 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule imaging in vivo: the dancing building blocks of the cell
Miguel
Coelho
,
Nicola
Maghelli
and
Iva M.
Tolić-Nørrelykke
*
Max Planck Institute of Molecular Cell Biology and Genetics, Pfotenhauerstrasse 108, 01307 Dresden, Germany. E-mail: tolic@mpi-cbg.de; Tel: +49 351 210 2691
First published on 22nd February 2013
Abstract
A cell can be viewed as a dynamic puzzle, where single pieces shuffle in space, change their conformation to fit different partners, and new pieces are generated while old ones are destroyed. Microscopy has become capable of directly observing the pieces of the puzzle, which are single molecules. Single-molecule microscopy in vivo provides new insights into the molecular processes underlying the physiology of a cell, allowing not only for visualizing how molecules distribute with nanometer resolution in the cellular environment, but also for characterizing their movement with high temporal precision. This approach reveals molecular behaviors normally invisible in ensemble measurements. Depending on the molecule, the process, and the cellular region studied, single molecules can be followed by conventional epifluorescence microscopy, or by illuminating only a thin region of the cell, as in Total Internal Reflection Fluorescence (TIRF) and Selective Plane Illumination Microscopy (SPIM), and by limiting the amount of detectable molecules, as in Fluorescence Speckle Microscopy (FSM) and Photo-Activation (PA). High spatial resolution can be obtained by imaging only a fraction of the molecules at a time, as in Photo-Activated Localization Microscopy (PALM) and Stochastic Optical Reconstruction Microscopy (STORM), or by de-exciting molecules in the periphery of the detection region as in Stimulated Emission-Depletion (STED) microscopy. Single-molecule techniques in vivo are becoming widespread; however, it is important to choose the most suited technique for each biological question or sample. Here we review single-molecule microscopy techniques, describe their basic principles, advantages for in vivo application, and discuss the lessons that can be learned from live single-molecule imaging.
Insight, innovation, integrationSingle molecule imaging in vivo provides new insights into the molecular processes in cells. This approach is important not only to map the distribution of single molecules inside the cell with high spatial resolution, but also to analyze the movement of these molecules and their interactions over time. Single molecule imaging in live cells reveals transient molecular interactions that cannot be identified by conventional biochemical methods. Moreover, single molecule imaging allows for direct measurements of parameters of molecular reactions, including the number of molecules, concentrations, reaction rate constants and diffusion coefficients. These parameters can be used in building mathematical models of intracellular processes. |
1. Introduction
The biology of the cell is a mixture of complex physico-chemical processes, a product of interactions between individual molecules. Inside the cell, single molecules travel through different compartments, interact and react, giving rise to higher-order macromolecular behaviors, which are vital for maintaining the cell architecture or responding to extracellular signals. Single-molecule microscopy is important not only to map the distribution of single molecules inside the cell with high spatial resolution, but also to analyze the movement of these molecules and their interactions over time. While molecular interactions have been extensively explored using biochemistry, single-molecule observations provide new information on their spatio-temporal dynamics, revealing transient molecular interactions that cannot be identified by conventional biochemical techniques.1 Moreover, single-molecule imaging allows for direct measurements of kinetic and dynamic parameters of molecular reactions, including the number of molecules, concentrations, reaction rate constants and diffusion coefficients.2While single-molecule imaging in vitro allows for studies of single molecules in controlled environments, this approach requires special substrates or imaging chambers, which are different from the cellular environment. Because the conditions reproduced in vitro differ from those inside the cell, cell-based approaches are required for understanding the localization and interactions of single molecules in vivo. To gain insight into the intracellular localization, fixation methods can be used to map the distribution of single molecules in the cell. However, observations on fixed cells do not yield information about the temporal dynamics of molecules, and artifacts may be induced by the fixation techniques. Contrary to imaging in vitro or in fixed cells, single-molecule imaging in vivo combines high spatial and temporal resolution to address the behavior of single molecules in their native environment.
In this review we focus on methods to label, visualize and follow single molecules in vivo. A chronological overview of the developments of single-molecule techniques and their applications in vivo is depicted in Fig. 1.
 | ||
| Fig. 1 Timeline of single-molecule microscopy developments and their initial in vivo applications. In red, on the left side of the timeline, important technological discoveries that allowed for studying single molecules in vivo are shown. The use of naturally occurring fluorescent proteins, like the green fluorescent protein (GFP) from Aequorea victoria46 to label proteins endogenously, provided a major breakthrough in single-molecule labeling in vivo. Fluorescent molecules were used for the first time in vitro in 1976 to label and detect a protein, γ-globulin, as it diffused through a thin layer of illumination.47 Nanoparticle tracking was used to monitor microtubule dependent cell movement in 1985.14a In 1988, in order to describe how enzymes convert energy into mechanical work, kinesins, force generating ATPases, attached to plastic beads, were tracked along microtubules in vitro with a 1–2 nm precision.14b In 1993, the technique of orthogonal-plane fluorescence optical sectioning (ORFOS), based on the original idea published in 1903 by Siedentopf & Zsigmondy, was put into practice using a cylindrical lens.48 Later in 2004, this concept developed into selective plane illumination microscopy (SPIM) used in the same year to visualize living samples.24 In 1997, fluorescent speckle microscopy (FSM) allowed for mixing fluorescent and non-fluorescent molecules, reducing the fluorescence background.26 Stimulated emission-depletion microscopy (STED), developed by Hell and collaborators in 2000, was able to achieve resolutions below the Abbe diffraction limit.37 In 2002, a variant version of GFP was engineered to be excitable only after photo-activation, so that only a small fraction of GFP molecules is made visible.31 The precise mapping of molecules inside the cell can be obtained by repeating this photo-activation and imaging procedure, as was shown in PALM and STORM.33 In blue, on the right side of the timeline, we show the in vivo breakthroughs and applications of single-molecule microscopy techniques. A description of these studies can be found in Table 1. | ||
2. Labeling: minimum invasion and high precision
The first step in studying single molecules is detection, using a specific marker attached to the molecule. Depending on the molecular species, its intracellular localization, and the process of interest, different types of probes and detection methods can be used. One of the most widely used detection techniques is fluorescence microscopy.Fluorescent labeling of molecules can be achieved by coupling a fluorescent dye to the target molecule using covalent chemistry or antibody affinity. This approach is commonly used for imaging of cultured cells, by microinjecting fluorescently labeled molecules into live cells or by using antibodies on fixed cells. Fluorescent markers should interfere minimally with the biological function of the target molecule. Alternatively, genetic manipulations can be used to express a modified copy of the molecule of interest fused to a fluorescent protein. The expression of the fused molecule can be either transient, as in the case of transfection with plasmids that do not get integrated into the genome, or stable, when the manipulation alters permanently the genome of the cell and of its daughter cells. Such genetic manipulations are used for live imaging of cells, tissues and even whole animals. Transient transfection is easier to perform, but the level of expression varies among cells and the implementation becomes more challenging with an increasing complexity of the sample, moving from cells to tissues and animals. Stable transfection, in contrast, can be used irrespective of the complexity of the sample and offers a more homogeneous level of expression among cells, but the creation of stably transfected cell lines and especially organisms may require a longer procedure due to, for example, a low rate of recombination of the modified gene into the genome.
Genetic labeling is highly specific, as only the labeled proteins are fluorescent, and by using different fluorescent molecules it is possible to visualize different protein species simultaneously. Moreover, several copies of a fluorescent molecule can be fused to a single protein, enhancing the signal. The most commonly used fluorescent labels are green fluorescent protein (GFP) and its derivatives.3
However, it is not always possible to directly label molecules by using a genetic approach, either because the targets are not proteins (e.g., sugars, nucleic acids, lipids) or when the imaging requirements are not met by the fluorescent protein (e.g., fast imaging over an extended time period). In these cases, synthetic dyes or fluorescent nanocrystals (quantum dots) conjugated to antibodies can be used to label single molecules.4 In general, synthetic dyes are brighter than genetically expressed fluorescent proteins, while quantum dots offer a better photostability. Dye labeling might induce toxicity and quantum dot labeling requires a complex protocol.5 Organic dyes such as rhodamine,6 Alexa fluorophores (350–750 nm)7 and cyanine dyes (Cy3 and Cy5)8 are commonly used to label molecules in vivo. In addition, organic dyes can be delivered to GFP-tagged proteins by using small and high-affinity antibodies (nanobodies), in order to combine the molecular specificity of genetic tagging with the high brightness of organic dyes.9
The presence of a large fluorescent protein may interfere with the function of the target molecule, e.g. in the case of actin.10 To circumvent this issue, it is possible to use smaller genetic tags consisting of a motif (e.g., tetra-cysteine tag) that binds a chemical dye to label the target protein. The HaloTag11 and the SNAP/CLIP tags12 can bind and activate different fluorescent dyes in vivo. Analogously, the Flash-tag can act as a linker to a fluorescent dye, having the advantage of being much smaller (12 aminoacids) than conventional fluorescent molecules (>200 aminoacids).13
To detect the signal from single molecules, fluorescent markers must be bright and photostable (see Glossary). The brightness is important for single-molecule imaging in vivo because the signal of a single molecule must be above the background fluorescence generated by other molecular species present in the cell, which is not a concern when imaging single molecules in vitro. In comparison with non-single molecule imaging in vivo, single-molecule imaging in vivo requires higher sensitivity to detect fainter signals originating from single molecules, and higher speed of imaging because the dynamics of a single molecule occurs at a faster time scale than the dynamics averaged over an ensemble of molecules.
Single molecules can also be tracked by using video-enhanced brightfield or Differential Interferometric Contrast (DIC) microscopy, eliminating problems related to photobleaching. In this case, colloidal or gold nanoparticles with a diameter of 20–100 nm are attached to the molecule by antibody conjugation14 and tracking is achieved by Single Particle Tracking (SPT) algorithms.14b,15 These algorithms rely on image processing methods, such as centroid calculation or Gaussian fitting, to determine the position of a particle with sub-pixel resolution.
3. Fishing out single molecules
Before drawing a conclusion from experiments performed on single molecules, one must be sure that the observed signal indeed originates from individual molecules. The strategies for designing control experiments depend on the labeling technique.If the target molecule is labeled with a fluorescent marker, one strategy is to compare the intensity of the measured fluorescence signal to a reference intensity obtained on single fluorescent molecules. Alternatively, the reference intensity can be obtained by studying the bleaching kinetics, together with the knowledge of the number of fluorescent molecules labeling the target molecule. The amplitude of the intensity decrease (bleaching step) corresponds to the intensity generated by a single fluorescent molecule.16
When using SPT to track single molecules, it is important to ensure that only one molecule binds the nanoparticle. This issue can be solved by careful control of the particles functionalization, leading to nanoparticles carrying, on average, one linker per particle.17 In addition, when particles are used to label molecules, the diffusion coefficient or the activity of the protein should be compared to a control where the target molecule is instead labeled with a small non-interfering organic dye.18
4. Chasing single molecules
4.1 TIRF: single-molecule movement in vivo
Total Internal Reflection Fluorescence (TIRF) microscopy relies on the selective illumination of a thin layer of the sample.19 By illuminating a coverslip using a laser beam at an incident angle greater than the critical angle, a non-propagating electromagnetic field, known as evanescent wave, is established at the coverslip–sample interface (Fig. 2a). The intensity of the evanescent wave decays exponentially with the distance from the interface. Therefore, efficient excitation of fluorescence is achieved only within a few hundred nanometers from the interface. As a result, only the fluorescent molecules that are in this thin layer will fluoresce: the background fluorescence, which originates mainly from out of focus molecules, is reduced. This translates into an increase of the signal-to-noise (S/N) ratio, allowing for detection of the weak fluorescence signal coming from single molecules.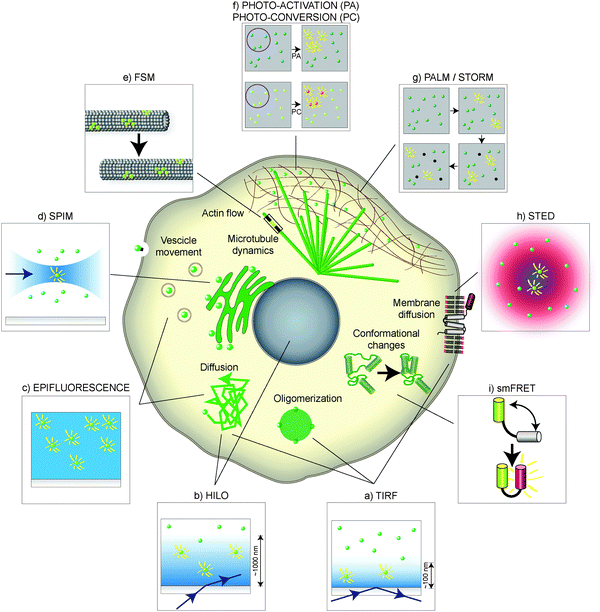 | ||
| Fig. 2 Single-molecule techniques in the cellular landscape. Depending on the region of the cell, or on the biological process under study, different single-molecule labeling and microscopy techniques can be used. Cell components are shown schematically in the middle, and the methods typically used to study them are enclosed in surrounding boxes. (a) TIRF is useful for fast imaging of single molecules close to the cell surface, addressing receptor dynamics, diffusion and oligomerization. (b) HILO, (c) widefield or epifluorescence and (d) SPIM can be used to image deeper inside the cell, allowing for visualization of single molecules in the nucleus and other compartments that are not accessible by TIRF. In a similar fashion, (e) FSM is useful to image single molecules in crowded environments, such as microtubule or actin networks. (f) Photo-activation (PA) and photo-conversion (PC) can be used to track a subpopulation of single molecules. Other techniques such as (g) PALM/STORM and (h) STED have a higher spatial resolution but their in vivo application is limited by the time resolution. (i) Single-molecule FRET (smFRET) allows for detection of conformational changes in the same molecule, as well as interaction between molecules, based on the light emitted when a donor and an acceptor fluorophore get in close proximity. | ||
TIRF can be used in vivo to investigate the dynamics of single molecules. For example, using TIRF single E-cadherin–GFP molecules were imaged for the first time in vivo.16a By comparing the movement of E-cadherin–GFP monomers and oligomers it was observed that the diffusion coefficient of oligomers at the membrane was lower than what was expected. Thus, the existence of a membrane skeleton, which would trap the E-cadherin oligomers lowering their mobility, was proposed. In another example, Cy3-labeled cAMP molecules were visualized using TIRF as they bound and got released from their receptors in the membrane of Dictyostelium.20 It was observed that Cy3–cAMP receptor complexes dissociated faster in the anterior than in the posterior region, unveiling the dynamic properties of receptors involved in chemotaxis.
The nature of the evanescent wave, on which TIRF microscopy relies, limits the analysis to cellular structures lying in close proximity to the coverslip–sample interface. If the coverslip–sample interface is illuminated slightly below the critical angle, the refracted beam propagates into the sample at a high inclination (Highly Inclined and Laminated Optical sheet, HILO, Fig. 2b), allowing for imaging single molecules several micrometers deep in the sample.21 The lateral (i.e., on a plane perpendicular to the optical axis) resolution is similar to the resolution that can be achieved using TIRF; however, the S/N ratio is lower compared to TIRF because of the increased thickness of the illuminated volume, resulting in more out-of-focus fluorescence. The penetration depth of HILO is limited by the increase in the thickness of the illumination beam to around 20 μm. Accessing regions that are further away from the coverslip requires other approaches.
4.2 Widefield microscopy and SPIM: seeing deeper into the cell
Widefield fluorescence microscopy, or epifluorescence, can be used to obtain single-molecule sensitivity in cases where the background fluorescence of the specimen is low and the number of fluorescent molecules is small (Fig. 2c). These conditions are often met in bacterial and yeast cells. The small thickness of these cells decreases the imaging volume, therefore reducing the background.Using widefield microscopy it was possible to image single fluorescently labeled Ash1 mRNA molecules in budding yeast. A cell-cycle dependent movement of the Ash1 mRNA molecule, defined by a quick translocation from the bud tip to the cell division site immediately prior to cytokinesis, was observed.22 This pioneering work showed the potential of using fluorescent proteins to label and track single molecules other than proteins inside the cell. Another application of widefield microscopy is to count single-molecule events. Stochastic bursts of protein production were monitored by imaging of a membrane-targeting peptide labeled with Venus, a fast folding fluorescent protein.23 It was observed that four copies of the protein were synthesized from a single burst of protein translation per cell cycle. Hence, real-time assays of single-molecule synthesis allow for precise quantification of single-cell gene expression.
Unlike TIRF, widefield microscopy excites fluorescence through the entire sample: the molecules that are not in the focal plane still emit light that contributes to the background fluorescence, lowering the S/N ratio and increasing the photobleaching. This prevents widefield microscopy from achieving single-molecule resolution when a high number of molecules are present in the imaging volume.
A useful approach to image deeper inside the sample while retaining a low background is to use orthogonal illumination, as in Selective Plane Illumination Microscopy (SPIM).24 Orthogonal illumination is achieved by illuminating the sample from one side with a thin sheet of light while collecting the fluorescence in the orthogonal direction (Fig. 2d). The thin sheet of light is typically achieved by focusing a Gaussian laser beam by means of a cylindrical lens. The illumination sheet is only a few micrometers thick, which minimizes the out-of-focus fluorescence and allows for observation of single molecules not only in regions inaccessible by TIRF inside cells, but also in tissues and embryos of animals.
Similarly to TIRF, HILO, and widefield microscopy, SPIM allows for characterization of fast processes, such as diffusion, and for studies of the dynamics of single molecules inside the cell. Only recently single-molecule imaging using SPIM has been achieved in vivo. To understand the behavior of the hrp36 ribonuclear protein inside messenger ribonucleoprotein particles, hrp36 labeled in vitro with the fluorescent molecule ATTO647-N was injected into the salivary glands of Chironomus tentans.25 By illuminating the larvae orthogonally, SPIM allowed for visualization and tracking of single messenger ribonucleoproteins 200 μm deep in the sample.
4.3 Imaging mosaics: speckle microscopy and photo-activation
An alternative approach to increase the S/N ratio to image single molecules in vivo consists of labeling only a small number of molecules belonging to a molecular species. This can be achieved by microinjecting a low number of fluorescently labeled molecules that will be diluted by the endogenous non-labeled version of the molecule in the cellular environment, by controlling gene expression, and by using photo-manipulation. These approaches are especially useful in dense regions of the cell, like the cytoskeleton, or to follow molecules synthesized at different times (pulse-chase experiments).The first implementation of this approach was termed Fluorescent Speckle Microscopy (FSM, Fig. 2e).26 Microinjecting fluorescently labeled tubulin in cells, which is incorporated together with non-fluorescent endogenous tubulin into microtubules, results in a speckled fluorescence of the microtubule lattice. The speckled pattern originating from single fluorescent tubulin molecules is a landmark that allows for visualizing microtubule growth, shrinkage, and sliding, for example of kinetochore microtubules in the spindle.27 Using the same technique, actin movement during filament turnover was monitored, showing that the actin filaments in the lamellipodium were mostly generated by polymerization away from the tip.28
In a similar approach, Photo-Activation (PA) and Photo-Conversion (PC) allow for the detection of a sub-population of molecules inside the cell (Fig. 2f). Both photo-activation and photo-conversion are induced by irradiating the molecules with a pulse of light at a specific wavelength. The irradiation induces a conformational change that switches the molecules from a non-fluorescent to a fluorescent state (photo-activation) or modifies the absorption and emission spectra of the fluorescent molecule (photo-conversion). In the first case, the photo-activated molecules can be detected on a background of dark, non-fluorescent molecules. In the second case, the photo-convertible molecules, such as PS-CFP2 (cyan-green) and Dendra2 (green-red),29 shift their emission spectrum towards red upon UV excitation.30 The use of photo-convertible proteins as labels has the advantage that fluorescent detection of both the unconverted and converted states is possible.
Lysosomal protein trafficking was studied in vivo using photo-activation. Photo-activated molecules of Igp120 (Igp120–PA-GFP), a lysosomal membrane protein, were found to traffic from the photo-activated to non-photo-activated lysosomes.31 This demonstrates that Igp120 is exchanged between lysosomes.
In a recent work, a photo-convertible tag was used to test whether de novo nuclear pore synthesis was the only source of nuclear pores in the daughter cell of budding yeast. Using photo-conversion of Nic96 (Nic96-2xDendra2), a nuclear pore protein, a population of “old pores” (photo-converted, red) in the mother cell was visually distinguished from the de novo synthesized pores in the daughter cell (unconverted, green). By differentially labeling old and new molecules one can compare their behavior. In this case, it was observed that nuclear pores that were present in the mother were transmitted to the daughter cell upon division.32
The methods described so far are useful for studying the movement of individual single molecules and molecular complexes. Yet, mapping a large number of molecules inside the cell with higher spatial resolution requires a different approach.
4.4 Super-resolution fluorescence microscopy: the molecular atlas of the cell
In the case of single molecules labeled with a fluorescent protein, the resolution is limited by the point-spread function (PSF) of the microscope, which is usually not smaller than a few hundreds of nanometers (diffraction limit). Two molecules that are closer than the diffraction limit cannot be distinguished. However, the accuracy in defining the position of an object can be much higher than the size of the object: for example, SPT of a micron-sized sphere can resolve its position with nanometer accuracy. This concept has been translated to the mapping of the position of single molecules.In Photo-Activated Localization Microscopy (PALM) and Stochastic Optical Reconstruction Microscopy (STORM), a small random fraction of photo-activatable or photo-convertible fluorophores, typically apart from each other by a distance larger than the diffraction limit, is activated at a time (Fig. 2g). Fitting a Gaussian distribution to the accumulated distribution of photons, and taking the center of the fitted Gaussian as the position of the molecule, allows for the localization of each molecule at a higher resolution than the diffraction limit.33 The positioning accuracy scales roughly with the square root of the total number of detected photons (see Glossary). Therefore, PALM and STORM are most suitable for observing molecules that move slowly or that are bound to cell compartments. Repeating the process by activating a different set of molecules at a time leads to a complete reconstruction of the positions of the molecules in the sample down to 10 nm resolution.
As an example, time-lapse PALM was performed with enhanced yellow fluorescent protein (EYFP)-labeled version of MreB, the actin homolog of Caulobacter crescentus. With this approach, it was possible to monitor MreB treadmilling with a spatial resolution of 30–40 nm, well below the diffraction limit.34 Two distinct MreB superstructures were identified, a quasi helix in the stalked cell and a midplane ring that forms before division. Recently, pair-correlation analysis was combined with PALM, which allowed for the determination of the nanoscale organization of membrane proteins with distinctive membrane anchoring and lipid partitioning features in COS-7 cells.35 In another study, STORM was used to detect single actin filaments and their three-dimensional ultrastructure and organization in COS-7 and BSC-1 cells.36
A conceptually different approach to circumvent the diffraction limit is to shape the PSF of the microscope in order to detect the fluorescence emitted by molecules that are in a region smaller than the diffraction limit. In Stimulated Emission-Depletion microscopy (STED), the PSF of the microscope is reduced by de-exciting the fluorescent molecules around a central excitation peak by using a doughnut shaped beam (Fig. 2h). By exploiting nonlinearity, the spatial extent of the central region of the depletion beam can be made smaller than the diffraction limit, leaving molecules excited only in this small volume.37
The reduction of the excitation volume achievable by STED was used to detect the intensity fluctuations of single diffusing lipids, in regions one order of magnitude below the diffraction limit. This study showed that sphingolipids and glycosylphosphatidylinositol (GPI)-anchored proteins are transiently (10–20 ms) trapped in cholesterol complexes in areas smaller than 20 nm.38 The decreased diffusion in these areas supports the existence of membrane nanodomains.
4.5 smFRET: conformational changes within a single molecule
Fluorescence Resonance Energy Transfer (FRET), also known as Förster Resonance Energy Transfer, is used to determine whether a labeled molecular pair gets close enough for energy to be transferred from a donor to an acceptor molecule39 (Fig. 2i). If the energy transfer occurs, the acceptor molecule is excited and emits a photon, indicating that the two molecules interacted. Single-molecule FRET (smFRET) can be used to detect changes in the conformation of a molecule, when both donor and acceptor are present in different domains of the same molecule.40 Changes in the conformation are measured by the changes in the emission spectra when tracking a single molecule. This technique was used to study the conformation of membrane fusion proteins in vivo, by labeling SNARE proteins (SNAP-25) with a FRET donor (Cy3 or A555) and acceptor (Cy5 or A647) and microinjecting them into cultured neuronal and kidney cells.41 Upon formation of the SNARE complex, the proteins underwent conformational changes when binding to the molecules in the membrane occurred, which was detected by an increase in the FRET signal. Moreover, the duration of individual interactions can be measured by fluctuations in the FRET signal.5. Lessons from single-molecule imaging
Depending on the biological question and the experimental sample, single-molecule imaging techniques in vivo can provide a deeper understanding of the mechanisms underlying the processes in the cell. In principle, epifluorescence would suffice to see single molecules: if the number of molecules is very low, the molecules do not move very fast, and the label is highly specific, with a good S/N ratio. However, when following a single molecule inside the cell one often encounters complications such as auto-fluorescence, high molecule density, limited illumination depth and quenching of the signal over time. It is therefore important to resolve the molecules spatially, either by imaging only a thin volume of the cell (TIRF and SPIM) or limiting the number of labeled molecules (FSM and PA/PC), or detecting only a subset of the labeled molecules inside the cell (PALM/STORM or STED). While TIRF is the most suited technique to achieve high temporal resolution (order of few ms), PALM/STORM and STED are more suited to precisely map the positions of single-molecules. HILO and SPIM can reach deeper into the sample, and by using FSM it is possible to pinpoint the localization of a molecule in a highly organized cellular structure, such as the cytoskeleton. Photo-activation and photo-conversion are real time pulse-chase techniques, where a selected subset of the molecules can be followed. If one is interested in determining the changes within a single molecule, smFRET allows for the detection of conformational changes in vivo. If, on the other hand, one is interested only in the localization pattern of the molecules, working on fixed samples is advantageous because it offers the possibility of imaging the sample for a longer time and with a stronger illumination, resulting in a better S/N ratio. PALM/STORM and STED yield better resolution on fixed than on live specimens. TIRF, HILO, epifluorescence microscopy and SPIM yield a similar resolution on live and fixed samples, whereas other techniques that rely on the movement or conformational changes of the molecules, such as FSM, photo-activation, photo-conversion and smFRET, are only suited for live-cell imaging. A synthetic presentation of all the single-molecule imaging techniques and examples of their in vivo applications are presented in Table 1.| Technique | Spatial resolution | Time resolution | Photo-toxicity | Measurable physical parameters | Cellular region | Compatible labels | Molecule imaged/label | In vivo applications | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TIRF | 200–250 nm | 5 ms | Low | Position and movement | Cover slip interface | FPs, organic dyes | E-cadherin–GFP | Oligomerization dynamics | 16a |
| cAMP–Cy3 | Chemotaxis | 20 | |||||||
| PHD–GFP | Membrane binding | 49 | |||||||
| Telenzepine–Cy3b | Membrane receptors | 50 | |||||||
| G-protein YFP–CIOH-Ras | Membrane microdomains | 51 | |||||||
| EPI | 200–250 nm | 5 ms | Medium | Position and movement | All | FPs, organic dyes, Q-dots, colloidal particles | Glycoprotein–gold | Membrane proteins | 52 |
| Gly-receptor-Qdot | Neuronal receptors | 53 | |||||||
| Viruses-Cy3/5 | Viral infection | 54 | |||||||
| SPIM | 200–250 nm | 5 ms | Low | Position and movement | All | FPs, organic dyes | Kinesin-Qdot | Molecular motors | 55 |
| Tsr-Venus | Protein synthesis | 23 | |||||||
| Hrp36-ATTO647N | Ribonuclear particles | 25 | |||||||
| FSM | 200–250 nm | ∼1 s | Medium | Position and movement | All | FPs, organic dyes | Tubulin–XRhodamine | Microtubule dynamics | 26 |
| β Actin–EGFP | Actin dynamics | 28 | |||||||
| Photo-activation/photo-conversion | 200–250 nm | ∼1 s | High | Position and movement | All | PA-FP, PC-FP, tetracysteine | Igp120–PA-GFP | Membrane diffusion | 31 |
| Connexin43-Flash/ReAsh | Gap junctions | 56 | |||||||
| Fibrillarin-Dendra2 | Nuclear transport | 29, 30 | |||||||
| Nic95-2xDendra2 | Nuclear pore segregation | 32 | |||||||
| Super resolution | 20 nm | ∼100 ms | High | Position | 5–10 μm from the cell surface | Organic dyes, FPs, PA-FP, PC-FP | MreB-PS-EYFP | Prokaryote cytoskeleton | 34 |
| Atto647N-PE and sphingomyelin | Membrane microdomains | 38 | |||||||
| FtsZ-mEos2 | Prokaryotic septum | 57 | |||||||
| smFRET | 200–250 nm (Donor–acceptor 1–10 nm) | ∼100 ms | Low | Position, movement and conformation | All | Donor–acceptor fluorophores | SNAP25-A555/A647 | Membrane protein folding | 41 |
The lessons that can be learned from single-molecule techniques, when compared with ensemble imaging methods, derive from analyzing movement, dwelling, interactions and conformational changes of single molecules and identifying how different subsets of the same molecular species contribute to their global function in the cell. For example, by quantifying the localization, movement and dwell time of single molecules in vivo it is possible to understand how they get targeted to the sites of their function. Based on this knowledge, it is possible to design genetic and biochemical perturbations, such as mutations and chemicals, that affect a specific single-molecule characteristic, from the molecular interaction affinity to the movement and localization pattern of the molecules. This approach allows for distinguishing between different mechanistic models, leading to a deeper understanding of how collective behavior arises from the interactions between single molecules.
6. Future directions of single-molecule imaging in cell biology
In light of recent technological advances, a number of biological processes can be directly visualized using microscopy. However, these are to some extent limited to certain regions of the cell, or to the cases in which a single molecule binds to a slower diffusing species or gets trapped in a larger structure.Single-molecule detection deep inside the tissue could benefit from the development of new fluorescent molecules emitting in the infrared, because most biological tissues are transparent at these wavelengths. Conversely, developing markers emitting in the deep UV or even at shorter wavelengths could enhance the positioning accuracy of TIRF or other methods accessing the cellular membrane.42
Together with the development of new fluorescent reporters, advances in the detection techniques that would allow for an increased image acquisition speed would facilitate studies of the movement of molecules from their synthesis to their incorporation into more complex structures, or while interacting with their substrates. An example of this is the use of super-registration. With this method, in which an internal registration signal is used to register spectrally different channels relative to each other, it is possible to measure fast transport of single molecules.43 In the era of quantitative biology, it would be important to develop high-throughput methodologies that allow for studying a high number of single-molecule events in real time, comparing the number, diffusion, and the binding of molecules for a large set of experimental conditions.44 This is especially important in drug design where the minimization of off-target secondary effects is desirable.
Light can also be used to control the behavior of single molecules. It would be useful to inactivate a molecule with a very precise spatio-temporal localization in the cell, and see how this would affect the process under study, especially in cases where constitutive genetic inactivation methods are not feasible, such as for genes affecting multiple processes or essential genes. Similar to Chromophore-Assisted Laser Inactivation (CALI),45 the development of methods to target a smaller volume of inactivation in vivo would facilitate a precise control of the number of inactivated molecules in a confined region of the cell. Such new developments in microscopy fostered by the need of in vivo quantification of molecular interactions will shed light on the life cycle of a molecule inside a cell.
Glossary
Fluorescence is a physical process in which molecules temporarily absorb energy, promoting electrons to a higher energy level (excitation), followed by the production of photons when the electrons decay to a lower energy level (emission). In fluorescence microscopy, the excitation is usually performed by irradiating the molecules with a light source (arc lamp, laser). A fluorescent molecule can be characterized by the following parameters:The excitation spectrum defines the efficiency of excitation of a fluorescent molecule as a function of the wavelength of the exciting light. The wavelength of the exciting light is usually chosen close to the maximum of the excitation spectrum.
The emission spectrum defines the intensity of the emitted fluorescence as a function of the wavelength. The wavelength of the emitted fluorescence is longer than the wavelength used for excitation, and the detection is performed close to the maximum of the spectrum. The difference in wavelength between the excitation and the emission maxima is termed Stokes shift.
Quantum yield defines the efficiency of the excitation process as the ratio between the number of emitted photons and the number of absorbed photons. Fluorescent molecules with a quantum yield close to 1 will efficiently convert the absorbed energy into photons.
Molar absorption, also known as extinction coefficient, defines how strongly a fluorescent molecule absorbs light as a function of the wavelength. The product of the quantum yield and the molar absorption, measured at the absorption maximum, is the brightness.
Lifetime is the average time elapsing between the excitation of a fluorescent molecule and the emission of a photon from that molecule. Typically of the order of nanoseconds, it is strongly influenced by the environment in which the fluorescent molecules are.
Photostability is the ability of a fluorescent molecule to undergo repeated cycles of absorption and emission keeping its chemical structure intact. It depends on the molecular species present in the surroundings of the fluorescent molecule.
Photo-activation, photo-conversion, photo-switching: the process by which a molecule, undergoing a conformational change of its structure, becomes fluorescent (photo-activation) or changes its absorption and/or emission spectrum (photo-conversion). The conformational change can be permanent or reversible (photo-switching), and is achieved by irradiating the molecules with a pulse of light at a specific wavelength (usually UV).
FRET
Fluorescence resonance energy transfer, also known as Förster resonance energy transfer, relies on the excitation of an acceptor fluorophore by a closely localized donor fluorophore. An efficient energy transfer happens if the emission spectrum of the donor molecule overlaps with the excitation spectrum of the acceptor, and if the two molecules are in close proximity. The distance at which the energy transfer has a 50% efficiency is termed the Förster critical radius, and has values typically between 2 nm and 10 nm.FSM
Fluorescent speckle microscopy consists of mixing fluorescent and non-fluorescent molecules. This can be achieved by introducing a low number of fluorescently-labeled molecules that will be diluted by the endogenous non-fluorescent version in the cellular environment, for instance by using microinjection or genetically by low-expression vectors.HILO
Highly inclined and laminated optical sheet microscopy employs illumination of the coverslip–sample interface slightly below the critical angle. As a consequence, the refracted beam propagates into the sample at a high inclination, i.e., almost parallel to the coverslip, allowing for imaging single molecules several microns deep in the sample.PALM
Photo-activation localization microscopy is a technique that achieves a spatial resolution higher than the diffraction limit. After activating a small fraction of molecules, their position is determined precisely by fitting a Gaussian function to the signal intensities. Repeating the process while activating a different set of molecules at a time allows for reconstruction of an image of the labeled structure with a spatial resolution of roughly 10 nm.PSF
Point-spread function of a linear optical system defines its resolving power. It can be thought of as the yield of the optical system when imaging a point source. The image of an object formed by a linear optical system consists of the convolution of its point-spread function with the actual object shape.SPIM
Selective plane illumination microscopy employs illumination of the sample from one side with a thin sheet of light and collection of the fluorescence signal in the orthogonal direction. It minimizes the out-of-focus fluorescence and allows for imaging with a high signal-to-noise ratio in regions deep in the sample.SPT
Single particle tracking is the observation of the motion of single molecules, yielding their trajectories in the cell. In a simplified picture, where each photon emitted by the tracked particle contributes to specify its position, the achievable tracking precision reduces the error of the average of the detected photon positions. Assuming n emitted photons, if the width of the imaging system PSF is ΔXPSF, then the standard error of the mean of the measured photon positions is ΔXPSF/√n.STED
Stimulated emission-depletion microscopy achieves super-resolution, i.e., a resolution below the diffraction limit, by reducing the point spread function of the microscope. This is accomplished by means of stimulated emission, in which fluorophores are switched off by additional irradiation with a de-excitation beam.TIRF
Total internal reflection fluorescence microscopy relies on the selective illumination of a thin layer of the sample. By illuminating a coverslip using a laser beam at an incident angle greater than the critical angle, an evanescent field is established at the coverslip–sample interface. The amplitude of the evanescent field decays exponentially with the distance from the interface. Therefore, efficient excitation of fluorescence is achieved only within a few hundred nanometers from the interface.Acknowledgements
We thank Nenad Pavin and Mariola Chacon for constructive comments on the manuscript, Ivana Šarić for the drawings, and members of the Tolić-Nørrelykke group for discussions.Notes and references
- I. Tinoco, Jr. and R. L. Gonzalez, Jr., Biological mechanisms, one molecule at a time, Genes Dev., 2011, 25, 1205–1231 Search PubMed.
- Y. Sako, M. Hiroshima, C. G. Pack, K. Okamoto, K. Hibino and A. Yamamoto, Live cell single-molecule detection in systems biology, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2012, 4, 183–192 Search PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, The fluorescent toolbox for assessing protein location and function, Science, 2006, 312, 217–224 CrossRef CAS.
- (a) U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Quantum dots versus organic dyes as fluorescent labels, Nat. Methods, 2008, 5, 763–775 CrossRef CAS; (b) N. Tomczak, D. Janczewski, D. Dorokhin, M. Y. Han and G. J. Vancso, Enabling biomedical research with designer quantum dots, Methods Mol. Biol., 2012, 811, 245–265 Search PubMed.
- C. Jing and V. W. Cornish, Chemical tags for labeling proteins inside living cells, Acc. Chem. Res., 2011, 44, 784–792 CAS.
- D. Magde, R. Wong and P. G. Seybold, Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: improved absolute standards for quantum yields, Photochem. Photobiol., 2002, 75, 327–334 CrossRef CAS.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao and W. Y. Leung, Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates, J. Histochem. Cytochem., 1999, 47, 1179–1188 CAS.
- L. A. Ernst, R. K. Gupta, R. B. Mujumdar and A. S. Waggoner, Cyanine dye labeling reagents for sulfhydryl groups, Cytometry, 1989, 10, 3–10 CrossRef CAS.
- J. Ries, C. Kaplan, E. Platonova, H. Eghlidi and H. Ewers, A simple, versatile method for GFP-based super-resolution microscopy via nanobodies, Nat. Methods, 2012, 9, 582–584 Search PubMed.
- R. E. Hosein, S. A. Williams, K. Haye and R. H. Gavin, Expression of GFP-actin leads to failure of nuclear elongation and cytokinesis in Tetrahymena thermophila, J. Eukaryotic Microbiol., 2003, 50, 403–408 Search PubMed.
- G. V. Los and K. Wood, The HaloTag: a novel technology for cell imaging and protein analysis, Methods Mol. Biol., 2007, 356, 195–208 CAS.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa, Jr., M. Kindermann, F. Beaufils and K. Johnsson, An engineered protein tag for multiprotein labeling in living cells, Chem. Biol., 2008, 15, 128–136 CrossRef CAS.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Specific covalent labeling of recombinant protein molecules inside live cells, Science, 1998, 281, 269–272 CrossRef CAS.
- (a) M. De Brabander, G. Geuens, R. Nuydens, M. Moeremans and J. De Mey, Probing microtubule-dependent intracellular motility with nanometre particle video ultramicroscopy (nanovid ultramicroscopy), Cytobios, 1985, 43, 273–283 Search PubMed; (b) J. Gelles, B. J. Schnapp and M. P. Sheetz, Tracking kinesin-driven movements with nanometre-scale precision, Nature, 1988, 331, 450–453 CrossRef CAS.
- (a) C. M. Anderson, G. N. Georgiou, I. E. Morrison, G. V. Stevenson and R. J. Cherry, Tracking of cell surface receptors by fluorescence digital imaging microscopy using a charge-coupled device camera. Low-density lipoprotein and influenza virus receptor mobility at 4 degrees C, J. Cell Sci., 1992, 101(Pt 2), 415–425; (b) M. K. Cheezum, W. F. Walker and W. H. Guilford, Quantitative comparison of algorithms for tracking single fluorescent particles, Biophys. J., 2001, 81, 2378–2388 CrossRef CAS.
- (a) R. Iino, I. Koyama and A. Kusumi, Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface, Biophys. J., 2001, 80, 2667–2677 CrossRef CAS; (b) M. Schuster, R. Lipowsky, M. A. Assmann, P. Lenz and G. Steinberg, Transient binding of dynein controls bidirectional long-range motility of early endosomes, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3618–3623 Search PubMed.
- F. Daumas, H. Mazarguil, C. Millot, A. Lopez and L. Salome, Probing functionalized gold colloids for single particle tracking experiments, Biochem. Biophys. Res. Commun., 2002, 295, 610–615 Search PubMed.
- A. Kusumi, H. Ike, C. Nakada, K. Murase and T. Fujiwara, Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules, Semin. Immunol., 2005, 17, 3–21 CrossRef CAS.
- D. Axelrod, Cell–substrate contacts illuminated by total internal reflection fluorescence, J. Cell Biol., 1981, 89, 141–145 CAS.
- M. Ueda, Y. Sako, T. Tanaka, P. Devreotes and T. Yanagida, Single-molecule analysis of chemotactic signaling in Dictyostelium cells, Science, 2001, 294, 864–867 CrossRef CAS.
- M. Tokunaga, N. Imamoto and K. Sakata-Sogawa, Highly inclined thin illumination enables clear single-molecule imaging in cells, Nat. Methods, 2008, 5, 159–161 CrossRef CAS.
- (a) E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Localization of ASH1 mRNA particles in living yeast, Mol. Cell, 1998, 2, 437–445 CrossRef CAS; (b) D. L. Beach, E. D. Salmon and K. Bloom, Localization and anchoring of mRNA in budding yeast, Curr. Biol., 1999, 9, 569–578 CrossRef CAS.
- J. Yu, J. Xiao, X. Ren, K. Lao and X. S. Xie, Probing gene expression in live cells, one protein molecule at a time, Science, 2006, 311, 1600–1603 CrossRef CAS.
- J. Huisken, J. Swoger, F. Del Bene, J. Wittbrodt and E. H. Stelzer, Optical sectioning deep inside live embryos by selective plane illumination microscopy, Science, 2004, 305, 1007–1009 CrossRef CAS.
- J. G. Ritter, R. Veith, A. Veenendaal, J. P. Siebrasse and U. Kubitscheck, Light sheet microscopy for single molecule tracking in living tissue, PLoS One, 2010, 5, e11639 Search PubMed.
- C. M. Waterman-Storer and E. D. Salmon, Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling, J. Cell Biol., 1997, 139, 417–434 CrossRef CAS.
- J. R. LaFountain, Jr., C. S. Cohan and R. Oldenbourg, Functional states of kinetochores revealed by laser microsurgery and fluorescent speckle microscopy, Mol. Biol. Cell, 2011, 22, 4801–4808 Search PubMed.
- N. Watanabe and T. J. Mitchison, Single-molecule speckle analysis of actin filament turnover in lamellipodia, Science, 2002, 295, 1083–1086 Search PubMed.
- D. M. Chudakov, S. Lukyanov and K. A. Lukyanov, Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2, Nat. Protocols, 2007, 2, 2024–2032 Search PubMed.
- N. G. Gurskaya, V. V. Verkhusha, A. S. Shcheglov, D. B. Staroverov, T. V. Chepurnykh, A. F. Fradkov, S. Lukyanov and K. A. Lukyanov, Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light, Nat. Biotechnol., 2006, 24, 461–465 CrossRef CAS.
- G. H. Patterson and J. Lippincott-Schwartz, A photoactivatable GFP for selective photolabeling of proteins and cells, Science, 2002, 297, 1873–1877 CrossRef CAS.
- A. Khmelinskii, P. J. Keller, H. Lorenz, E. Schiebel and M. Knop, Segregation of yeast nuclear pores, Nature, 2010, 466, E1 Search PubMed.
- (a) E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Imaging intracellular fluorescent proteins at nanometer resolution, Science, 2006, 313, 1642–1645 CAS; (b) M. J. Rust, M. Bates and X. Zhuang, Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM), Nat. Methods, 2006, 3, 793–795 CrossRef CAS.
- J. S. Biteen, M. A. Thompson, N. K. Tselentis, G. R. Bowman, L. Shapiro and W. E. Moerner, Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP, Nat. Methods, 2008, 5, 947–949 CrossRef CAS.
- P. Sengupta, T. Jovanovic-Talisman, D. Skoko, M. Renz, S. L. Veatch and J. Lippincott-Schwartz, Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis, Nat. Methods, 2011, 8, 969–975 Search PubMed.
- K. Xu, H. P. Babcock and X. Zhuang, Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton, Nat. Methods, 2012, 9, 185–188 Search PubMed.
- T. A. Klar, S. Jakobs, M. Dyba, A. Egner and S. W. Hell, Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8206–8210 CrossRef CAS.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorff, A. Schonle and S. W. Hell, Direct observation of the nanoscale dynamics of membrane lipids in a living cell, Nature, 2009, 457, 1159–1162 CrossRef.
- R. B. Sekar and A. Periasamy, Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS.
- K. Truong and M. Ikura, The use of FRET imaging microscopy to detect protein–protein interactions and protein conformational changes in vivo, Curr. Opin. Struct. Biol., 2001, 11, 573–578 CrossRef CAS.
- J. J. Sakon and K. R. Weninger, Detecting the conformation of individual proteins in live cells, Nat. Methods, 2010, 7, 203–205 Search PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Creating new fluorescent probes for cell biology, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS.
- D. Grunwald and R. H. Singer, In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport, Nature, 2010, 467, 604–607 CrossRef.
- D. M. Owen, D. Williamson, C. Rentero and K. Gaus, Quantitative microscopy: protein dynamics and membrane organisation, Traffic, 2009, 10, 962–971 CrossRef CAS.
- K. Jacobson, Z. Rajfur, E. Vitriol and K. Hahn, Chromophore-assisted laser inactivation in cell biology, Trends Cell Biol., 2008, 18, 443–450 CrossRef CAS.
- O. Shimomura, F. H. Johnson and Y. Saiga, Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea, J. Cell. Comp. Physiol., 1962, 59, 223–239 CrossRef CAS.
- T. Hirschfeld, Optical microscopic observation of single small molecules, Appl. Opt., 1976, 15, 2965–2966 CrossRef.
- A. H. Voie, D. H. Burns and F. A. Spelman, Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens, J. Microsc., 1993, 170, 229–236 Search PubMed.
- S. Matsuoka, M. Iijima, T. M. Watanabe, H. Kuwayama, T. Yanagida, P. N. Devreotes and M. Ueda, Single-molecule analysis of chemoattractant-stimulated membrane recruitment of a PH-domain-containing protein, J. Cell Sci., 2006, 119, 1071–1079 Search PubMed.
- M. J. Schaaf, W. J. Koopmans, T. Meckel, J. van Noort, B. E. Snaar-Jagalska, T. S. Schmidt and H. P. Spaink, Single-molecule microscopy reveals membrane microdomain organization of cells in a living vertebrate, Biophys. J., 2009, 97, 1206–1214 Search PubMed.
- J. A. Hern, A. H. Baig, G. I. Mashanov, B. Birdsall, J. E. Corrie, S. Lazareno, J. E. Molloy and N. J. Birdsall, Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2693–2698 CrossRef CAS.
- M. P. Sheetz, S. Turney, H. Qian and E. L. Elson, Nanometre-level analysis demonstrates that lipid flow does not drive membrane glycoprotein movements, Nature, 1989, 340, 284–288 CrossRef CAS.
- M. Dahan, S. Levi, C. Luccardini, P. Rostaing, B. Riveau and A. Triller, Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking, Science, 2003, 302, 442–445 CrossRef CAS.
- (a) M. Lakadamyali, M. J. Rust, H. P. Babcock and X. Zhuang, Visualizing infection of individual influenza viruses, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9280–9285 CrossRef CAS; (b) M. J. Rust, M. Lakadamyali, F. Zhang and X. Zhuang, Assembly of endocytic machinery around individual influenza viruses during viral entry, Nat. Struct. Mol. Biol., 2004, 11, 567–573 Search PubMed.
- S. Courty, C. Luccardini, Y. Bellaiche, G. Cappello and M. Dahan, Tracking individual kinesin motors in living cells using single quantum-dot imaging, Nano Lett., 2006, 6, 1491–1495 CrossRef CAS.
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Multicolor and electron microscopic imaging of connexin trafficking, Science, 2002, 296, 503–507 CrossRef CAS.
- G. Fu, T. Huang, J. Buss, C. Coltharp, Z. Hensel and J. Xiao, In Vivo Structure of the E. coli FtsZ-ring Revealed by Photoactivated Localization Microscopy (PALM), PLoS One, 2010, 5, e12680 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |