertig and S. Diez, Small, 2006, 2, 1090–1098 CrossRef CAS.
- T. Korten and S. Diez, Lab Chip, 2008, 8, 1441–1447 RSC.
- C. T. Lin, M. T. Kao, K. Kurabayashi and E. Meyhofer, Nano Letters, 2008, 8, 1046 Search PubMed.
- M. Raab and W. O. Hancock, Biotechnol. Bioeng., 2008, 99, 773 CrossRef.
- C. M. Soto, B. D. Martin, K. E. Sapsford, A. S. Blum and B. R. Ratna, Anal. Chem., 2008, 80, 5433–5440 CrossRef CAS.
- A. Agarwal, P. Katira and H. Hess, Nano Lett., 2009, 9, 5 CrossRef.
- T. Fischer, A. Agarwal and H. Hess, Nat. Nanotechnol., 2009, 4, 162–166 CrossRef CAS.
- A. Carroll-Portillo, M. Bachand, A. C. Greene and G. D. Bachand, Small, 2009, 5, 1835–1840 CrossRef CAS.
- L. Rios and G. D. Bachand, Lab Chip, 2009, 9, 1005–1010 RSC.
- R. Martinez-Neira, M. Kekic, D. Nicolau and C. G. dos Remedios, Biosens. Bioelectron., 2005, 20, 1428–1432 CrossRef CAS.
- P. Katira and H. Hess, Nano Lett., 2010, 10, 567–572 CrossRef CAS.
- J. D. Pardee and J. A. Spudich, Methods Cell Biol., 1982, 24, 271–289 CrossRef CAS.
- M. Sata, S. Sugiura, H. Yamashita, S. Momomura and T. Serizawa, Circ. Res., 1993, 73, 696–704 CrossRef CAS.
- S. J. Kron, Y. Y. Toyoshima, T. Q. P. Uyeda and J. A. Spudich, Methods Enzymol., 1991, 196, 416 Search PubMed.
- D. L. Coy, M. Wagenbach and J. Howard, J. Biol. Chem., 1999, 274, 3671 CrossRef.
- A. Hyman, D. Drechsel, D. Kellogg, S. Salser, K. Sawin, P. Steffen, L. Wordeman and T. Mitchison, Methods Enzymol., 1991, 196, 478–485 CAS.
- M. Sundberg, J. P. Rosengren, R. Bunk, J. Lindahl, I. A. Nicholls, S. Tågerud, P. Omling, L. Montelius and A. Månsson, Anal. Biochem., 2003, 323, 127–138 CrossRef CAS.
- , B. Nitzsche, V. Bormuth, C. Bräuer, J. Howard, L. Ionov, J. Kerssemakers, T. Korten, C. Leduc, F. Ruhnow, S. Diez, in Microtubules, in vitro, ed. W. Leslie and J. C. John, Academic Press, 2010, vol. Volume 95, pp.247–271 Search PubMed.
- J. D. Dignam, R. M. Lebovitz and R. G. Roeder, Nucleic Acids Res., 1983, 11, 1475–1489 CrossRef CAS.
- A. Månsson and S. Tågerud, Anal. Biochem., 2003, 314, 281–293 CrossRef.
- W. O. Hancock and J. Howard, J. Cell Biol., 1998, 140, 1395–1405 CrossRef CAS.
- E. Homsher, B. Kim, A. Bobkova and L. S. Tobacman, Biophys. J., 1996, 70, 1881–1892 CrossRef CAS.
- F. Zhang, M. W. A. Skoda, R. M. J. Jacobs, R. A. Martin, C. M. Martin and F. Schreiber, J. Phys. Chem. B, 2011, 111, 251–259 CrossRef.
- J. Helenius, G. Brouhard, Y. Kalaidzidis, S. Diez and J. Howard, Nature, 2006, 441, 115–119 CrossRef CAS.
- H. Hess, J. Clemmens, C. Brunner, R. Doot, S. Luna, K.-H. Ernst and V. Vogel, Nano Lett., 2005, 5, 33 CrossRef.
- R. Dixit, J. L. Ross, Y. E. Goldman and E. L. Holzbaur, Science, 2008, 319, 1086–1089 CrossRef CAS.
- V. Schaller, C. Weber, C. Semmrich, E. Frey and A. R. Bausch, Nature, 2010, 467, 73–77 CrossRef CAS.
- S. Taira, Y.-Z. Du, Y. Hiratsuka, K. Konishi, T. Kubo, T. Q. P. Uyeda, N. Yumoto and M. Kodaka, Biotechnol. Bioeng., 2006, 95, 533–538 CrossRef CAS.
- N. Albet-Torres, M. J. Bloemink, T. Barman, R. Candau, K. Frölander, M. A. Geeves, K. Golker, C. Herrmann, C. Lionne, C. Piperio, S. Schmitz, C. Veigel and A. Månsson, J. Biol. Chem., 2009, 284, 22926–22937 CrossRef CAS.
- M. Balaz and A. Månsson, Anal. Biochem., 2005, 338, 224–236 CrossRef CAS.
- M. Balaz, M. Sundberg, M. Persson, J. Kvassman and A. Månsson, Biochemistry, 2007, 46, 7233–7251 CrossRef CAS.
- E. Homsher, F. Wang and J. R. Sellers, American Journal of Physiology-Cell Physiology, 1992, 262, C714–C714 CAS.
- K. J. Böhm, R. Stracke and E. Unger, Cell Biol. Int., 2000, 24, 335–341 CrossRef.
- R. Seetharam, Y. Wada, S. Ramachandran, H. Hess and P. Satir, Lab Chip, 2006, 6, 42 RSC.
- N. Albet-Torres and A. Mansson, Langmuir, 2011, 27, 7108–7112 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2lc41099k |
| ‡ These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2013 |