Direct determination of Cu isotope ratios in dried urine spots by means of fs-LA-MC-ICPMS. Potential to diagnose Wilson's disease
Martín
Resano
*a,
Maite
Aramendía
b,
Luis
Rello
c,
Mª Luisa
Calvo
c,
Sylvain
Bérail
d and
Christophe
Pécheyran
d
aUniversity of Zaragoza, Faculty of Sciences, Department of Analytical Chemistry, Pedro Cerbuna 12, Zaragoza, E-50009, Spain
bCentro Universitario de la Defensa-Academia General Militar de Zaragoza, Carretera de Huesca s/n, 50090, Zaragoza, Spain
cDepartment of Clinical Biochemistry, “Miguel Servet” Universitary Hospital, Paseo Isabel La Católica 1-3, 50009, Zaragoza, Spain
dLCABIE, IPREM UMR 5254, CNRS – Université de Pau et des Pays de l'Adour, 64053, Pau cedex 9, France
First published on 25th October 2012
Abstract
This work investigates the potential of a 257 nm femtosecond (fs) laser ablation (LA) device operating at a high repetition rate (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz) coupled to a multicollector (MC)-ICPMS to develop a method for the direct determination of Cu ratios in dried urine spots, prepared by deposition of urine (300 μL) onto precut clinical filter paper discs (16 mm diameter). The sampling capabilities offered by the fs LA system, permitting ablation of 150 μm thick coronas in the rim area of the filter, together with the use of admixed Ni as an internal standard, the proper optimization of the MC-ICPMS conditions (e.g., use of pseudo high-resolution mode to avoid interferences) and the use of a data processing approach particularly suitable for short transient signals (linear regression fit) enabled analysis of real urine samples with precision values around 500 ppm (RSD) for urinary Cu contents of a few hundred μg L−1. The methodology developed could prove to be useful for implementing screening protocols to detect Wilson's disease (WD), a well-known disorder related to Cu metabolism. In fact, the use of this analytical methodology permitted us to observe significant differences between (i) untreated WD patients and (ii) WD patients that are under treatment and control samples. This work represents the first time that determination of 65Cu/63Cu ratios has been used in the context of WD research, and serves as a proof of principle, suggesting that Cu isotope analysis could help in developing earlier and more reliable means to diagnose WD.
000 Hz) coupled to a multicollector (MC)-ICPMS to develop a method for the direct determination of Cu ratios in dried urine spots, prepared by deposition of urine (300 μL) onto precut clinical filter paper discs (16 mm diameter). The sampling capabilities offered by the fs LA system, permitting ablation of 150 μm thick coronas in the rim area of the filter, together with the use of admixed Ni as an internal standard, the proper optimization of the MC-ICPMS conditions (e.g., use of pseudo high-resolution mode to avoid interferences) and the use of a data processing approach particularly suitable for short transient signals (linear regression fit) enabled analysis of real urine samples with precision values around 500 ppm (RSD) for urinary Cu contents of a few hundred μg L−1. The methodology developed could prove to be useful for implementing screening protocols to detect Wilson's disease (WD), a well-known disorder related to Cu metabolism. In fact, the use of this analytical methodology permitted us to observe significant differences between (i) untreated WD patients and (ii) WD patients that are under treatment and control samples. This work represents the first time that determination of 65Cu/63Cu ratios has been used in the context of WD research, and serves as a proof of principle, suggesting that Cu isotope analysis could help in developing earlier and more reliable means to diagnose WD.
1. Introduction
Cu is an essential element for the proper functioning of several metaloenzymes with oxidase activity. In this way, Cu takes part in energy production (cytochrome C oxidase), iron metabolism (ceruloplasmin), antioxidant functions (superoxidase dismutase), connective tissue formation (lysyl oxidase), neurotransmitter metabolism (dopamine β-hydroxylase and monoamine oxidase), spermine oxidase, melanin formation (tyrosinase) and intermediate metabolism (uricase, benzylamine oxidase and diamine oxidase). Approximately 50% of dietary Cu is absorbed in the duodenum and proximal small intestine, and the process is facilitated by the complexing of Cu with amino acids. In plasma, approximately 75–90% of Cu is bound to ceruloplasmin, a ferroxidase whose function is essential for iron metabolism. The non-bound-ceruloplasmin plasma Cu, loosely bound to α-2 macroglobulin, albumin and amino acids (mainly histidine), participates in the interchangeable fraction of Cu between tissues.1The most prominent disorders related to Cu metabolism are the genetically determined Wilson's disease (WD), Menkes disease and occipital horn's syndrome (a mild form of Menkes disease).1 Although Menkes disease is usually fatal in the early infancy, WD can be treated if diagnosed before the onset of symptoms, when irreversible damage has not occurred yet. Being the most prevalent of the three (its incidence is approximately 1 in 30000), WD diagnosis is a field of extensive research.2
In short, WD is caused by mutations in the ATP7B gene, which encodes the ATP7B protein that is essential for Cu metabolism in hepatocytes. This protein facilitates the transfer of Cu ions to the main plasma Cu protein mentioned before, ceruloplasmin, and its release into the bloodstream, as well as the removal of excess Cu by secreting it into bile. Both functions of ATP7B are impaired in WD patients. As a consequence, Cu accumulates in the liver tissue, among other organs, while ceruloplasmin is still secreted but in a form that lacks Cu, named apoceruloplasmin, which is rapidly degraded in the bloodstream.3
From an analytical point of view, this means that patients of WD show low levels of both Cu and ceruloplasmin in blood and serum. Conversely, they show relatively higher contents of Cu in urine. This could open possibilities for diagnosis, but superimposed intervals have been observed between controls and affected patients, especially in young infants, due to the immaturity of the liver.3–5 There is still no totally reliable test to diagnose this disease and the golden standard is a liver biopsy.6,7 The lack of a near 100% sensible marker has prevented the implementation of a well-established neonatal screening protocol, in contrast to other genetic diseases that are less prevalent and less tractable. Still, investigation of potential screening procedures for WD diagnostic is a topic of great interest in the field of paediatrics.8,9
Instead of relying on elemental concentration, another possibility worth exploring is isotopic analysis. Taking into account that isotope fractionation typically accompanies (bio)chemical reactions,10 it could be expected that the protein ATP7B would interact with the two natural occurring isotopes of Cu (63Cu and 65Cu) with slightly different yields, which may be hard to measure in the case of well-functioning proteins operating with an efficiency close to 100%. However, the defective ATP7B protein found in WD patients shows a much lower efficiency for Cu incorporation into ceruloplasmin, as discussed before, and hence, the preferential presence of one of the two natural occurring Cu isotopes in biological fluids could become more apparent for WD patients with respect to people with low levels of Cu due to other reasons (for example, premature infants or adults following bariatric surgery procedures).11 In this case, the development of a proper methodology for the determination of Cu isotope ratios may open new possibilities for early detection of WD.
