 Open Access Article
Open Access ArticleVisualizing viral assemblies in a nanoscale biosphere†
Brian L.
Gilmore‡
a,
Shannon P.
Showalter‡
a,
Madeline J.
Dukes
b,
Justin R.
Tanner
a,
Andrew C.
Demmert
a,
Sarah M.
McDonald
a and
Deborah F.
Kelly
*a
aVirginia Tech Carilion Research Institute, Virginia Tech, Roanoke, Virginia 24016, USA. E-mail: debkelly@vt.edu; Fax: 1-540-985-3373; Tel: 1-540-526-2031
bApplications Science, Protochips, Inc., Raleigh, North Carolina 27607, USA. E-mail: madeline@protochips.com; Fax: 1-919-341-2748; Tel: 1-919-341-2612
First published on 19th November 2012
Abstract
We present a novel microfluidic platform to examine biological assemblies at high-resolution. We have engineered a functionalized chamber that serves as a “nanoscale biosphere” to capture and maintain rotavirus double-layered particles (DLPs) in a liquid environment. The chamber can be inserted into the column of a transmission electron microscope while being completely isolated from the vacuum system. This configuration allowed us to determine the structure of biological complexes at nanometer-resolution within a self-contained vessel. Images of DLPs were used to calculate the first 3D view of macromolecules in solution. We refer to this new fluidic visualization technology as in situ molecular microscopy.
Introduction
Understanding the properties of molecular machines is a common goal of biologists and engineers. Our knowledge is currently limited by the inability to study dynamic structures at high-resolution. Transmission electron microscopy (TEM) allows us to peer into the world of molecules while approaching near-atomic resolution.1–3 However, biological machines must be fixed in order to enter the vacuum system of an electron microscope. This is typically done by freezing specimens in a thin layer of vitreous ice.4,5 Although ice preserves the features of biological complexes, it also arrests molecular machinery and limits the information we seek to acquire.Recent advances in the development of materials such as graphene6,7 and silicon nitride8 provide new opportunities for imaging chemical processes in real-time. These new materials can be exploited as environmental chambers for performing experiments in situ or “inside” the EM column.9,10 In conjunction with new microfluidic based specimen holders,11 scientists have observed the growth of materials12 and living cells engulfing nanoparticles.13 One inherent limitation with this technique is that motion occurs when entities are freely diffusing in solution results in poor structural resolution.
Affinity Capture devices14 consist of functionalized silicon nitride microchips that bind specifically and with high affinity to protein complexes, thereby tethering them in a fluidic chamber. These devices can be produced using commercially available microchips (Protochips, Inc., Raleigh, NC) and form the imaging platform for a liquid flow holder. Two of these chips are tightly sealed together with nanometer spacers to accommodate samples in a self-contained “nanoscale biosphere”. Transparent windows (50 nm thick) etched into the chips allow the electron beam to penetrate the liquid chamber for imaging purposes.
Our present work utilizes this new technology to capture rotavirus double-layered particles (DLPs) onto functionalized microchips decorated with antibodies against the viral capsid protein, VP6. Native, rotavirus DLPs were examined in solution within a microfluidic chamber using TEM. Here we show the first high-resolution images of biological assemblies contained entirely within liquid. We refer to this technical innovation as in situ molecular microscopy.
