Compact zwitterion-coated iron oxide nanoparticles for in vitro and in vivo imaging†
He
Wei
,
Oliver T.
Bruns
,
Ou
Chen
and
Moungi G.
Bawendi
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: mgb@mit.edu
First published on 5th October 2012
Abstract
We have recently developed compact and water-soluble zwitterionic dopamine sulfonate (ZDS) ligand coated superparamagnetic iron oxide nanoparticles (SPIONs) for use in various biomedical applications. The defining characteristics of ZDS-coated SPIONs are small hydrodynamic diameters, low non-specific interactions with fetal bovine serum, the opportunity for specific labeling, and stability with respect to time, pH, and salinity. We report here on the magnetic characterization of ZDS-coated SPIONs and their in vitro and in vivo performance relative to non-specific interactions with HeLa cells and in mice, respectively. ZDS-coated SPIONs retained the superparamagnetism and saturation magnetization (Ms) of as-synthesized hydrophobic SPIONs, with Ms = 74 emu g−1 [Fe]. Moreover, ZDS-coated SPIONs showed only small non-specific uptake into HeLa cancer cells in vitro and low non-specific binding to serum proteins in vivo in mice.
Insight, innovation, integrationOur newly developed compact and water-soluble zwitterionic dopamine sulfonate (ZDS) ligand coated superparamagnetic iron oxide nanoparticles (SPIONs) are superparamagnetic with a high saturation magnetization of 74 emu g−1 [Fe], which renders ZDS-coated SPIONs suitable for potential magnetic resonance imaging applications. Furthermore, ZDS-coated SPIONs exhibit small non-specific cellular uptake into HeLa cancer cells and low non-specific affinity toward mouse serum. These in vitro and in vivo data therefore suggest that when conjugated with peptides, aptamers, or proteins, functionalized ZDS-coated SPIONs can serve as an attractive tool for imaging, sensing, and hyperthermia. |
Introduction
The high saturation magnetization, superior chemical stability, and potentially minimized toxicity of superparamagnetic iron oxide nanoparticles (SPIONs) render them as interesting contrast agents for improving the sensitivity of in vitro and in vivo magnetic resonance imaging (MRI).1 Uniformly sized SPIONs are often synthesized in organic solvent so that they are hydrophobic and thus non-soluble in aqueous systems of biomedical interest.2 Therefore, modifying the surface of SPIONs by ligand exchange has been essential for endowing SPIONs with hydrophilic properties and for demonstrating their potential use in various biomedical applications.3 Polyethylene glycol modified (PEGylated) dopamine derivatives have served as attractive ligands to render SPIONs hydrophilic and to alleviate non-specific binding to proteins;4 however, a significant increase of the effective hydrodynamic diameter (HD) can occur due to these PEGylated ligands, which may in turn affect the endocytotic behavior of these nanoparticles (NPs) into cells, restrict access to confined spaces, or hinder renal elimination.5 In our prior paper,6 we reported the design and synthesis of a compact and water-soluble zwitterionic dopamine sulfonate (ZDS) ligand with strong binding affinity to SPIONs. The resulting bio-compatible ZDS-coated SPIONs demonstrated minimized HDs, low non-specific interactions with fetal bovine serum in vitro, specific labeling of biotin receptors through conjugation to streptavidin, and stability with respect to time, pH, and salinity. For potential in vitro and in vivo MRI as well as specific targeting and imaging applications, there remains a need for the newly developed ZDS-coated SPIONs to be further investigated in terms of their magnetic properties and non-specific interactions with cells and in live mice.Materials and methods
Chemicals and analysis
All chemicals unless indicated were obtained from Sigma Aldrich and used as received. Air-sensitive materials were handled in an Omni-Lab VAC glove box under a dry nitrogen atmosphere with oxygen levels <0.2 ppm. All solvents were spectrophotometric grade and purchased from EMD Biosciences. TEM images of the iron oxide NPs were obtained with a JEOL 200CX electron microscope operated at 120 kV and HR-TEM images of the iron oxide NPs were obtained with a JEOL 2010 electron microscope operated at 200 kV. Magnetism measurements were performed on a Quantum Design MPMS-XL superconducting quantum interference device. Cell imaging was recorded using a microscope manufactured by Carl Zeiss.Synthesis of iron oxide NPs
Maghemite (Fe2O3) magnetic NPs were prepared using a method modified from the literature.7 As an example, for the synthesis of 6 nm NPs, 400 μL of Fe(CO)5 was added to 1.91 mL of oleic acid (≥99% by GC) in the mixture of 5.7 mL 1-octadecene and 14.3 mL 1-hexadecene at 100 °C. The temperature was increased at a rate of 2 °C min−1 to a final temperature of 290 °C, at which it was held constant for 1 h. After the mixture was cooled to room temperature, 0.32 g of (CH3)3NO was added as an oxidizing agent.8 The mixture was heated to 130 °C for 2 hours and was then quickly heated to 275 °C for 15 min. After cooling, adding ethanol, and centrifuging, the supernatant was discarded, and the precipitated NPs were then redispersed and stored in hexane.NP ligands
Zwitterionic dopamine sulfonate (ZDS, MW: ∼300 g mol−1) and thiol-terminated catechol-derivative (TD, MW: ∼850 g mol−1) ligands were synthesized according to the protocol in the Supporting Information of our prior paper.6 Dextran (Dex) ligands (MW: ∼6000 g mol−1) were obtained from Sigma Aldrich. 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] ammonium salt (PEG-lipid, MW: ∼2800 g mol−1) in chloroform solution was purchased from Avanti Polar Lipids.Ligand exchange of iron oxide NPs
ZDS-coated iron oxide NPs (ZDS) and TD/ZDS-coated iron oxide NPs (TD/ZDS-NPs) were prepared according to the protocol in the Supporting Information of our prior paper.6 For the preparation of dextran-coated iron oxide NPs (Dex-NPs), ethanol was added into 25 μL of NPs in hexane stock solution up to the point of turbidity. ∼1 mg of dry pellet was obtained by the followed centrifugation and decantation. Then 2-[2-(2-methoxyethoxy)ethoxy]acetic acid (MEAA) coated iron oxide NPs (MEAA-NPs) were prepared according to protocol in the Supporting Information of our prior paper.6 In 0.6 mL of DMF plus 0.3 mL of DI water, the as-synthesized MEAA-NPs pellet was dispersed and 50 mg dextran ligand was added. Then the mixture was stirred at 70 °C for 8 hours and precipitated by adding 5 mL of acetone. Centrifugation at 5800 RPM for 3 min yielded a clear supernatant, which was discarded. The pellet was then dispersed in 1 mL of phosphate buffer saline (PBS) 1X and sonicated (Branson 3510) for 20 min. The sample was further purified using a dialysis filter (NMWL = 50 kDa, 1 time) in order to wash away excess dextran ligand.Dynamic light scattering and pH stability measurements
Light-scattering analysis was performed using a Malvern Instruments Nano-ZS90. pH was varied from 6.0 to 8.5 with 0.5 increments with all NP samples at the same concentration. Each autocorrelation function (ACF) was acquired for ∼10 s, and averaged for ∼2 min per measurement. Hydrodynamic diameters were obtained from a volume-weighted size distribution data analysis and reported as the average of ten measurements (error bars in Fig. S1b (ESI†) were the standard deviations of the values given by the ten parallel measurements).
Encapsulation of iron oxide NPs into PEG-lipid micelles.
Ethanol was added into 25 μL of NPs in hexane stock solution up to the point of turbidity. Centrifugation and decantation yielded ∼1 mg of dry pellet, to which 0.5 mL of chloroform and 375 μL of PEG-lipid in chloroform (25 mg mL−1) were added. Vortexing at 2000 RPM for 1 min yielded a homogeneous solution, which was dried in vacuo. The pellet was then dispersed in 2 mL PBS 1X and ultra-sonicated (Microson ultrasonic cell disruptor) for 5 min. The sample was further purified using a dialysis filter (NMWL = 50 kDa, 1 time).Iron determination
Iron standards (1000 μg mL−1, Ultra Scientific) were separately diluted to 2 μg mL−1, 1.6 μg mL−1, 0.8 μg mL−1, 0.4 μg mL−1, 0.2 μg mL−1, and 0.04 μg mL−1 by using volumetric flasks. The above standards and blank (DI water) were transferred into a 96-well plate. Acetate buffer (2 mol L−1, pH = 4.8, with 10% ascorbic acid) and bathophenanthroline (BPT) were then added in succession. Pink-red color, from the formation of BPT–iron complex, gradually developed and was allowed to saturate before the absorbance of each well was measured at 540 nm by a plate reader (BioTek, SYNERGY 4). A calibration curve (Fig. 2d) was determined from the average of three parallel measurements, yielding the linear fit Abs = 0.05178 + 0.2887 × CFe (R2 = 0.998), where Abs is the absorbance at 540 nm and CFe is the iron concentration in μg mL−1. Hydrophobic NPs, MEAA-coated NPs, and TD/ZDS-NPs were dried in vacuo and then digested by 6 mol L−1 hydrochloric acid. Iron determination of NPs followed the same protocol as that of iron standards and the results were averaged from three parallel measurements.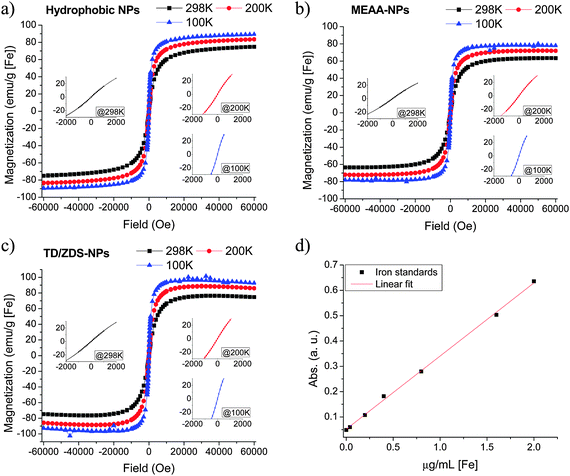 | ||
| Fig. 2 SQUID curves of (a) hydrophobic NPs, (b) MEAA-NPs, and (c) TD/ZDS-NPs (inset: magnified SQUID curves near zero field); (d) calibration curve of iron determination. | ||
Cellular uptake into HeLa cells
HeLa cells were obtained from American Type Culture Association. The cells were cultured in full medium (DMEM medium supplemented with 10% fetal bovine serum and 100 IU mL−1 penicillin–streptomycin). The cell density was determined using a hemocytometer prior to any experiments, after which, approximately 10 million cells were washed in full medium, centrifuged at 1000 RPM for 5 min, and then redispersed in 10 mL pre-warmed full medium. For each assay, freshly prepared HeLa cell suspensions were used with a density of ∼106 cells per mL. A BD Falcon 12-well transparent assay plate (Fisher Scientific, Pittsburgh, PA) was utilized with an 18 mm pre-sterilized glass slide in each well. An aliquot of 800 μL pre-warmed full medium was added to each well. Afterwards, 200 μL of the HeLa cell suspension (approximately 0.2 million cells) was added to each well. The 12-well transparent assay plate was then incubated at 37 °C for 5 min, followed by the addition of iron oxide NP in full medium solution to yield the specified concentrations. The iron oxide NP solution was then mixed gently with the cell suspension by pipet tips before the cells were cultured in a cell incubator for 24 hours (37 °C, 5% CO2). Each well of HeLa cells was washed using 1 mL PBS 1X three times and then fixed at room temperature for 30 min using 1 mL 2% paraformaldehyde. A Prussian blue iron-staining solution was freshly prepared by mixing equal volume of 2% hydrochloric acid aqueous solution and 2% potassium ferrocyanide(II) trihydrate. The intracellular iron content of the fixed HeLa cells was stained by incubation with 1 mL Prussian blue iron-staining solution at 37 °C for 30 min before the cells were washed with 1 mL PBS 1X (twice). The micro cover glass slips bearing HeLa cells were mounted onto microscope slides separately, dried at room temperature, and finally imaged by a Carl Zeiss microscope. A slightly blue background was shown in Fig. 3a, presumably caused by the small amount of residual Fe-containing salts which can form from FeCl3 in the cell media.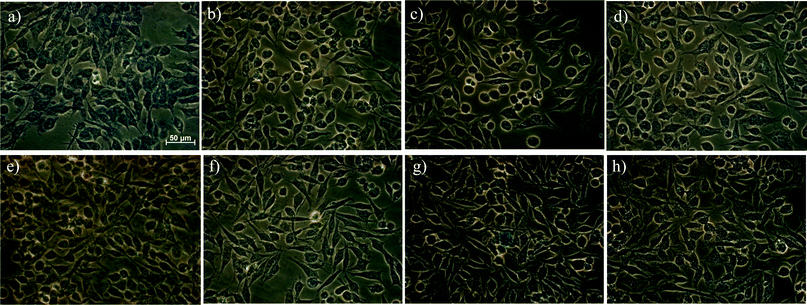 | ||
| Fig. 3 Iron uptake into HeLa cells determined using Prussian blue after 24 hours incubation: (a) FeCl3 (400 μg mL−1), (b) control, (c) Dex-NPs (400 μg mL−1), (d) Dex-NPs (40 μg mL−1), (e) ZDS-NPs (400 μg mL−1), (f) ZDS-NPs (40 μg mL−1), (g) TD/ZDS-NPs (400 μg mL−1), (h) TD/ZDS-NPs (40 μg mL−1). | ||
In vivo stability test
Male FVB mice were purchased from Charles River Laboratories International, Inc., and housed in an AAALAC-accredited facility in the Division of Comparative Medicine at MIT. All mice were studied according to an approved institutional protocol. Mice were anesthetized by intraperitoneal injection of a mixture of Ketamine and Xylazine. For in vivo NP stability tests, different NPs in PBS 1X solution were injected through the tail vein. After 10 min, blood was taken by cardiac puncture. For serum formation, the blood was left for 15 min at room temperature to coagulate, followed by centrifugation at 3000 RPM for 6 min. The supernatants (i.e. serums) were collected and further centrifuged at 13000 RPM for 10 min before they were separately injected into a SuperoseTM 6 (GE Healthcare, 10/300 GL) size-exclusion column via a gel-filtration chromatography machine (Amersham Biosciences, AKTAprime plus). With a flow rate of 0.5 mL min−1 PBS 1X, the serums were eluted and their fractions were collected at 1 min intervals within the retention time from 15 min to 53 min. To each fraction (0.5 mL), 50 μL of concentrated hydrochloric acid (∼12 mol L−1) was added, and the mixture was incubated at 60 °C for half an hour in order to digest iron oxide NPs. Centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM for 10 min yielded a clear supernatant, which was collected (white precipitates, presumably acidified serum proteins, were discarded). In order to determine the iron concentration of each fraction, 100 μL of the supernatant was transferred to a 96-well plate, where 100 μL of acetate buffer (2 mol L−1, pH = 4.8) and 50 μL of bathophenanthroline (BPT, 1 mg mL−1) were then added in succession. After the pink-red color developed and saturated, the absorbance of each well was measured by a plate reader (BioTek, SYNERGY 4) at 540 nm. The small iron concentration peak of serum from control mouse in Fig. 4b is presumably caused by iron-containing proteins in the mouse serum.
000 RPM for 10 min yielded a clear supernatant, which was collected (white precipitates, presumably acidified serum proteins, were discarded). In order to determine the iron concentration of each fraction, 100 μL of the supernatant was transferred to a 96-well plate, where 100 μL of acetate buffer (2 mol L−1, pH = 4.8) and 50 μL of bathophenanthroline (BPT, 1 mg mL−1) were then added in succession. After the pink-red color developed and saturated, the absorbance of each well was measured by a plate reader (BioTek, SYNERGY 4) at 540 nm. The small iron concentration peak of serum from control mouse in Fig. 4b is presumably caused by iron-containing proteins in the mouse serum.
 | ||
| Fig. 4 (a) Schematic of in vivo stability test (see Materials and Methods), (b) in vivo stability chromatograms of NPs injected into mice, and (c) in vivo stability chromatograms of NPs injected into mice and NPs in PBS 1X (data in panel b are included in panel c, while in panel b the data are not normalized and in panel c the data are normalized by area). | ||
Results and discussion
Hydrophobic iron oxide NPs were synthesized from the thermal decomposition of Fe(CO)5 in a mixture of 1-octadecene (ODE) and 1-hexadecene (HDE) solvent in the presence of native oleic acid ligands and trimethylamine N-oxide oxidizing reagent.2,8 Different sizes of monodisperse NPs can be produced by adjusting the solvent boiling point, keeping both the concentration of Fe(CO)5 and oleic acid constant as well as the growth time.9 Changing the boiling point is achieved by changing the volume ratio of ODE to HDE. For the NPs used in this study, a 2:5 (by volume) mixture of ODE:HDE was used. Transmission electron microscopy (TEM) shows that the NPs are monodisperse (Fig. 1a) with an inorganic particle diameter of 5.3 ± 0.3 nm (Fig. 1b). After the NPs were ligand-exchanged from oleic acid to zwitterionic dopamine sulfonate (ZDS), dynamic light scattering (DLS) measurement reveals that the ZDS-coated iron oxide NPs (ZDS-NPs) in aqueous buffer saline have a narrow size distribution (inset of Fig. S1a, ESI†) with a hydrodynamic diameter (HD) of ∼9.5 nm at pH = 7.5, indicating that the ZDS ligand contributes ∼2 nm to the overall radius. Moreover, the HD of ZDS-NPs is insensitive to pH ranging from 6.0 to 8.5, indicating good colloidal stability over physiological pHs (Fig. S1b, ESI†). The size change induced by the ZDS ligand and the pH stability is consistent with our prior study;6 when combined with the recent advance in the synthesis of uniform and extremely small-sized iron oxide nanoparticles,10 ZDS-coated iron oxide NPs would have the potentials to meet the requirement of HD for renal elimination.5 Moreover, for imaging and sensing purposes, it is also desirable to minimize the thickness of the ligand shell in order that magnetic resonance signals from the inorganic core of iron oxide NPs can be maximized under the same hydrodynamic diameter, which is usually determined by the size limits for successful penetration and targeting in confined spaces.11
In order to characterize the magnetic behavior and saturation magnetization of as-synthesized SPIONs, the SQUID curves of hydrophobic NPs were measured at four different temperatures with the magnetic field ranging from −6 to 6 T.12 After determining the mass of iron in the hydrophobic NPs (see Materials and Methods), the saturation magnetization (Ms) of hydrophobic NPs at room temperature was found to be ∼74 emu g−1 [Fe] (Fig. 2a), which compares reasonably well with the bulk value for maghemite of 106 emu g−1 [Fe] (i.e. 74 emu g−1 [Fe2O3]).13 Moreover, the inset of Fig. 2a shows that there are no hysteresis loops at zero field for hydrophobic NPs measured at 298 K, 200 K, or 100 K, indicating that the hydrophobic NPs are superparamagnetic at these temperatures.14 In contrast, for hydrophobic NPs measured at 5 K, a hysteresis loop is observed (inset of Fig. S2a, ESI†). This is consistent with the blocking temperature of maghemite NPs, which is ∼30 K for 5.5 nm maghemite NPs.15 To further study the influence of ligand exchange and purification processes on Ms, we also performed SQUID measurements on 2-[2-(2-methoxyethoxy)ethoxy]acetic acid (MEAA) coated iron oxide NPs (MEAA-NPs) and TD/ZDS-coated iron oxide NPs (TD/ZDS-NPs). Interestingly, the Ms of MEAA-NPs and TD/ZDS-NPs at room temperature were found to be ∼63 and ∼74 emu g−1 [Fe] (Fig. 2b and c), respectively, which were close to the Ms value of hydrophobic NPs. These results therefore suggest that the Ms of as-synthesized SPIONs are largely insensitive to the heating, precipitation, sonication, and dialysis steps which were involved in our ligand exchange and purification processes.16 In addition, these results indicate that our newly designed TD/ZDS ligand coating is not likely to significantly change the magnetic behavior of as-synthesized SPIONs, which makes TD/ZDS a promising ligand coating for SPIONs used in potential MRI applications.10,17
As ZDS-NPs and TD/ZDS-NPs are newly developed SPION systems, it is important to evaluate their non-specific uptake into cancer cells before they can potentially be used for specific targeting and imaging applications when functionalized by antibodies, peptides, or aptamers.18 Therefore, we investigated the cellular uptake of ZDS-NPs and TD/ZDS-NPs into HeLa cells in a period of 24 hours and compared the results with that of FeCl3 and a well-established SPION system, i.e. dextran-coated iron oxide NPs (Dex-NPs).19 After iron staining by Prussian blue, Fig. 3a shows blue intracellular signal detected for HeLa cells incubated with 400 μg mL−1 FeCl3, indicating intracellular Fe. In contrast, control HeLa cells that were not incubated with any iron source show minimal intracellular Fe (Fig. 3b). Similarly, HeLa cells incubated with Dex-NPs, ZDS-NPs, and TD/ZDS-NPs, also show minimal intracellular Fe (Fig. 3c–h). The only exception is the blue ∼20 μm spot in the middle of Fig. 3g, presumably caused by aggregation of cross-linked TD/ZDS-NPs, which have free thiol groups on their surface.20 Generally, the non-specific cellular uptake of Dex-NPs, ZDS-NPs, or TD/ZDS-NPs was much less than that of FeCl3. More importantly, although ZDS (MW: ∼300 g mol−1) and TD (MW: ∼850 g mol−1) ligands are relatively small compared to dextran (MW: ∼6000 g mol−1), no significant differences between Dex-NPs, ZDS-NPs, and TD/ZDS-NPs were found in terms of non-specific cellular uptake into HeLa cells. As Dex-NPs have served as a well-established SPION platform for molecular imaging, diagnostics, and therapeutics,21 ZDS-NPs and TD/ZDS-NPs with low non-specific cellular uptake also have the potential to be used for specific targeting and imaging studies.22
To further study the in vivo imaging potential of ZDS-NPs, we performed an in vivo stability test, which compared the sizes of NPs injected in mice to those incubated with PBS 1X (see Materials and Methods). We used gel-filtration chromatography with a size-exclusion column, in which the retention times of NPs have an inverse relationship with the sizes of NPs23 to fractionize serum and/or NPs. Iron determination was then performed on each fraction using bathophenanthroline (BPT). The chromatograms of serum and/or NPs were then plotted as iron concentration (IC) versus retention time. The non-normalized data in Fig. 4b shows that the peak intensity of IC in serum from sample mice (injected with NPs) is about an order higher than that from the control mouse, indicating that BPT-based iron determination serves as a good method for analyzing the size distribution of NPs after incubation in vivo. After data normalization, Fig. 4c shows that in PBS 1X both iron oxide NPs encapsulated in PEG-lipid micelles (PEG-NPs, Fig. 4c, blue lines) and ZDS-NPs (Fig. 4c, pink lines) are nearly monodisperse with retention times of 27 and 35 min, respectively. The size of PEG-NPs injected in a mouse was almost the same as the size of PEG-NPs in PBS 1X (Fig. 4c, black lines), showing a nearly monodisperse distribution with a peak at 27 min. The size distribution of ZDS-NPs injected in a mouse slightly shifted to a new peak at 32 min (Fig. 4c, cyan lines), which is only a 3 min different from that of ZDS-NPs in PBS 1X, and corresponds to a size increase of ∼4 nm in diameter.24 As expected PEG-NPs show minimal non-specific affinity toward serum proteins.25 These data also show that ZDS-NPs have low non-specific interactions with serum proteins in vivo and consequently that they are suitable for in vivo experiments, which is consistent with our prior in vitro results.6 The small size of ZDS ligands (MW: ∼300 g mol−1) compared to PEG-lipid ligands (MW: ∼2800 g mol−1) ensures that ZDS-NPs have a significantly reduced size compared to PEG-NPs (Fig. 4c).
Conclusion
In conclusion, we have investigated the magnetic behavior as well as the in vitro and in vivo non-specific interactions of compact zwitterionic dopamine sulfonate (ZDS) ligand coated superparamagnetic iron oxide nanoparticles. We find that the TD and ZDS ligand does not change the superparamagnetism and Ms (74 emu g−1 [Fe]) of hydrophobic NPs; moreover, ZDS-coated NPs have a small non-specific cellular uptake into HeLa cells after 24 hours incubation, which is similar to dextran-coated NPs. In vivo stability tests show low non-specific binding affinity for ZDS-coated NPs toward serum proteins and a smaller hydrodynamic size than NPs encapsulated in PEG-lipid micelles. These properties render ZDS-NPs and TD/ZDS-NPs suitable for in vitro and in vivo applications, where peptides, aptamers, or proteins could be conjugated for imaging and sensing,26 and when combined with an alternating magnetic field, the unaltered Ms of ligand exchanged NPs (compared to hydrophobic NPs) could maximize the magneto-thermal effect of the as-synthesized hydrophobic NPs and thus enable ZDS-NPs and TD/ZDS-NPs to serve as a powerful sensitizer for hyperthermia treatment.27Acknowledgements
This work was supported by the MIT-Harvard NIH Center for Cancer Nanotechnology Excellence Grant (1U54-CA119349), an NIH National Cancer Institute Grant (R01-CA126642), the ARO through the Institute for Soldier Nanotechnologies (W911NF-07-D-0004), and the NSF through a Collaborative Research in Chemistry Program (CHE-0714189). O.T.B. receives an EMBO Longterm Fellowship. We thank Yunfei Zhang for her contribution in cellular uptake experiments and Dr Dorthe M. Eisele as well as Dr Cliff R. Wong for thoughtful discussions. H.W. thanks Dr Yong Zhang for his assistance with acquiring TEM images and Dr Shaoyan Chu for discussions on SQUID measurements.References
- M. G. Harisinghani, J. Barentsz, P. F. Hahn, W. M. Deserno, S. Tabatabaei, C. H. van de Kaa, J. de la Rosette and R. Weissleder, N. Engl. J. Med., 2003, 348, 2491 CrossRef.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. Bin Na, J. Am. Chem. Soc., 2001, 123, 12798 CrossRef CAS.
- (a) N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575 RSC; (b) Y. W. Jun, J. H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122 CrossRef CAS; (c) A. H. Latham and M. E. Williams, Acc. Chem. Res., 2008, 41, 411 CrossRef CAS.
- (a) C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938 CrossRef CAS; (b) E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042 CrossRef CAS; (c) S. Peng, C. Wang, J. Xie and S. H. Sun, J. Am. Chem. Soc., 2006, 128, 10676 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Kandapallil, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS.
- H. Wei, N. Insin, J. Lee, H. S. Han, J. M. Cordero, W. H. Liu and M. G. Bawendi, Nano Lett., 2012, 12, 22 CrossRef CAS.
- N. Insin, J. B. Tracy, H. Lee, J. P. Zimmer, R. M. Westervelt and M. G. Bawendi, ACS Nano, 2008, 2, 197 CrossRef CAS.
- K. Woo, J. Hong, S. Choi, H. W. Lee, J. P. Ahn, C. S. Kim and S. W. Lee, Chem. Mater., 2004, 16, 2814 CrossRef CAS.
- T. D. Schladt, T. Graf and W. Tremel, Chem. Mater., 2009, 21, 3183 CrossRef CAS.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624 CrossRef CAS.
- M. Howarth, K. Takao, Y. Hayashi and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7583 CrossRef CAS.
- B. Chertok, B. A. Moffat, A. E. David, F. Q. Yu, C. Bergemann, B. D. Ross and V. C. Yang, Biomaterials, 2008, 29, 487 CrossRef CAS.
- C. J. Serna and M. P. Morales, Surf. Colloid Sci., 2004, 17, 27 CrossRef CAS.
- (a) C. R. Lin, R. K. Chiang, J. S. Wang and T. W. Sung, J. Appl. Phys., 2006, 99, 08N710 CrossRef; (b) D. J. Zhang, K. J. Klabunde, C. M. Sorensen and G. C. Hadjipanayis, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 14167 CrossRef CAS.
- M. J. Martinez-Perez, R. de Miguel, C. Carbonera, M. Martinez-Julvez, A. Lostao, C. Piquer, C. Gomez-Moreno, J. Bartolome and F. Luis, Nanotechnology, 2010, 21, 465707 CrossRef CAS.
- W. Wu, Q. G. He and C. Z. Jiang, Nanoscale Res. Lett., 2008, 3, 397 CrossRef CAS.
- (a) J. S. Choi, J. H. Lee, T. H. Shin, H. T. Song, E. Y. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015 CrossRef CAS; (b) O. T. Bruns, H. Ittrich, K. Peldschus, M. G. Kaul, U. I. Tromsdorf, J. Lauterwasser, M. S. Nikolic, B. Mollwitz, M. Merkell, N. C. Bigall, S. Sapra, R. Reimer, H. Hohenberg, H. Weller, A. Eychmuller, G. Adam, U. Beisiegel and J. Heeren, Nat. Nanotechnol., 2009, 4, 193 CrossRef CAS; (c) B. Freund, U. I. Tromsdorf, O. T. Bruns, M. Heine, A. Giemsa, A. Bartelt, S. C. Salmen, N. Raabe, J. Heeren, H. Ittrich, R. Reimer, H. Hohenberg, U. Schumacher, H. Weller and P. Nielsen, ACS Nano, 2012, 6, 7318 CrossRef CAS.
- (a) J. H. Gao, H. W. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097 CrossRef CAS; (b) D. Yoo, J. H. Lee, T. H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863 CrossRef CAS.
- C. Tassa, S. Y. Shaw and R. Weissleder, Acc. Chem. Res., 2011, 44, 842 CrossRef CAS.
- W. H. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530 CrossRef CAS.
- K. A. Kelly, S. Y. Shaw, M. Nahrendorf, K. Kristoff, E. Aikawa, S. L. Schreiber, P. A. Clemons and R. Weissleder, Integr. Biol., 2009, 1, 311 RSC.
- (a) H. W. Gu, K. M. Xu, C. J. Xu and B. Xu, Chem. Commun., 2006, 941 RSC; (b) D. N. Ho, N. Kohler, A. Sigdel, R. Kalluri, J. R. Morgan, C. J. Xu and S. H. Sun, Theranostics, 2012, 2, 66 CrossRef CAS; (c) H. L. Karlsson, P. Cronholm, J. Gustafsson and L. Moller, Chem. Res. Toxicol., 2008, 21, 1726 CrossRef CAS.
- U. I. Tromsdorf, O. T. Bruns, S. C. Salmen, U. Beisiegel and H. Weller, Nano Lett., 2009, 9, 4434 CrossRef CAS.
- (a) H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS; (b) C. Wong, T. Stylianopoulos, J. A. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426 CrossRef CAS.
- (a) H. Ai, C. Flask, B. Weinberg, X. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. M. Gao, Adv. Mater., 2005, 17, 1949 CrossRef CAS; (b) S. Tong, S. J. Hou, B. B. Ren, Z. L. Zheng and G. Bao, Nano Lett., 2011, 11, 3720 CrossRef CAS; (c) W. J. M. Mulder, G. J. Strijkers, G. A. F. Van Tilborg, D. P. Cormode, Z. A. Fayad and K. Nicolay, Acc. Chem. Res., 2009, 42, 904 CrossRef CAS.
- (a) T. Atanasijevic, M. Shusteff, P. Fam and A. Jasanoff, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14707 CrossRef CAS; (b) N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6, 675 CrossRef CAS; (c) T. J. Yoon, H. Lee, H. L. Shao, S. A. Hilderbrand and R. Weissleder, Adv. Mater., 2011, 23, 4793 CrossRef CAS; (d) D. Shangguan, Y. Li, Z. W. Tang, Z. H. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. Y. J. Yang and W. H. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838 CrossRef CAS.
- J. H. Lee, J. T. Jang, J. S. Choi, S. H. Moon, S. H. Noh, J. W. Kim, J. G. Kim, I. S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ib20142a |
| This journal is © The Royal Society of Chemistry 2013 |