In this context, the development of multicollector inductively coupled plasma mass spectrometry (MC-ICPMS) has brought a new dimension to the field of isotopic analysis. This technique is characterized by lower requisites in terms of sample preparation when compared with more traditional techniques (e.g., thermal ionization mass spectrometry, TIMS), thus offering higher sample throughput, while its precision still rivals that of TIMS (down to 0.001% RSD in the best of cases), and it is significantly superior (a factor of 50–100) to that attainable by means of quadrupole-based ICPMS or sector field ICPMS.10 Moreover, the higher efficiency of the ICP to produce positively charged ions from nearly all elements in the periodic table has triggered the exploration of new isotopic systems, opening research possibilities in many new fields apart from those in which the use of isotopic analysis was already well-established (e.g., geochemistry and forensics). That includes the development of medical and biological applications,11–14 although the presence of this type of instrument in clinical labs is still limited, probably owing to its cost and the need for expert technicians.
In any case, for neonatal screening purposes, the methodology to be developed should profit from the screening programs for metabolic diseases already implemented throughout the world. Thus, to gain broad diffusion, the analytes must be assayed in the biological samples typically used, that is, blood and/or urine collected on standardized filter paper.8,9 The use of these clinical specimens offers many advantages, particularly in terms of logistics, such as the possibility to preserve samples at room temperature and to transport them over large distances by regular mail for analysis at referral centers, which makes them ideal for the application of screening schemes.15
It is the purpose of this work to investigate a method for direct isotopic analysis of Cu in urine samples adsorbed and dried on clinical filter papers (the so-called dried urine spots, DUS), in an attempt to establish potential differences for WD patients, which, to the best of our knowledge, have not been reported before. In order to achieve this goal, a MC-ICPMS coupled to a UV high-repetition-rate femtosecond laser ablation (fs-LA) system will be deployed. This coupling may combine the high precision typical of MC-ICPMS, clearly required to appreciate significant differences in this case, with the flexible sampling possibilities of this new UV fs-LA device, needed to obtain sufficient sensitivity. The selection of urine and not blood for this first study is based on the relatively less complex nature of the sample.
2. Experimental
2.1. Instrumentation
All measurements were carried out on a Nu Plasma HR-MC-ICPMS instrument (Nu instruments, Wrexham, UK) coupled to a Lambda 3 femtosecond laser ablation system (Nexeya SA, Canejan, France). This laser is fitted with a diode-pumped Yb:KGW crystal laser source (HP2, Amplitudes Systèmes, Pessac, France) delivering 360 fs pulses. Three wavelengths can be selected: 1030 nm (fundamental), 515 nm (2nd harmonic) and 257 nm (4th harmonic). The 257 nm wavelength was used in this study. Interestingly, the laser source operates within a wide range of repetition rate (1–100 kHz) and low energy (from 200 μJ per pulse below 1 kHz to 1.5 μJ per pulse at 100 kHz at this wavelength), which represents a new approach in analytical applications where high energy and low repetition rate are commonly used. The laser beam is focused with a 100 mm objective, and it can be rapidly moved (up to 2 m s−1) with high repositioning precision owing to a 2D galvanometric scanning module fitted to the optical line. The optical field covered by the laser beam is 16 × 16 mm2. Further details for a previous and similar model (operating in the IR region only) of this laser ablation system are described elsewhere.16,17The LA-ICPMS coupling was carried out using a 10 m long antistatic tube (PTFE electroconductive, Fisher Bioblock, Illkirch Cedex, France) of 6 mm external diameter and 4 mm internal diameter (Legris SA) into the ICP torch, using He as the carrier gas. A two-inlet torch was used to mix the laser-generated aerosol together with a liquid aerosol (nebulized by means of a pneumatic 200 μL min−1 micro-concentric nebulizer combined with a mini-cyclonic Cinnabar spray chamber) before introduction into the plasma.18,19 This dual-flow introduction system enables easy optimization of the MC-ICPMS by nebulizing a suitable solution for tuning. Furthermore, during laser ablation analyses, the plasma was kept under wet conditions by the continuous nebulization of a Ni standard solution (1 mg L−1 in 0.14 M HNO3), which is used to correct for mass bias.