Materials and methods
Preparation of functionalized microchips
Affinity Capture devices were prepared and decorated with nickel-nitrilotriacetic acid (Ni-NTA) lipid layers as indicated in the ESI.† The lipid-labeled chips were incubated for 1 min at room temperature with 4 μl aliquots of His-tagged Protein A (0.01 mg ml−1) (Abcam, Cambridge, MA) in buffer containing 50 mM Hepes, pH 7.5, 150 mM NaCl, 10 mM MgCl2 and 10 mM CaCl2. The excess solution was blotted away, and the chips were incubated with 4 μl aliquots of 0.01 mg ml−1 VP6-specific guinea pig polyclonal antisera (#53963) for 1 min at room temperature. For cryo-EM experiments, C-flat grids (Protochips, Inc., Raleigh, NC) containing 2 μm holes with 1 μm spacing between holes were used in place of silicon nitride chips.Preparation of EM specimens
Aliquots (3 μl) of purified DLPs (0.1 mg ml−1 in 50 mM Hepes, pH 7.5, 150 mM NaCl, 10 mM MgCl2 and 10 mM CaCl2) were added to antibody-labeled silicon nitride microchips or C-flat grids and incubated for 2 min. Following the incubation step, specimens were prepared by 3 different methods: 1) maintained completely in solution while placed in the in situ specimen holder, or 2) negatively stained and dried upon the chips, or 3) plunged into liquid ethane on C-flat grids after a 3-second blotting interval using a FEI Mach III Vitrobot (FEI Company, Hillsboro, OR). In situ specimens were prepared by adding the DLP sample (3 μl) onto the antibody labeled chips then spiking the wet chip with a small volume (∼0.3 μl) of contrast reagent (0.2% w/v, uranyl formate). The final pH of the DLPs in buffer solution containing contrast reagent was 7.2. The contrast reagent did not fix the specimens and was used to ensure proper visualization of DLPs for subsequent image processing routines. The wet chips, containing DLPs in solution and contrast reagent were then sealed in the chamber by the addition of another microchip. The entire assembly was positioned into the tip of a Poseidon TEM specimen holder (Protochips, Inc., Raleigh, NC). The holder containing the liquid samples was maintained at room temperature prior to transfer to the TEM. The DLP samples remained fully hydrated in the TEM during the course of the experiments. We periodically tested for the presence of solution in the chamber by exposing a region until bubbles formed. Representative images of bubble formation in 150 nm of liquid can be found in Fig. 1 (a–c) of the (ESI).† Bubble formation in the liquid chamber supports our claim that rotavirus particles were contained in solution and were not dried nor fixed. Details on DLP preparation and image processing routines can be found in the ESI. Negatively stained control specimens were prepared as described in the ESI. Frozen specimens used for comparison purposes were transferred to a 626 Gatan holder and maintained at −180 °C prior to inserting into the TEM.Results and discussion
Capturing viral complexes using affinity devices
To engineer a system for capturing biological assemblies in a microfluidic chamber (Fig. 1a), we coated silicon nitride microchips with a lipid biofilm doped with functionalized Ni–NTA lipids and DLPC filler lipids. His-tagged protein A was added to the Ni–NTA-coated microchips to serve as an adaptor molecule to bind to polyclonal antibodies against VP6.15 The antibodies against VP6 were added to the protein A-containing chips (Fig. 1b), as a means of mimicking an antibody affinity column. This technique was used previously to purify macromolecular complexes from mammalian cell lysates.16 Rotavirus DLPs were prepared using established protocols17 and contained the viral protein subunits VP1, VP2, VP3 and VP6 according to SDS-PAGE analysis (Fig. 2a, ESI†). To assess the overall recruitment of particles using this technique, we added DLPs (0.1 mg ml−1) to microchips decorated with protein A/antibodies and viewed the resulting samples in negative stain using TEM.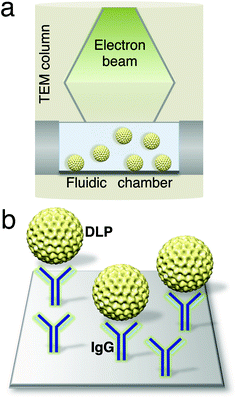 | ||
| Fig. 1 Schematic of a microfluidic chamber (a) that accommodates biological assemblies (yellow) in solution while inside a TEM column. (b) Magnified view of the chamber surface decorated with IgG antibodies (blue) against the DLP outer capsid protein, VP6. | ||
DLP samples were examined using a FEI Spirit BioTwin TEM equipped with a tungsten filament and operating at 120 kV. Rotavirus DLPs bound to the decorated microchips in adequate quantities for single particle analysis using 0.2% uranyl formate (Fig. 2c, ESI†). Negative control samples lacking VP6-specific antibodies failed to recruit DLPs (Fig. 2b, ESI†). DLPs in buffer solution were unaffected by the change in pH (from 7.5 to pH 7.2) upon the addition of contrast reagent over the length of the experiments (Fig. 3, ESI†). Images were recorded under low-dose conditions (∼5 electrons/Å2) for all samples with a final sampling of 10 Å/pixel and processed using the SPIDER software package.18 Individual particles were selected from the images using the WEB interface associated with SPIDER. Selected particles were windowed into individual panels of 110 × 110 pixels and subjected to 10 cycles of multi-reference alignment. Each round of alignment was followed by principal component analysis and K-means classification. References used for the first alignment cycle were randomly selected from the raw images. Averages of the DLPs in negative stain had diameters of 80–94 nm and showed features with visible icosahedral symmetry (Fig. 5a, ESI†).
Using this system, the detection limit of 0.03 mg ml−1 was determined by the amount of DLPs present on the chip that would still allow for image processing routines. Overall, these results indicated that affinity microchips coated with antibodies against the VP6 protein could specifically recruit DLPs for quantification involving statistical image processing analysis and 3D reconstruction routines.
In situ molecular microscopy
We applied the affinity capture strategy to produce DLP specimens that were tethered in a liquid environment for in situ imaging. A low concentration of contrast reagent was added to the liquid chamber to enhance downstream image processing. This reagent did not fix the native DLPs unlike traditional staining procedures. The holder containing the intact liquid chamber was inserted into the TEM column and examined under low-dose conditions (∼5 electrons/Å2 for each exposure). To ensure the liquid environment did not flatten the DLPs in a manner similar to dried negatively stained specimens, we recorded untilted and tilted images at 30° for both liquid specimens (Fig. 4a, ESI†) and negatively stained specimens (Fig. 4b, ESI†). No distortion due to flattening were detected in the tilted liquid specimens while flattening was noted in the tilted stained specimens, presumably due to air-drying. Images of rotavirus particles suspended in liquid were recorded at 30,000x for processing routines.Individual virus particles were selected from images of DLPs in liquid (Fig. 2a) and subjected to the same classification procedures as our negatively stained samples (Fig. 5a, ESI†). Projection averages of particles in solution (Fig. 5b, ESI†) are more sharply defined than in dried negatively stained specimens. Averages of liquid specimens also appeared to exhibit a variety of different views in comparison to the limited number of views displayed by the negatively stained averages (compare Fig. 5b and 5a, ESI†). To determine the overall structure DLPs in solution, we calculated 3D reconstructions using the RELION software package.19
 | ||
| Fig. 2 Individual DLPs imaged in solution (a) have structural heterogeneity and show a variety of views. Scale bar is 300 nm. (b) Multiple 3D volumes were calculated from the original reconstruction (purple) of DLPs in solution. Percentages for the number of particles contained in each structure are indicated below each volume: 65% (light blue), 23% (pink), 7% (grey) and 5% (yellow). Dynamic volumes are reduced by 50% of the original volume. Reconstructions were visualized using the program Chimera.22 Each DLP volume is ∼80 nm in diameter. | ||
The image stack derived from the SPIDER package was imported into RELION and a reference map for the rotavirus DLP structure20 was downloaded from the website (http://emlab.rose2.brandeis.edu/rotavirusdlp) for the Grigorieff laboratory. Refinement parameters in RELION included a pixel size of 10 Å, an initial model low-pass filtered of the initial model to 80 Å and a regularisation parameter of T = 4 while enforcing icosahedral symmetry over an angular search space of 7.5 degrees. The refinement procedure was iterated for 15 cycles outputting a single reconstruction (Fig. 2b, purple) having a resolution of ∼25 Å. The density map was in good agreement with the reference model.
Heterogeneous structures in solution
We used the RELION program to assess the level of structural heterogeneity present in the viral assemblies. Following the first five rounds of refinement, statistical values indicated the presence of four variant structures in the image stack. We refined each of the structures independently for an additional 10 iterations. The resulting 3D volumes were masked to 94 nm with a major structure containing 65% of the particles (blue in Fig. 2b) and another variant containing 23% (pink in Fig. 2b). Two additional reconstructions contained only 7% (grey in Fig. 2b) and 5% (yellow in Fig. 2b) of the total particles present in the image stack. The fact that we were able to determine four individual reconstructions for DLPs in solution suggested that they exhibited some degree of structural heterogeneity, as they were not fixed.The reconstruction representing the majority of the particles (65%, blue in Fig. 2b) best resembles the initial model that was derived from previous cryo-EM experiments. This main reconstruction has strongly defined symmetry axes with distinctive crevices and more rigid features. The reconstruction containing 23% of the particles (pink in Fig. 2b) showed blurring in surface crevices that are much more shallow than in the initial model. The minor reconstructions contained only 7% and 5% of the particles (grey and yellow in Fig. 2b, respectively). These particles have ill-defined crevices and symmetry axes exhibiting a greater degree of heterogeneity. The individual particles constituting these averages were visually assessed and determined to contain no major distortions or damage (data not shown). We attribute a majority of the heterogeneity seen in the liquid reconstructions to 1) particles being located in different thicknesses of liquid, 2) Brownian motion and 3) beam-induced motion.21 There is also a remote possibility that heterogeneity is caused by the presence of contrast reagent or by subtle differences in biological stages/conformations. However, it is unlikely that the contrast reagent played a major role as the particles were fully contained in solution during imaging. Contrast reagents contribute to heterogeneity in single particle reconstructions as a result of air-drying the specimens after applying the reagents, similar to Fig. 4b, ESI.†
Cryo-EM of rotavirus assemblies
To determine how reconstructions of DLPs in liquid correlate to those of frozen-hydrated samples, we collected images of ice-embedded DLPs. Specimens were prepared using the same experimental parameters that we used for the in situ experiments, substituting C-flat grids for microchips. Images of DLPs (Fig. 3) were processed using SPIDER and projection averages of the frozen specimens (Fig. 5c, ESI†) revealed a similar degree of quality as averages of the liquid specimens. The same refinement and reconstruction procedures were implemented in RELION to calculate a 3D volume. In contrast to what was found for the in situ specimens, a single, statistically significant population was present in the image stack of ice-embedded DLPs, yielding a ∼24 Å reconstruction (Fig. 3, inset). The cryo reconstruction was similar to the major structure identified in the liquid experiments. These results are consistent with the idea that frozen specimens of macromolecular complexes are less heterogeneous in comparison to liquid specimens. Nonetheless, the concept of heterogeneity among macromolecules in liquid versus ice needs to be further validated. | ||
| Fig. 3 Image of vitrified DLPs and 3D reconstruction of vitrified DLPs (inset) exhibit icosahedral symmetry (view down the 3-fold axis). The 3D volume is highly similar to the initial liquid reconstruction (Fig. 2b, purple). DLP reconstruction is ∼80 nm in diameter. Scale bar is 300 nm. | ||
Conclusions
In this study, we developed a new technology to determine the first 3D structure of macromolecules fully contained within a liquid environment. In situ molecular microscopy, utilizing Affinity Capture devices, expands upon our previous work to view the first biological complexes in solution using transmission electron microscopy.14 The technical advancements, presented here, provide for the first time a useful strategy to produce specimens in solution that are suitable for single particle image processing routines and 3D reconstruction calculations. Employing these new tools, we found that the viral assemblies imaged in solution have a greater degree of structural heterogeneity than those fixed in vitreous ice. As such, we envision that this new technology may be applied to “live-EM imaging” of molecular mechanisms, such as viral assembly pathways and viral entry into host cells. This exciting new frontier would allow investigators to examine biological processes in solution with a remarkable new level of resolution.Acknowledgements
The authors thank Dr Michael Friedlander, Director of the Virginia Tech Carilion Research Institute, for supporting our endeavors and Dr John Patton of the National Institute of Allergy and Infectious Diseases at NIH for generously providing the VP6 antisera used in these studies.References
- Z. H. Zhou, Curr. Opin. Struct. Biol., 2008, 18, 218–228 CrossRef CAS.
- Y. Cheng and T. Walz, Annu. Rev. Biochem., 2009, 78, 723–742 CrossRef CAS.
- M. Wolf, R. L. Garcea, N. Grigorieff and S.C. Harrison, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6298–6303 CrossRef CAS.
- J. Dubochet, Q. Rev. Biophys., 1988, 21, 129–228 CrossRef CAS.
- J. Frank, Q. Rev. Biophys., 2009, 42, 139–158 CrossRef CAS.
- S. Chen, W. Cai, R. D. Piner, J. W. Suk, Y. Wu, Y. Ren, J. Kang and R. S. Ruoff, Nano Lett., 2011, 11, 3519–3525 CrossRef CAS.
- J. M. Yuk, J. Park, P. Ercius, K. Kim, D. J. Hellenbusch, M. F. Crommie, J. Y. Lee, A. Zettl and A. P. Alivisatos, Science, 2012, 336, 61–64 CrossRef CAS.
- E. A. Ring, D. B. Peckys, M. J. Dukes, J. P. Baudoin and N. de Jonge, J. Microsc., 2011, 243, 273–283 CrossRef CAS.
- N. de Jonge, D. B. Peckys, G. J. Kremers and D. W. Piston, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2159–2164 CrossRef CAS.
- N. de Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695–704 CrossRef CAS.
- E. A. Ring and N. de Jonge, Microsc. Microanal., 2010, 16, 622–629 CrossRef CAS.
- J. E. Evans, K. L. Jungjohann, N. D. Browning and I. Arslan, Nano Lett., 2011, 11, 2809–2813 CrossRef CAS.
- D. B. Peckys and N. de Jonge, Nano Lett., 2011, 11, 1733–1738 CrossRef CAS.
- K. Degen, M. Dukes, J. R. Tanner and D. F. Kelly, RSC Adv., 2012, 2, 2408–2412 RSC.
- L. S. Silvestri, Z. F. Taraporewala and J. T. Patton, J. Virol., 2004, 78, 7763–7774 CrossRef CAS.
- D. F. Kelly, D. Dukovski and T. Walz, J. Mol. Biol., 2010, 400, 675–681 CrossRef CAS.
- P. Bican, J. Cohen, A. Charpilienne and R. Scherrer, J. Virol., 1982, 43, 1113–1117 CAS.
- J. Frank, M. Radermacher, P. Penczek, J. Zhu, M. Ladjadj and A. Leith, J. Struct. Biol., 1996, 116, 190–199 CrossRef CAS.
- S. H. Scheres, J. Mol. Biol., 2012, 415, 406–418 CrossRef CAS.
- X. Zhang, E. Settembre, C. Xu, P. R. Dormitzer, R. Bellamy, S. C. Harrison and N. Grigorieff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1867–1872 CrossRef CAS.
- A. F. Brilot, J. Z. Chen, A. Cheng, J. Pan, S. C. Harrison, C. S. Potter, B. Carragher, R. Hendersen and N. Grigorieff, J. Struct. Biol., 2012, 177, 630–637 CrossRef CAS.
- T. D. Goddard, C. C. Huang and T. E. Ferrin, J. Struct. Biol., 2007, 157, 281–287 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2lc41008g |
| ‡ These authors contributed equally to the work |
| This journal is © The Royal Society of Chemistry 2013 |