Measuring conditions on both instruments were adjusted for maximum sensitivity and stability, plasma robustness and minimum influence of interferences on the analyte signals and are summarized in Table 1.
| Laser ablation conditions | |
|---|---|
| Laser ablation system | Lambda 3 |
| Wavelength | 257 nm |
| Pulse duration | 360 fs |
| Repetition rate | 10000 Hz |
| Spot diameter (airy 1/e2) | 8 μm |
| Energy/fluence | 9.5 μJ/19 J cm−2 |
| Scan speed (y axis) | 10 mm s−1 |
| Ablation strategy | Corona (150 μm thick), made with 20 concentric circles |
| Transport gas | He, 500 mL min−1 |
| Nu MC-ICPMS | |
|---|---|
| RF power | 1300 W |
| Instrument resolution | Pseudo-high resolution ∼5700 (edge resolved power) |
| Source slit width | 0.05 mm |
| Alpha 1 slit | 80 mA |
| Alpha 2 slit | 90 mA |
| Integration time | 5 s |
| Plasma gas flow rate | 13.0 L min−1 |
| Auxiliary gas flow rate | 0.80 L min−1 |
| Nebulizer pressure | 28.8 psi |
| Faraday cup configuration | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Collector | H7 | H6 | H5 | H4 | H3 | H2 | H1 | Ax | L1 | L2 | IC0 | L3 | IC1 | IC2 | L4 |
| m/z | 65 | 63 | 62 | 61 | |||||||||||
A graphite furnace atomic absorption spectrometer (GFAAS) Analytik Jena (Jena, Germany) ZEEnit 700 with Zeeman correction was used for determining the Cu levels in the urine samples.
A Sartorius (Goettingen, Germany) model BP211D analytical balance with a precision of 10−5 g was used for all weighing.
2.2. Samples and standards
Clincheck® Urine Control Level II, a reference material used to check the performance of the analytical method developed, was obtained from Recipe chemicals + Instruments GmbH (Munich, Germany). This control is provided as a lyophilized material, which, once reconstituted with 10 mL of ultrapure water, contains certified quantities of several elements, including Cu.
Whatman no. 903 paper cards (formerly Schleicher & Schuell) (Lot W-092) were obtained from Whatman International Ltd. (Maidstone, Kent, UK). This is one of the two commercial paper sources registered by the FDA as Class II medical devices.20 16 mm diameter discs were punched out of these totally empty cards (no inked) using a manual paper punch (Artemio, Wavre, Belgium).
For the preparation of the DUS samples, 300 μL aliquots of each of the urine samples were directly applied with a micropipette onto 16 mm diameter filter paper discs placed on disposable PS weighing boats. Samples were left to dry at room temperature for at least 4 h. Once dried, the paper discs containing the samples were kept in sealed plastic bags at room temperature until analysis.
2.3. Procedure for LA-MC-ICPMS analysis of the samples
The parameters used for the analysis of the samples are summarized in Table 1. The samples were conveniently fixed in the ablation cell using a small amount of double-sided tape attached to their bottoms. 5 replicate measurements (five coronas, see Section 3.2.) were carried out per sample. For every replicate measurement, a signal of approximately 100 second duration was obtained. A Ni solution was always simultaneously nebulized, such that Ni could be used as an internal standard for correction of mass discrimination. Note that the isotopic composition of this Ni solution was not certified, which rules out the absolute determination of 65Cu/63Cu ratios. This was beyond the scope of this study. However this approach allowed the comparison of the Cu isotope ratios for the different samples.In order to calculate Cu isotope ratios from these signals, a linear regression slope (LRS) method, where the signal intensities for 65Cu were plotted against the signal intensities for 63Cu, was used. The slope of the regression curve provides the raw 65Cu/63Cu ratio, which is later corrected for mass bias applying Russell's exponential law.21 More details on this procedure are provided elsewhere.22–24
3. Results and discussion
3.1. Considerations on sensitivity and sample deposition
One of the main challenges of the topic evaluated in this work is the relatively low analyte content found in the urine of the patients subject to investigation. The actual Cu levels were determined prior to LA-MC-ICPMS analysis by means of the standard approach (using graphite furnace atomic absorption spectrometry25). The level of copper for WD patients that were following treatment with penicillamine (a chelating agent) was significantly higher, ranging from 128 to 699 μg L−1, as could be expected because addition of this chelating agent helps in freeing Cu ions from the organs in which they are retained when suffering from WD (e.g., liver).6,7,26 However, the values for WD patients that were not yet following any treatment ranged between 34 and 73 μg L−1, and for healthy persons similar or lower values are expected.Although the MC-ICPMS deployed in this work is fitted with two ion counters, it is necessary to stress that its detector configuration does not permit to use them for the monitoring at m/z 63 and 65 simultaneously. Faraday cups have to be used instead, with the subsequent loss in sensitivity. Moreover, at these low levels, the contribution of interfering species may be significant, and, in particular, the contribution of 40Ar23Na+ to the Cu signal at a mass-to-charge (m/z) of 63 should be monitored, as NaCl is present in urine at the 1000 mg L−1 level.12
In order to minimize these problems, one of the key aspects may be the way in which urine is deposited onto the clinical filter paper, giving rise to the dried urine spot (DUS) sample. If urine is simply deposited on a relatively large filter paper card, as it is typically done for other biological samples such as blood, urine will spread over a significantly larger area than blood. As a result, the amount of immobilized sample per area unit will be lower, seriously degrading the sensitivity attainable by means of LA-ICPMS. In a recent paper aiming at multielemental analysis of DUS samples carried out in our lab, this issue was explored in detail and it was concluded that the best way to cope with it was to deposit the urine sample onto small precut paper pieces, thus limiting the area where the urine is allowed to expand.27 Therefore, in this work, Whatman no. 903 paper cards were precut in paper discs of 16 mm and 300 μL of urine was deposited on each of them and left to dry. These amounts correspond to the maximum urine volume that the sample's surface tension retains on the paper discs, thus providing the highest possible sample concentration in the DUS specimens after a single drying cycle (≈150 μL urine per cm2) which is about 10–12 times higher than the concentration obtained when urine is deposited on big-sized paper cards.
In addition to providing improved sensitivity, this solution is simple to follow even in an unsupervised context (e.g., any patient could do it by him/herself using a disposable Pasteur pipette), it is straightforward in comparison with other approaches (e.g., deposition and drying of urine multiple times) and prevents chromatographic effects from occurring, as the urine sample is not allowed to freely migrate on the cellulose support. It was demonstrated in the above-mentioned work27 that the distribution of metals, including Cu, is more homogenous using the approach described before, even though there is still a significantly higher concentration (approx. a factor of 2) in the rim of the precut filter. In fact, this aspect could be used in our favor in the current work, as long as no spatial isotopic fractionation occurs on the paper surface.
Finally, it is important to mention that the Whatman no. 903 paper cards used in this work were blank cards containing no ink at all. It is often the case that the specimens used in the hospitals are inked to indicate the areas of preferred deposition. In our experience, this ink may contain some Cu impurities,27 so it is preferable to use blank cards whenever possible.
3.2. Laser ablation-MC-ICPMS monitoring of the samples and potential interferences
Based on the Cu concentration previously detected in our samples, the absolute mass of copper deposited on filters was ranging within 38 ng to 210 ng (i.e. from 0.19 ng mm−2 to 1.05 ng mm−2) for WD patients treated with penicillamine, and from 10 ng to 22 ng (i.e. from 0.05 ng mm−2 to 0.11 ng mm−2) for untreated WD patients. Due to the low signal sensitivity of the MC-ICPMS fitted with Faraday cups, such low concentrations result in poor statistics, limiting precision and accuracy on the 65/63Cu isotope ratio. Thus, an appropriate ablation strategy must be used to achieve sufficiently high signal intensities. Use of a femtosecond LA unit may be of great importance for this application. This type of laser provides well-known advantages for isotopic analysis, as its shorter pulse duration has been shown to drastically reduce thermal effects and minimize isotope fractionation and matrix dependencies in comparison to laser pulses in the nanosecond range.28,29 Moreover, use of femtosecond laser ablation typically enhances signal intensity and stability, limiting the production of signal spikes due to larger particles, eventually leading to improvements in precision and accuracy compared to nanosecond laser ablation.30,31There are different types of fs-LA units in the market32–34 but for this particular application perhaps the one that has been described by Pécheyran et al. in a number of works16,17,19 is the best suited. Instead of using high energies (>1 mJ per pulse) and low repetition rates (1–20 Hz), as most common devices, this unit is based on the use of a relatively low-energy laser unit (up to 200 μJ per pulse at 257 nm). Thus, in order to keep a sufficiently high fluence, the laser spot used must be small (around 10 μm). However, this drawback can be compensated by combining a fast beam scanning system with the high repetition rate of the laser (up to 100000 Hz). In other words, the small craters corresponding to the laser beam spot size (8 μm in our case) are repeated in such a way that they can be overlapped, while the beam and the sample are continuously displaced at constant rates. This strategy permits ablation of large areas in a reduced time, resulting in short transient signals, which dramatically enhance the signal-to-background ratios. This approach has proven to be satisfactory in increasing the sensitivity in relatively similar applications to the one studied in this work (e.g., analysis of metals in gels19). Moreover, this unit permits high flexibility regarding the sampling of the urine filter because any desired geometrical form can be used in practice (lines, craters, coronas, etc.).
The laser used in the current work represents an upgrade from the device described in those previous works,16,17,19,35,36 as it operates in the UV region and not in the IR region. This, in principle, should be beneficial to further reduce fractionation28 but its fundamental operating idea is the same. Thus, ablation of the DUS samples was evaluated using different geometries, as shown in Fig. 1A.
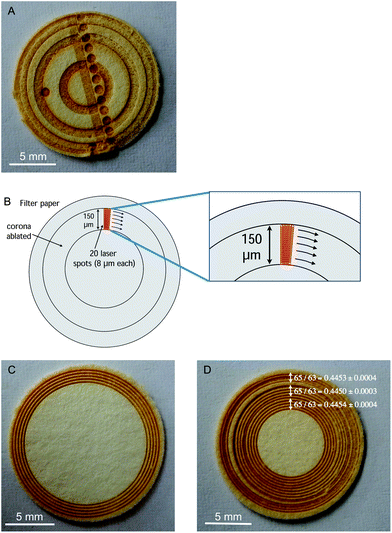 | ||
| Fig. 1 (A) Example of DUS sample ablated using different sampling strategies (craters, coronas, rasters, etc.); (B) strategy for ablation of a 150 μm thick corona by means of the fast translation of a small laser spot (8 μm). For construction of the corona, 20 concentric circular lines were ablated; (C) example of DUS sample ablated at five different locations (5 replicates) using the corona sampling approach; (D) example of DUS sample ablated at 15 locations using the corona sampling approach, showing the spatially resolved 65Cu/63Cu ratios obtained. Uncertainties (error bars) are expressed as 2SD on the average value obtained for five different replicates. | ||
From these preliminary optimizations, different conclusions could be drawn: (a) ablation of the areas close to the rim offered the best sensitivity (a factor of two compared with the central area), as expected; (b) the ratios obtained deviated significantly from the expected ratios (natural ratio for Cu,37 approx. 0.44613 for 65Cu/63Cu), and they are consistently biased low, contrary to what can be expected if instrumental mass bias would be the dominant factor to explain these inaccuracies. There are indications of matrix-effects on the plasma (depletion of the Ni signal when the material ablated arrived to the plasma), which could contribute to some extent to the effect observed; however, the influence of 40Ar23Na+ on 63Cu+ is probably very significant for this sample, which explains the results achieved; (c) carrying out very prolonged ablation was not recommended, as the signal drops if the ablation reaches the bottom of the filter paper.
Aspects (a) and (c) can be easily addressed owing to the potential of the LA unit for spatially resolved analysis. In the end, it was decided to ablate relatively small coronas 150 μm thick (difference between inner and outer radius), which are constructed by fast rotation of an 8 μm spot (using 20 concentric lines per corona, as illustrated in Fig. 1B), under the conditions shown in Table 1. This ablation strategy allowed ablation of approx. 7 mm2 in about 100 s. The depth of the coronas varies with the radius (the shorter the radius the deeper the crater to ablate the same amount) but it is always in the range of 100–150 μm, while the thickness of the paper is about 400–500 μm. Thus, the entire depth of the filter was never completely ablated to achieve the best sensitivity and to prevent contamination from the double-sided tape. Fig. 1C shows a DUS specimen after ablation of 5 of these coronas.
As for issue (b), in order to compensate for both instrumental as well as for matrix-dependent non-spectral mass discrimination, use of Ni as an internal standard (IS) was evaluated.21,38 In order to do so, the simultaneous aspiration of a Ni solution that reaches the plasma together with the laser aerosol is required. This strategy results in a wet plasma, which is in principle more robust than a dry one.39–41 Selection of Ni instead of Zn, another element often selected as an IS for Cu isotope analysis by MC-ICPMS,42–44 was preferred as the Ni ionization potential is more similar to that of Cu, and Ni is also less prone to suffer from contamination issues than Zn.12,45
On the other hand, the MC-ICPMS instrument used in this work offers the possibility to enhance the spectral resolution by using a high-resolution entrance slit (narrower) while still using a low resolution detector slit. Therefore, the peaks from the interference and the analyte are not totally resolved, as keeping a flat top peak shape is required to maintain the best isotopic precision,46 but there is a wide window wherein only the analyte ion contributes to the signal intensity and where the signal is still flat. This pseudo high-resolution mode of operation is well-known10,38,46 and has been used successfully before for Cu isotope analysis.47 In the current case, the optimum position for measuring was evaluated before every sample measurement, and the 63Cu+ signal was measured approximately 0.053 m/z away (drifted to a lower mass) from the central position that would have been selected in the absence of any potential overlap.
The success of these correction strategies can be evaluated from the results presented in Table 2. For this purpose, several measurements were carried out either by aspirating a 40 μg L−1 Cu solution while a blank filter paper was being ablated, or by ablating a Cu solution that was deposited onto the filter paper and dried (see Section 2.2.2.), and prepared in such a way that a similar Cu signal was obtained in both cases. Moreover, additional experiments in which a 2500 mg L−1 NaCl solution was also deposited onto a filter paper previously spiked with a Cu solution were also performed.
| Cu introduction | Mass resolution | No NaCl | With NaCl | Cu “natural” ratio37 | |
|---|---|---|---|---|---|
| Uncorrected measurement | Ni as IS | Ni as IS | |||
| Solution | Low resolution | 0.46950 ± 0.00005 | 0.44630 ± 0.00010 | 0.42775 ± 0.00180 | 0.44613 |
| Solution | Pseudo high res | 0.46935 ± 0.00015 | 0.44655 ± 0.00014 | 0.44448 ± 0.00048 | 0.44613 |
| Deposition on filter | Pseudo high res | 0.46880 ± 0.00024 | 0.44606 ± 0.00021 | 0.44499 ± 0.00044 | 0.44613 |
As can be appreciated, when working at low mass resolution, ablation of a blank filter does not seem to represent any significant negative effect. There were no indications of matrix effects (the signal for the Ni spike was very constant whether the filter paper was simultaneously ablated or not), the value for the Cu solution, after correction for instrumental mass bias, is close to the expected natural ratio, and the precision is rather good (approx. 100 ppm RSD for a signal of 63Cu approx. 1 V). However, if the filter ablated contains NaCl the situation is very different. Clear indications of matrix effects can be observed on the Ni signal intensity, precision deteriorates by a factor of 20 and a bias of more than 4% appears. This problem can be minimized working in pseudo high-resolution, as described before. This working mode represents a significant decrease in sensitivity (approx. 10-fold), which typically results in a lower precision in the absence of the interfering species but, when NaCl is present, the bias observed significantly decreases and the precision is 4–5 times better. Similar results are obtained when Cu is not introduced from the solution but rather ablated from the filter paper, thus opening possibilities for analysis of real samples.
Therefore, this approach of ablating coronas and monitoring 63,65Cu off-peak was used in further work.
3.3. Calculating the isotope ratio when dealing with short transient signals
The results obtained so far were considered as promising but there is still a margin for further improvement depending on the way in which transient signals are treated.Fig. 2A shows a typical signal obtained when a DUS sample is ablated following the corona shape strategy. 5 different replicates are obtained. As can be seen, the signal is relatively stable (it decreases a bit when ablating inner areas, as expected) and the introduction of a matrix into the plasma is accompanied by signal suppression, as clearly indicated by the drop in the Ni signal intensity value.
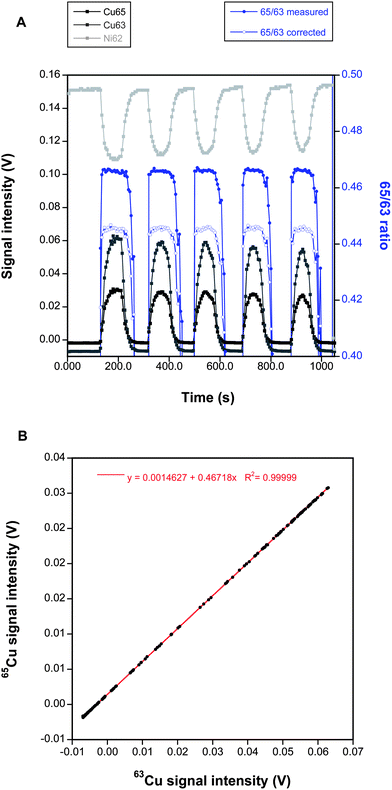 | ||
| Fig. 2 (A) Example of a time-resolved signal obtained when ablating a DUS sample (16 mm diameter) at five different locations (starting from the rim) using the corona sampling approach. The uncorrected and corrected (using Ni as an internal standard) 65Cu/63Cu ratios calculated for every measurement available are also plotted against the right y-axis. (B) 65Cu vs.63Cu plot for the same raw measurements shown in (A). | ||
Now, the traditional way to process this data would be to use a point-by-point method where the 65/63 ratio for every measurement (1 value every 5 seconds) is calculated using the signal intensities (blank-corrected) for 65Cu and 63Cu. The 62/61 ratio can be calculated also in the same way, and Russell's exponential law for mass bias correction can be finally applied to obtain the corrected 65/63 ratio for every data point. This is illustrated in Fig. 2A where all 65/63 values are plotted. Obviously, the approach seems to work to some extent as the corrected ratio provides a value much closer to the natural one than the uncorrected measure. Lastly, the final 65/63 ratio can be estimated as the average obtained for the duration of every transient signal, which can be calculated after setting the limits where every measurement is to be integrated. Normally, the stable flat top portion of every 65/63 signal replicate is integrated, although there is always some uncertainty as to how to set these limits, which may affect the result obtained.
However, an alternative approach was proposed by Fietzke et al.,22,23 specifically designed for the short transient signals typically obtained by means of LA-MC-ICPMS. The idea is to plot the signal intensities for 65Cu against the signal intensity for 63Cu, and the slope of the regression curve directly provides the raw 65Cu/63Cu ratio, which can be later mass bias corrected applying Russell's exponential law using the average value of the 62Ni/61Ni ratio measured during the same period. This simple approach provides significant advantages in this situation as it uses all the data, avoiding any subjective influence that may occur by setting the integration limits. But perhaps its main advantage is that each data point contributes to the fit depending on its signal intensity (high intensity values will influence more the final slope than low intensity values), while in the classical approach all points contribute to the same extent (unless they are excluded). Fig. 2B shows the application of this approach to the same data points used for Fig. 2A. As can be seen, a remarkably good linear fit is attained, and the slope provides the uncorrected 65Cu/63Cu ratio. In practice, in order to obtain an estimation of the imprecision, the signals obtained for ablation of every corona were processed separately in the current work, thus obtaining five replicates per sample.
In order to establish which processing procedure would provide the best results, a urine sample from a WD patient was deposited on 5 different filter paper discs and LA-MC-ICPMS analysis was carried out on different sessions, using both approaches for data calculation. The results are shown in Fig. 3. The internal precision39 obtained by means of both approaches is similar (RSD values ranging between 200 and 500 ppm for 5 replicates), but the external precision obtained by means of the linear regression approach (LRS) is a factor of 5 better (RSD 540 vs. 2480 ppm), as the values obtained by means of the point-by-point (PBP) approach deviate more between sessions. This difference is statistically significant at the 95% confidence level (Fvalue = 20.810; Fcritical = 6.388). Results obtained by means of the LRS method are thus more consistent, and this approach was selected for analysis of the samples. These results also serve to provide an idea of the reproducibility of the method for real urine sample analysis.
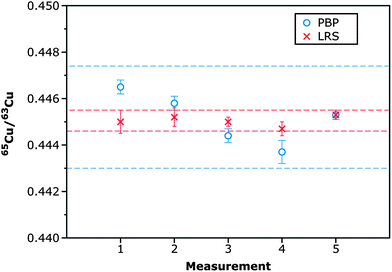 | ||
| Fig. 3 65Cu/63Cu ratios corrected for mass discrimination using Ni as an internal standard obtained after analysis of five different DUS specimens in which the same urine sample was deposited, following the procedure described in Section 2.2.2. The individual values obtained were processed according to either a point-by-point (PBP) approach or a linear regression slope (LRS) method. Internal precision values (error bars) are expressed as 2SD on the average value obtained for the five different replicates (n = 5) carried out for every sample. External precision values (dotted lines) for every processing approach are expressed as 2SD on the average value obtained for the five samples analyzed. | ||
Finally, the possible influence of the sampling position on the 65Cu/63Cu ratio obtained was also investigated. As can be seen in Fig. 1D, there does not seem to be any significant difference. Thus, ablating coronas on the edge of the filter is preferred in order to attain the best possible sensitivity.
3.4. Analysis of the samples
The results obtained in the previous section seem promising enough to attempt analysis of the real samples. It can be stated that even though we have been reporting isotope ratios so far, our goal was not to report absolute values (a certified isotope reference material for Ni should be used otherwise) but rather to establish if there is any significant difference between WD patients and healthy persons. Thus, the results are expressed as delta values, i.e. as a relative difference (in per mil) versus a reference. There is no information on this point in the literature and thus, there is no idea on what precision level should be needed for the task. Analysis of the samples was undertaken, as described in Section 2.3.The results obtained are shown in Fig. 4. Samples analyzed can be divided into three categories: (i) the ones selected as controls (“Normal” samples) corresponding to a urine reference sample (Clincheck® Level II), and a sample from a patient who did not suffer from WD but had a Cu level (44 μg L−1) similar to that found in WD patients before any treatment. Therefore, the ratio experimentally obtained for this last sample was selected as a reference to calculate deltas for the rest of the samples (it thus represents the zero delta value); (ii) samples of people suffering from WD that are following a treatment with penicillamine (labelled as under treatment); and (iii) samples of people suffering from WD that were not yet following any treatment, as the same samples analyzed in this work were collected at the time of diagnosis.
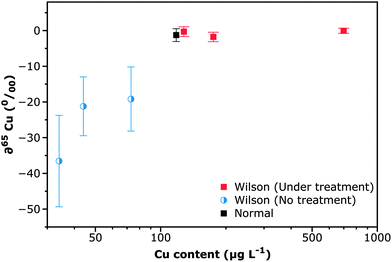 | ||
| Fig. 4 Concentration vs. isotopic information plot for the urine samples of the different groups of individuals considered in the investigation. Isotopic values are expressed as deltas, i.e. as a relative difference (in per mil) versus a reference sample (urine from a person who does not suffer from WD but shows a similar Cu level to that found in untreated WD patients, 44 μg L−1). Analysis was carried out as described in Section 2.3. Uncertainties (error bars) are expressed as 2SD on the average value obtained for the five different replicates (n = 5) carried out for every sample. The Cu level in urine samples was determined by GFAAS analysis. As a typical normal sample, a Clincheck Level II reference urine sample is shown. WD patients following treatment were those receiving penicillamine. | ||
The so-called normal samples tend to show a value close to the natural abundance, as expected. Interestingly, those samples of people with WD that are following a treatment with penicillamine also show a very similar ratio, leading to low delta values (−0.06 to −1.8‰) that overlap with zero when considering uncertainties. This could be explained as addition of this chelating agent is intended to free copper ions from the organs in which they are retained. Thus, these patients are practically excreting all Cu that they ingest through urine (their Cu levels in urine are therefore higher), and therefore there is no indication of fractionation in the Cu ratio. However, those WD patients that are not following any treatment yet show a significantly 65/63 lower ratio, leading to delta values in the −20 to −35‰ range, indicating that there is fractionation between the Cu that is metabolized and naturally excreted through urine, and the Cu that is ingested, as a consequence of this disease. It can be stressed that Cu isotopic analysis permits us to clearly separate these last samples from the one used as a reference (delta = 0), while if only elemental analysis is carried out that reference (Cu 44 μg L−1) would be classified as potentially coming from a WD patient as well.
Hence, even though the number of samples available for investigation was small, it is the author's opinion that this work serves as a proof of principle confirming that isotope analysis may help in differentiating WD patients, and, eventually, may be a potent tool for early diagnosis of this disease.
It can also be noticed from the figure that the uncertainty of the measured ratios varies with the Cu content, reaching RSD values up to 2000–6000 ppm for samples of WD patients that were not treated yet. This is a direct consequence of the low signal intensity obtained for these samples, which was as low as 10 mV only. Note that when considering the larger corona (15–14.7 mm diameter) for these WD samples, the amount of copper introduced into the plasma during the 100 s ablation was lower than 250 pg. Here, for such low signals the LRS data reduction is particularly efficient compared to the conventional “point-by-point” method,22 improving precision at a signal level close to the LOD, as demonstrated by Epov et al.24
Thus, future efforts should be directed to develop methodologies to further improve sensitivity (e.g., using a MC-ICPMS device permitting us to achieve sufficient high-resolution to avoid 63Cu and 40Ar23Na overlap at a lower sensitivity cost38).
4. Conclusions
The current work explores the potential of an approach based on: (a) deposition of urine (300 μL) onto precut (16 mm) clinical filter papers and drying; and (b) direct Cu isotopic analysis of the resulting samples by laser ablation, using a 257 nm femtosecond device operating at a high repetition rate (10000 Hz), coupled to MC-ICPMS, for the establishment of a possible pattern to detect WD patients.
The results obtained indicate that, after proper optimization of (i) the sampling strategy (ablation of small coronas in the rim area of the filter), (ii) the MC-ICPMS conditions (working in pseudo high-resolution mode to avoid spectral overlap) and (iii) the way to process the resulting transient signal (use of a linear regression approach), it was possible to observe differences in the measured 65Cu/63Cu ratio for WD patients that were yet untreated (65/63 ratio biased low), in comparison with those that were under treatment with a chelating agent, which approximately showed the natural abundance value, as found for control samples.
These results hint that this procedure could be the basis of a non-invasive method for screening of WD, only requiring a few hundreds of μL of urine from the patient, while permitting easy storage and transportation of specimens to a referral center.
Acknowledgements
This work has been funded by the Spanish Ministry of Science and Innovation (Project CTQ2009-08606) and the Government of Aragón (Departamento de Ciencia, Tecnología y Universidad del Gobierno de Aragón y Fondo Social Europeo).Notes and references
- H. Kodama and C. Fujisawa, Metallomics, 2009, 1, 42–52 RSC.
- M. L. Schilsky, Biochimie, 2009, 91, 1278–1281 CrossRef CAS.
- J. D. Gitlin, Gastroenterology, 2003, 125, 1868–1877 CrossRef.
- K. H. Schulpis, T. Karakonstantakis, S. Gavrili, C. Costalos, E. Roma and I. Papassotiriou, Clin. Chem., 2004, 50, 1253–1256 CAS.
- A. M. Sutton, A. Harvie, F. Cockburn, J. Farquharson and R. W. Logan, Arch. Dis. Child., 1985, 60, 644–651 CrossRef CAS.
- European Association for the study of the liver, J. Hepatol., 2012, 56, 671–685 Search PubMed.
- E. A. Roberts and M. L. Schilsky, Hepatology, 2008, 47, 2089–2111 CrossRef CAS.
- W. A. Gahl, Clin. Chem., 2008, 54, 1941–1942 CAS.
- C. A. Kroll, M. J. Ferber, B. D. Dawson, R. M. Jacobson, K. A. Mensink, F. Lorey, J. Sherwin, G. Cunningham, P. Rinaldo, D. Matern and S. H. Hahn, Mol. Genet. Metab., 2006, 89, 134–138 CrossRef CAS.
- Isotopic Analysis: Fundamentals and Applications Using ICP-MS, ed. F. Vanhaecke and P. Degryse, Wiley-VCH, Weinheim, 2012 Search PubMed.
- F. Albarède, P. Telouk, A. Lamboux, K. Jaouen and V. Balter, Metallomics, 2011, 3, 926–933 RSC.
- F. Larner, M. Rehkämper, B. J. Coles, K. Kreissig, D. J. Weiss, B. Sampson, C. Unsworth and S. Strekopytov, J. Anal. At. Spectrom., 2011, 26, 1627–1632 RSC.
- L. Van Heghe, E. Engström, I. Rodushkin, C. Cloquet and F. Vanhaecke, J. Anal. At. Spectrom., 2012, 27, 1327–1334 RSC.
- K. Hotz, H. Augsburger and T. Walczyk, J. Anal. At. Spectrom., 2011, 26, 1347–1353 RSC.
- J. C. Rockett, G. M. Buck, C. D. Lynch and S. D. Perreault, Environ. Health Perspect., 2004, 112, 94–104 CrossRef.
- E. Ricard, C. Pécheyran, G. Sanabria Ortega, A. Prinzhofer and O. F. X. Donard, Anal. Bioanal. Chem., 2011, 399, 2153–2165 CrossRef CAS.
- C. Pécheyran, S. Cany, P. Chabassier, E. Mottay and O. F. X. Donard, J. Phys.: Conf. Ser., 2007, 59, 112–117 CrossRef.
- G. Ballihaut, L. Tastet, C. Pécheyran, B. Bouyssiere, O. Donard, R. Grimaud and R. Lobinski, J. Anal. At. Spectrom., 2005, 20, 493–499 RSC.
- G. Ballihaut, F. Claverie, C. Pécheyran, S. Mounicou, R. Grimaud and R. Lobinski, Anal. Chem., 2007, 79, 6874–6880 CrossRef CAS.
- J. V. Mei, S. D. Zobel, E. M. Hall, V. R. de Jesús, B. W. Adam and W. H. Hannon, Bioanalysis, 2010, 2, 1397–1403 CrossRef CAS.
- F. Vanhaecke, L. Balcaen and D. Malinovsky, J. Anal. At. Spectrom., 2009, 24, 863–886 RSC.
- J. Fietzke, V. Liebetrau, D. Günther, K. Gürs, K. Hametner, K. Zumholz, T. H. Hansteen and A. Eisenhauer, J. Anal. At. Spectrom., 2008, 23, 955–961 RSC.
- J. Fietzke, M. Frische, T. H. Hansteen and A. Eisenhauer, J. Anal. At. Spectrom., 2008, 23, 769–772 RSC.
- V. N. Epov, S. Berail, M. Jimenez-Moreno, V. Perrot, C. Pecheyran, D. Amouroux and O. F. X. Donard, Anal. Chem., 2010, 82, 5652–5662 CrossRef CAS.
- A. A. Almeida and J. L. F. C. Lima, At. Spectrosc., 2001, 22, 324–330 CAS.
- M. Van Caillie-Bertrand, H. J. Degenhart, I. Luijendijk, J. Bouquet and M. Sinaasappel, Arch. Dis. Child., 1985, 60, 652–655 CrossRef CAS.
- M. Aramendía, L. Rello, F. Vanhaecke and M. Resano, Anal. Chem., 2012, 84, 8682–8690 CrossRef.
- I. Horn and F. von Blanckenburg, Spectrochim. Acta, Part B, 2007, 62, 410–422 CrossRef.
- J. J. Gonzalez, D. Oropeza, X. Mao and R. E. Russo, J. Anal. At. Spectrom., 2008, 23, 229–234 RSC.
- K. Ikehata, K. Notsu and T. Hirata, J. Anal. At. Spectrom., 2008, 23, 1003–1008 RSC.
- J. González, S. H. Dundas, C. Yi Liu, X. Mao and R. E. Russo, J. Anal. At. Spectrom., 2006, 21, 778–784 RSC.
- J. Koch and D. Günther, Anal. Bioanal. Chem., 2007, 387, 149–153 CrossRef CAS.
- F. Vanhaecke, M. Resano, J. Koch, K. McIntosh and D. Günther, J. Anal. At. Spectrom., 2010, 25, 1259–1267 RSC.
- J. Koch and D. Günther, Appl. Spectrosc., 2011, 65, 155A–162A CrossRef CAS.
- F. Claverie, B. Fernández, C. Pécheyran, J. Alexis and O. F. X. Donard, J. Anal. At. Spectrom., 2009, 24, 891–902 RSC.
- B. Fernández, F. Claverie, C. Pécheyran and O. F. X. Donard, Trends Anal. Chem., 2007, 26, 951–966 CrossRef.
- M. Berglund and M. E. Wieser, Pure Appl. Chem., 2011, 83, 397–410 CrossRef CAS.
- L. Yang, Mass Spectrom. Rev., 2009, 28, 990–1011 CrossRef CAS.
- M. Aramendía, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2010, 25, 390–404 RSC.
- M. Resano, M. P. Marzo, R. Alloza, C. Saénz, F. Vanhaecke, L. Yang, S. Willie and R. E. Sturgeon, Anal. Chim. Acta, 2010, 677, 55–63 CrossRef CAS.
- C. O'Connor, B. L. Sharp and P. Evans, J. Anal. At. Spectrom., 2006, 21, 556–565 RSC.
- C. N. Maréchal, P. Télouk and F. Albarède, Chem. Geol., 1999, 156, 251–273 CrossRef.
- S. Graham, N. Pearson, S. Jackson, W. Griffin and S. Y. O'Reilly, Chem. Geol., 2004, 207, 147–169 CrossRef CAS.
- S. E. Jackson and D. Günther, J. Anal. At. Spectrom., 2003, 18, 205–212 RSC.
- W. Li, S. E. Jackson, N. J. Pearson, O. Alard and B. W. Chappell, Chem. Geol., 2009, 258, 38–49 CrossRef CAS.
- F. Vanhaecke and L. Moens, Anal. Bioanal. Chem., 2004, 378, 232–240 CrossRef CAS.
- G. H. Fontaine, K. Hametner, A. Peretti and D. Günther, Anal. Bioanal. Chem., 2010, 398, 2915–2928 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |