Common pitfalls in nanotechnology: lessons learned from NCI's Nanotechnology Characterization Laboratory
Rachael M.
Crist
,
Jennifer Hall
Grossman
,
Anil K.
Patri
,
Stephan T.
Stern
,
Marina A.
Dobrovolskaia
,
Pavan P.
Adiseshaiah
,
Jeffrey D.
Clogston
and
Scott E.
McNeil
*
Nanotechnology Characterization Laboratory, Advanced Technology Program, SAIC-Frederick, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD 21702. E-mail: ncl@mail.nih.gov
First published on 14th June 2012
Abstract
The Nanotechnology Characterization Laboratory's (NCL) unique set-up has allowed our lab to handle and test a variety of nanoparticle platforms intended for the delivery of cancer therapeutics and/or imaging contrast agents. Over the last six years, the NCL has characterized more than 250 different nanomaterials from more than 75 different investigators. These submitted nanomaterials stem from a range of backgrounds and experiences, including government, academia and industry. This has given the NCL a unique and valuable opportunity to observe trends in nanoparticle safety and biocompatibility, as well as note some of the common mistakes and oversights of nanoformulation. While not exhaustive, this article aims to share some of the most common pitfalls observed by the NCL as they relate to nanoparticle synthesis, purification, characterization and analysis.
Insight, innovation, integrationNanotechnology has the potential to radically change the way we treat cancer. While traditional chemotherapeutics typically deliver around 0.1% of the injected drug to the tumor site, nanoparticle therapies have the potential to deliver greater amounts to the tumor by the proposed enhanced permeation and retention effect while minimizing toxicity. However, due to the many nuances and intricacies of formulating nanoparticles and understanding their in vivo profiles, few nanotherapies have reached the clinical market. This article aims to improve researchers' understanding of development and formulation of nanoparticles by divulging some of the common oversights made in nanotherapy development. |
Introduction
The promise of cancer nanotechnology–increased treatment efficacy with decreased toxicities and side effects–has triggered a huge interest in the field and an outpouring of research projects aimed at generating the newest and most cutting-edge therapies. Unfortunately, the science behind nanotechnology is not always as straightforward as it can be for small molecules, and researchers are still learning more about the intricacies of nanoscience every day. The National Cancer Institute's Nanotechnology Characterization Laboratory (NCL), founded in 2004, has spent the last eight years studying nanomaterials of all types, and their related complexities, to increase understanding and educate nano-researchers worldwide in an effort towards developing better and more effective therapies.The NCL is part of the National Cancer Institute's Alliance for Nanotechnology in Cancer. As opposed to being a research organization focused on the development of just one nanoparticle platform, the NCL has had a unique opportunity to examine hundreds of different nanoparticles from nearly a hundred different sources (commercial manufacturers, academic labs, contract research organizations (CROs), large pharmaceutical companies, small biotech companies, and various government organizations). Via an application process, the NCL accepts nanoparticles of all platform types that have demonstrated a potential therapeutic or diagnostic benefit towards cancer; the majority of NCL's submissions are for intravenously administered cancer treatments. The NCL staff consists of a wide range of scientists with varying backgrounds to fully characterize and understand each nanomaterial. The submitted nanomaterials are then subjected to a range of experimental analyses including physicochemical characterization, assessment of batch-to-batch consistency and nanoparticle stability, sterility and endotoxin quantification, in vitro blood contact properties, in vitro cytotoxicity and immune cell function assays, and in vivo pharmacokinetics and toxicity profiling. The majority of assays run by the NCL have been tailored for intravenously administered formulations and have been shown to work for a variety of nanoparticle platforms. The bulk of these protocols have been published previously.1,2
To date, the NCL has characterized more than 250 different nanomaterials from more than 75 different investigators. It is from these experiences the NCL would like to share some of the more commonly observed pitfalls of nanoparticle formulation. Through the use of several case studies from the NCL, this article will highlight various aspects of nanoparticle synthesis, purification, characterization and analysis, discuss common missteps and oversights, and provide insight into how they can be avoided. While many of the examples that follow may seem trivial or obvious, it is likely due to the complexity of nanoformulation that they continue to be problematic. These issues are being brought into the public domain with the hope that researchers will be able minimize these missteps in their efforts to develop better formulations.
Sterility and endotoxin
One of the first screens the NCL performs on a nanomaterial submission is for sterility and endotoxin content. At an early stage of development, these can be less important; however, as the formulation progresses to the stage of preclinical assessment (the point at which the NCL usually becomes involved), where one begins to assess immunotoxicity and in vivo outcomes, sterility and endotoxin contamination become very important. Endotoxin, or lipopolysaccharide (LPS), is a component of the cell wall of Gram-negative bacteria and is omnipresent. High levels of endotoxin in a nanoformulation can cause immunostimulatory reactions and can mask the true biocompatibility of the formulation.3 The US Pharmacopeia has established limits on the amount of endotoxin allowed in intravenously-administered clinical formulations as 5 Endotoxin Units (EU)/kg body weight/h, and for those delivered intrathecally, the limit is 0.2 EU/kg/hour.4A recent snapshot of samples coming into the NCL for preclinical characterization revealed more than one-third had contamination. Of 75 samples (nanoparticles and their components) submitted to the NCL over a one year period, seven samples were contaminated with bacteria and 26 samples had high endotoxin levels which required purification or re-manufacture before proceeding with analysis. While there are various methods to remove endotoxin,5–8 it is generally recommended to take the necessary precautions to avoid endotoxin contamination altogether. This requires working under sterile conditions throughout the synthesis and purification procedure, as well as ensuring any reagents and materials used are sterile and endotoxin free. Many consumers assume their commercial reagents should be free of endotoxin; many commercial starting materials, however, often contain endotoxin. Another common misconception from many researchers is that water from their purification systems should be free of endotoxin; this too is often not the case. Endotoxin contamination can also reside in equipment used in the synthetic procedure, including tubing lines which are often hard to access. Some nanoparticles are also known to be very “sticky” with regards to endotoxin and can accumulate endotoxin throughout synthesis and processing.9 To help reduce endotoxin levels, Limulus amoebocyte lysate (LAL)-grade or pyrogen-free water can be substituted in buffers and dispersing media. Starting materials from commercial sources can be screened for endotoxin. Equipment can be tested for the presence of endotoxin by rinsing and testing the wash samples. Other simple approaches to reducing endotoxin levels during synthesis include working in biological safety cabinets instead of chemical fume hoods, depyrogenation of glassware, ensuring use of only sterile filters, and overall good aseptic technique.10
Endotoxin is generally measured by the Limulus amoebocyte lysate (LAL) assay. There are three different LAL variants: the chromogenic assay, the turbidity assay, and the gel-clot assay.11 Both the chromogenic and turbidity assays are also available in kinetic and end-point formats. Often times however, nanoparticles can interfere with the LAL assays, leading to ambiguous or deceptive results.12 Therefore, it is critical to always perform the appropriate inhibition and enhancement controls (IEC) when running LAL assays.13 Common types of interference include colored nanoformulations (e.g. fullerenes) which can interfere with the chromogenic LAL assay, turbid formulations (e.g. nanoemulsions) which can interfere with turbidity assay, and nanoparticles filtered with cellulose-based filters which can create-false positives. Cellulose-based filters contain beta-glucans which are also detected by the LAL assay. In the case of interference with one LAL assay, a second LAL method can generally be employed. NCL routinely applies two different LAL formats to each nanoparticle being evaluated for endotoxin contamination. The purpose of this screening is to estimate consistency in LAL findings from different formats as a measure of assay interference not otherwise detected. The decision tree for choosing an appropriate LAL format for a given nanoformulation has been described earlier.13 To account for false positives with cellulose-based filters, LAL assays can be run in the presence of Glucashield buffer, which negates the beta-glucan contribution. Alternatively, a recombinant Factor C assay can also distinguish between endotoxin and beta-glucan contamination.14 Other techniques to assess endotoxin include commercial fluorescent dyes, mass spectrometry, or other physical methods. Each of these alternative methods has certain advantages and limitations over LAL, the main limitation being these methods detect the physical presence of endotoxin rather than the biological activity. Since biological activity is responsible for toxicities experienced with endotoxin contamination, findings should be verified with functional tests such as the rabbit pyrogen test (RPT) or macrophage activation test. The in vivo rabbit pyrogen test confirms the presence of pyrogenic substrates including endotoxin, but is only a qualitative assay, is more expensive, and requires larger quantities of material. The macrophage activation test is a quantitative assay, but like the RPT it can respond to non-endotoxin pyrogens. Advantages and limitations for standard versus alternative methods of endotoxin detection, as well as challenges with endotoxin detection and quantification in engineered nanomaterials are reviewed in detail elsewhere.15
Physicochemical characterization
Without proper and adequate physicochemical characterization (PCC) of a nanoparticle, in vivo toxicity results may be misleading and ultimately meaningless. The NCL is often asked which assays are the most important in analyzing a nanomaterial. The answer, unfortunately, is not always straightforward, and what may apply to one formulation may not necessarily apply to another. There are, however, a few key parameters important for most any formulation: size/size distribution, shape, charge, composition, purity, and stability. How must one measure these parameters? That too depends on the formulation, the intended clinical route of administration, and a host of other factors. Unlike small molecule therapeutics, where one can use two or three techniques (such as nuclear magnetic resosnance (NMR), mass spectrometry (MS) and liquid chromatography (LC)) to determine the composition and purity, nanomaterials require more complex measurements and analyses in addition to the traditional methods to completely determine their attributes.The NCL has found that many researchers will use commercial materials at face value, i.e. use manufacturer specifications of a material without analyzing the material themselves. A recent study using commercially acquired silver nanoparticles revealed significant variability from the manufacturer-stated sizes.16 Dynamic light scattering (DLS) measurements of nominally 20, 40, 60, and 80 nm silver nanoparticles from a commercial source revealed average size values of 34, 38, 65, and 91 nm, respectively. Transmission electron microscopy (TEM) size measurements differed much more significantly; TEM-measured sizes were 36, 56, 78, and 107 nm, respectively. Measurement results and uncertainties vary depending upon the measurement technique and assay conditions, so it is important to inquire with the manufacturer as to their particular measurement techniques. Of course, not all manufacturers' reported values are necessarily inaccurate, but it is in the researchers' best interest to always characterize materials before proceeding with synthesis and more expensive functionalization and biological testing.
For the most relevant and clinically meaningful PCC data, characterization must be performed under biologically relevant conditions. There are several parameters, including size and charge, which vary with dispersing medium and the microenvironment. From a recent study, a gold colloid solution exhibited the reported nominal size when analyzed by DLS, TEM, or atomic force microscopy (AFM). However, when the particles were incubated with human plasma, the DLS-reported size nearly doubled, while TEM and AFM measurements yielded the same nominal sizes as measured in phosphate-buffered saline (PBS).17 This is an extremely important point, because as has been reported, changes in the size and charge of a particle could alter the material's biodistribution, toxicity, or immunological profile.18–20
Having a thorough understanding of the composition of your nanoformulation, as obvious as that may sound, is another critical point. Not knowing how much active pharmaceutical ingredient (API) is in a formulation, and how much of it is free versus encapsulated, will lead to dosing miscalculations and flawed estimates of the IC50 values in vitro and maximum tolerated dose (MTD) in vivo. Another common mistake observed in understanding a material's composition is erroneously assuming the presence, covalent attachment, and/or quantitation of surface ligands (functionalized groups, coatings, targeting moieties, etc.). These are not always straightforward questions to address. Many times protocols cannot simply be pulled from the literature, but rather assays have to be specifically developed for each formulation. Nevertheless, these are all critical parameters and deserve the extra attention at an early stage of nanoparticle characterization. It can be disheartening to realize after a multitude of failed experiments that your nanoformulation is not what you expected. A good case study illustrating this point involved the NCL attempting to quantify a targeting moiety on an iron oxide nanoparticle. In comparing the targeted and non-targeted formulations, both materials appeared practically identical in size when evaluated by DLS, TEM and AFM, and appeared to have identical (within the measurement errors) surface charges and iron content. None of these measures, however, are expected to necessarily differentiate the formulation with or without the targeting ligand. To more accurately assess targeting ligand concentration, the NCL requested a variant of the nanoparticle which used a radioactive isotope within the targeting ligand. A simple gel filtration analysis of this formulation, coupled with scintillation counting, was then able to prove the radioactive component (i.e., the targeting ligand) was not covalently attached to the nanoparticle. Analysis of the gel filtration fractions found the fractions containing the targeting ligand (as measured by scintillation counting) did not overlap with fractions containing nanoparticle (as measured by DLS; Fig. 1). Where other methods may fail to completely assess a particle's composition, radioactivity can be a valuable tool. And, when multiple components of a nanoformulation are separately labeled (i.e. the API and the vehicle platform are labeled with non-overlapping radiolabels), radioactivity can be used to assess the breakdown and biodistribution patterns of the individual components in vivo.21 A similar approach can be used with UV-Vis or other detection methods as appropriate, since radiolabeling is not necessarily a simple process for some and may require facilities, instrumentation and personnel to handle radiolabeled material.
 | ||
| Fig. 1 FPLC analysis of an iron oxide nanoparticle with a radioactively labeled targeting ligand. The formulation was injected into a gel filtration column and fractions were collected over 1 min (1 mL) intervals. Each fraction was then assessed for the presence of nanoparticles by DLS (red line) and for the presence of the targeting ligand by scintillation counting (blue line). There is minimal overlap of the two curves, suggesting the ligand dissociated from the nanoparticle and was not covalently attached. The use of radioactive labels can be a tremendously beneficial tool in assessing not only nanoparticle integrity in vitro, but also nanoparticle biodistribution in vivo. | ||
Residual manufacturing components
Related to proper and thorough PCC, one must consider the potential for residual manufacturing components, which may be toxic, in their nanoformulations. Various surfactants, peptizing agents, or other constituents are often used in the synthesis and purification of nanoparticles, but are not generally intended to be in the final product. Purification strategies that involve washing, buffer exchanges, extractions, or filtration steps are intended to remove these unwanted constituents but often are not 100% effective.Gold nanorods, for example, are often prepared using the cationic surfactant cetyltrimethylammonium bromide (CTAB) and purified using the anionic peptizing agent polystyrenesulfonate (PSS). CTAB is not a biologically compatible compound, and if not completely removed, the formulation can exhibit cytotoxic effects that may be erroneously attributed to the nanoparticle.22 In this example, a simple in vitro cytotoxicity assay was able to differentiate the cytotoxic component from the nanoparticle. Cell viability [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MTT] and membrane integrity assays (lactate dehydrogenase; LDH) revealed cytotoxicity of the total formulation. However, when the gold nanorods were separated from the buffer through a simple filtration step and re-analyzed, cytotoxicity was only seen in the buffer-containing supernatant fraction. The gold nanorods (in the pellet fraction) did not exhibit cytotoxicity (Fig. 2).23 The cytotoxicity was ultimately attributed to residual CTAB-PSS complexes in the formulation which were removed with additional exchanges using fresh PSS. This type of analysis is a quick and simple way to distinctly assess components in the buffer versus the nanoparticle itself, and has been used for a variety of nanoparticles, including separating potentially cytotoxic free silver ions (permeate) from silver nanoparticles (retentate).24
 | ||
| Fig. 2 Permeate/Retentate comparison by an in vitro cytotoxicity assay. The gold nanorod (GNR) solution exhibited in vitro cytotoxicity when assessed by the MTT assay in LLC-PK1 cells. In order to differentiate cytotoxicity stemming from the nanorods versus a component of the buffer, the GNR solution was filtered to separate the two species. Each was then re-assessed by the MTT assay, and all cytotoxicity was attributed to the buffer. | ||
Failing to thoroughly characterize a material has plagued the nanotechnology field for many years, oftentimes leading to misjudgments about the toxicities of nanoparticles. For example, several early studies with fullerene (C60) attributed the observed toxicities to the C60 itself.25 However, later studies attributed much of those toxicities to residual solvent used in the preparation of the fullerene samples.26 To properly attribute nanotoxicities, it is imperative that materials are free of toxic contaminants that might confound results.
Biocompatibility of components
While the above section discussed the consequences of using biologically non-compatible reagents in the synthesis and purification and incomplete removal of these reagents/residues prior to finalization of the nanoformulation, this section discusses the importance of using biologically compatible components as part of the formulation. With the rapidly evolving complexity of nanoparticles, it is important to consider the potential impact of every component within a formulation, including non-API components.Indeed, the NCL has seen examples of toxicity stemming from the non-API/non-essential components of a formulation. One example involved a liposome formulation encapsulating a nickel-chelated API. In vitro, both the Ni-only control liposome and Ni-API liposome showed similar trends in inducing cytotoxicity and IL-8 cytokine induction. In vivo, the Ni-only control liposome was more toxic than the free API and showed significant Ni-related toxicity (Fig. 3). With cancer drugs, one has to accept a certain degree of toxicity from the API, but NCL recommended trying alternative chelates (other than nickel) to reduce the toxicity of the entire formulation. Another example of toxicity stemming from a non-API component of a formulation was seen with folic acid-conjugated liposomes. In vitro, the folic acid-conjugated liposomes induced activation of the complement system to a level comparable to that seen with cobra venom factor, whereas the control, non-folic acid-conjugated liposome, did not appreciably activate the complement system. It was ultimately determined the linker used to conjugate folic acid to the liposome was the probable culprit of complement activation.
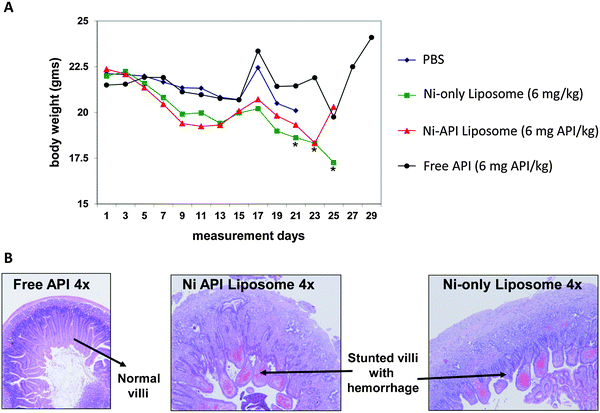 | ||
| Fig. 3 In vivo analysis reveals toxicity from Nickel, a non-API component of the formulation. In vivo analysis of the nanoformulation using an LS174T xenograft in seven week old athymic nu/nu female mice shows significant body weight loss for the Ni-only and Ni-API liposomes (A) and chronic necrotizing inflammation in the duodenum (B). Both analyses show the Ni-containing fractions are more toxic than the free API. *Significantly different from control p ≤ 0.05 (Dunnett test). | ||
In both examples above, the source of toxicity stemmed from a non-API component of the formulation. In the first example, nickel did not appear to be essential for the mechanism of therapeutic action. In fact, a survey of the literature shows precedence for nickel-related toxicity.27–32 An alternate metal chelate could have avoided this toxicity. In the second example, the linker serves no unique biological function either, and could be swapped out for a more biocompatible linker to reduce total toxicity. Several studies have been published which highlight immunocompatibility issues with linkers, including generation of anti-PEG antibodies in response to methoxyPEG exposure,33 complement activation in response to hydroxyPEG,34 and production of antipeptide-IgG antibodies in response to polyoxyethylene-based linkers.35
Batch-to-batch consistency
When considering production of nanomaterials for eventual use in the clinic, there must be a means of validating each production lot, i.e. assessing batch-to-batch consistency. Just as there was no simple answer as to which PCC methods to use for every nanoparticle, there is no panacea for batch-to-batch quality control analysis either. To properly assess batch-to-batch consistency, one must consider not only the PCC aspects of their formulation (size, aggregation, charge, etc.), but also biological activity, drug release rates, or a number of other factors all of which may be specific to each individual formulation. Identifying critical parameters that might affect the formulation early on and monitoring these parameters on multiple batches is a good practice and could minimize hassles later on.The NCL recently published a case study that highlighted issues observed with failed batch-to-batch consistency.36 The NCL had performed a variety of PCC and in vitro experiments, as well as in vivo toxicology studies on a PEGylated gold nanoparticle. Toxicology results from the first experiment (batch 1) showed severe inflammatory lung lesions, which, according to our collaborator (the developer of the nanomedicine), was unexpected and inconsistent with their previous results. The NCL repeated the toxicology study using another batch of material (batch 2); these results showed only minimal to mild lung inflammation. As a first assessment, the NCL compared the batches' size by dynamic light scattering and transmission electron microscopy, and surface charge by zeta potential measurement. However, there were no discernible differences between the batches revealed by these analyses. A more in depth analysis eventually revealed a difference in the degree of PEG coating on each batch: batch 1 showed a greater degree of plasma protein binding as compared to batch 2 when analyzed by 2-D polyacrylamide gel electrophoresis (PAGE), suggesting the PEG was dissociating from the particle over time. The hydrophilicity, i.e. the density of the PEG coating on a particle, can be critical as it will influence both biodistribution and toxicity profiles.
Nanoparticles will inherently have some degree of polydispersity associated with them, whether that is in terms of size, aggregation, biological activity, etc. This makes it important to thoroughly assess all parameters on every batch of material made. This not only will allow for an assessment of batch-to-batch variability, but will also afford a range of acceptable measures for lot release.
Nanoparticle in vivo stability
As noted above, nanoparticle stability is important in regulating biodistribution and toxicity; however, finding the appropriate measures to assess stability is not always straightforward. As was the case in the previous example, particle stability cannot always be assessed by the simple measures of size and charge. For assessment of the formulation discussed above, an assay had to be optimized in which nanoparticles were separated from the dispersing medium and PEG concentration was monitored in the supernatant fraction, i.e. to assess dissociation of PEG from the nanoparticle.Another NCL project involving a nanoemulsion platform which encapsulated a hemolytic peptide also serves as a good example of the importance of monitoring nanoparticle stability. When intact, the nanoparticle shielded the peptide, and prevented severe toxicities. However, when the nanoemulsion was added to whole blood, the particle rapidly disassociated, releasing the peptide, which in turn caused severe hemolysis (Fig. 4). This translated to almost immediate death (<10 min) for mice injected with the peptide-loaded nanoemulsion. In this case, particle stability as assessed by size monitoring in buffer, did not foretell the dramatic instability of the formulation in whole blood. This again highlights the importance of finding the appropriate assay for each individual formulation. An assay to monitor stability under biologically relevant conditions would have predicted this instability and could have saved time, money, and animal lives lost in performing the costly, failed in vivo studies.
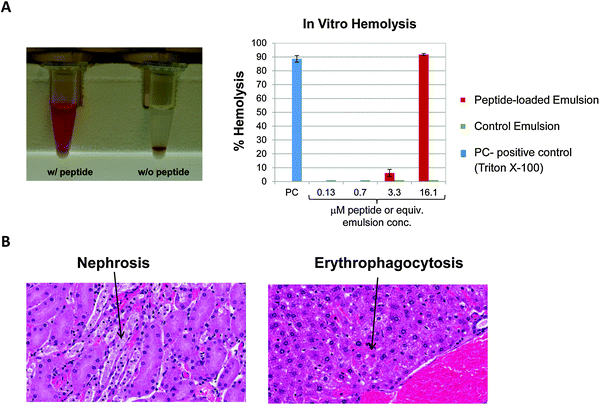 | ||
| Fig. 4 Nanoparticle instability under physiologically relevant conditions. (A) In vitro, the particle precipitated almost immediately upon introduction into whole blood and showed significant in vitro hemolysis (>80%). The nanoparticle can be seen as a white pellet in the bottom of the tube, and the red supernatant indicates the presence of plasma-free hemoglobin. (B) In vivo significant nephrosis and erythophagocytosis was observed, culminating in shock which resulted in almost immediate death for animals. | ||
Drug release rates
The benefit offered by some platforms used for solubilization/formulation purposes only, to minimize excipient related toxicity (such as Abraxane37,38), are valid. In these cases, the drug may be released immediately upon administration. Many nanoformulations, however, use the carrier platform to alter the biodistribution of the drug to minimize target organ toxicity,39 improve bioavailability, and increase drug release at the intended site of action. In these cases, drug release rates are very important and often relate to the overall in vivo safety, efficacy, and stability of a nanoformulation. The nanoformulation should be optimized such that the API is not released prematurely, before reaching the target site, or released too slowly such that it does not produce an effect on tumors. As outlined in a recent publication,40 there are two techniques employed most often in assessing nanoparticle stability and drug release in vivo. Dual radioactive labels, where both the nanoparticle platform and the drug component are labeled with different radioisotopes, can be used to track the biodistribution of each of these components within blood and tissues. If radioactivity is undesired or unachievable, complimentary chemical or biological assays to monitor both platform and drug components can offer the same evaluation. For example, the NCL employed complimentary analysis techniques to monitor the in vivo profile of a TNF-conjugated gold nanoparticle. For this formulation, the TNF could be monitored by an enzyme-linked immunosorbent assay (ELISA), while gold concentration was monitored by inductively coupled plasma-mass spectrometry (ICP-MS).40Often times, the rate of drug release is not assessed until in vivo studies for pharmacokinetics are conducted. In vitro assays which monitor drug release in whole blood can be predictive of in vivo stability, and can be done for a fraction of the cost and time of in vivo studies;21,41 however, one must be cautious not to make the assumption that in vitro results will directly translate to in vivo outcomes.
Conclusion
Nanotechnology is a complex science, and nanoparticles are rapidly evolving to more complex formulations that incorporate not only drugs, but also hydrophilic coatings, targeting ligands, antibodies, imaging agents and more. And, the expertise required to successfully generate an efficacious nanomedicine is also escalating. Not only must one understand the chemistry for initial formulation of the particle, but also have the knowledge to fully comprehend its biodistribution, metabolism and pharmacokinetics, toxicological profile and immunological consequences. The NCL was established to help researchers fill these gaps and educate the research community on proper and thorough characterization of nanomaterials. After more than 250 different nanoparticle analyses, the NCL has prepared this article to highlight some of the more common missteps in nanoparticle design. These cautionary notes include:(1) Sterility and endotoxin contamination must be addressed before investigation of immunological properties or in vivo studies begin. As many nanoparticles can interfere with the traditional LAL assay, be sure to run all appropriate controls when assessing endotoxin concentration.
(2) Always thoroughly characterize a nanoparticle, and do so under biologically relevant conditions. Never assume a manufacturer's specifications are the full story.
(3) Consider the biological consequence of every component that is used in the manufacture and purification process. If a non-biologically compatible reagent must be used during synthesis or purification, develop an appropriate assay to assess its presence after the removal process.
(4) Screen for the biocompatibility of every component, including linkers and non-API components that will be formulated into the nanoparticle. If nontoxic or more biocompatible non-API components are available, use them.
(5) Assess batch-to-batch consistency at an early stage. If batch-to-batch consistency is unresolved, results from assays using multiple lots of material may be misleading and erroneous.
(6) Find an appropriate assay to assess nanoparticle stability under biologically relevant conditions.
(7) Consider the drug release rate of a particle before delving into costly animal studies. In vitro assays may be predictive, but they do not always correlate with in vivo results. A properly designed small scale in vivo study to monitor nanoparticle stability and drug release rates can be beneficial with respect to both time and money.
This is by no means meant to be an exhaustive set of everything a nanomedicine developer needs to consider, but merely a representation of some of the most common issues encountered by the NCL. It is our hope that researchers will be able to use this information and prevent these same costly and time consuming miscalculations in their own research. After all, we are all working towards the same goal—improved therapies through nanotechnology.
Acknowledgements
The NCL would like to thank the technical staff for assistance with all of the projects highlighted in the article: Chris McLeland, Tim Potter, Barry Neun, Sarah Skoczen, Matt Hansen, Sonny Man, and Jamie Rodriguez. The NCL also appreciates the support of SAIC-Frederick, Inc.'s Electron Microscopy Lab and Laboratory Animal Sciences Program. Finally, the NCL would like to express our gratitude to all the collaborators submitting nanoparticles to the NCL. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.References
- Available from http://ncl.cancer.gov.
- Characterization of nanoparticles intended for drug delivery, in Methods in Molecular Biology, ed. S. McNeil, Humana Press, 2011, vol. 697 Search PubMed.
- Endotoxin in Health and Disease, ed. H. Brade, S. M. Opal, S. N. Vogel and D. C. Morrison, Marcel Dekker, Inc., New York, 1999 Search PubMed.
- Standard Procedure 〈85〉 Bacterial Endotoxins Test, 2007, The United States Pharmacopeial Convention, Rockville, MD 20852, USA Search PubMed.
- G. Csako, R. J. Elin, H. D. Hochstein and C. M. Tsai, Physical and biological properties of U.S. standard endotoxin EC after exposure to ionizing radiation, Infect. Immun., 1983, 41, 190–196 CAS.
- P. O. Magalhaes, A. M. Lopes, P. G. Mazzola, C. Rangel-Yagui, T. C. Penna and A. Pessoa, Jr., Methods of endotoxin removal from biological preparations: a review, J. Pharm. Pharm. Sci., 2007, 10, 388–404 Search PubMed.
- D. P. Nerkar, L. G. Govekar, U. S. Kumta and A. Sreenivasan, Radiation induced alterations in the endotoxin of S. typhimurium, Int. J. Radiat. Biol., 1977, 32, 259–266 CrossRef CAS.
- D. Petsch and F. B. Anspach, Endotoxin removal from protein solutions, J. Biotechnol., 2000, 76, 97–119 CrossRef CAS.
- R. Darkow, T. Groth, W. Albrecht, K. Lutzow and D. Paul, Functionalized nanoparticles for endotoxin binding in aqueous solutions, Biomaterials, 1999, 20, 1277–1283 CrossRef CAS.
- R. J. Coté, in Current Protocols in Cell Biology, John Wiley & Sons, Inc., 2001 Search PubMed.
- B. W. Neun and M. A. Dobrovolskaia, Detection and quantitative evaluation of endotoxin contamination in nanoparticle formulations by LAL-based assays, in Characterization of nanoparticles intended for drug delivery, ed. S. McNeil, Humana Press, 2011, vol. 697, pp. 121–130 Search PubMed.
- M. A. Dobrovolskaia, B. W. Neun, J. D. Clogston, H. Ding, J. Ljubimova and S. E. McNeil, Ambiguities in applying traditional Limulus Amoebocyte Lysate tests to quantify endotoxin in nanoparticle formulations, Nanomedicine, 2010, 5, 555–562 CrossRef CAS.
- M. A. Dobrovolskaia, D. R. Germolec and J. L. Weaver, Evaluation of nanoparticle immunotoxicity, Nat. Nanotechnol., 2009, 4, 411–414 CrossRef CAS.
- K. U. Alwis and D. K. Milton, Recombinant factor C assay for measuring endotoxin in house dust: comparison with LAL, and (1–>3)-beta-D-glucans, Am. J. Ind. Med., 2006, 49, 296–300 CrossRef CAS.
- M. A. Dobrovolskaia and S. E. McNeil, Endotoxin and Engineered Nanomaterials in Handbook of Immunological Properties of Engineering Nanomaterials, World Scientific Publishing Co., in press, est. winter, 2012 Search PubMed.
- J. Zheng, J. Clogston, A. Patri, M. Dobrovolskaia and S. McNeil, Sterilization of silver nanoparticles using standard gamma irradiation procedure affects particle integrity and biocompatibility, J. Nanomedic. Nanotechnol., 2011, S5, 001 Search PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Interaction of colloidal gold nanoparticles with human blood: effects on particle size and analysis of plasma protein binding profiles, Nanomed.: Nanotechnol., Biol. Med., 2009, 5, 106–117 CrossRef CAS.
- M. A. Dobrovolskaia, A. K. Patri, T. M. Potter, J. C. Rodriguez, J. B. Hall and S. E. McNeil, Dendrimer-induced leukocyte procoagulant activity depends on particle size and surface charge, Nanomedicine, 2012, 7, 245–256 CrossRef CAS.
- M. A. Dobrovolskaia, A. K. Patri, J. Simak, J. B. Hall, J. Semberova, S. H. De Paoli Lacerda and S. E. McNeil, Nanoparticle Size and Surface Charge Determine Effects of PAMAM Dendrimers on Human Platelets in vitro, Mol. Pharmaceutics, 2012, 9, 382–393 CrossRef CAS.
- H. Kobayashi and M. W. Brechbiel, Dendrimer-based macromolecular MRI contrast agents: characteristics and application, Mol. Imaging, 2003, 2, 1–10 CrossRef CAS.
- B. S. Zolnik, S. T. Stern, J. M. Kaiser, Y. Heakal, J. D. Clogston, M. Kester and S. E. McNeil, Rapid distribution of liposomal short-chain ceramide in vitro and in vivo, Drug Metab. Dispos., 2008, 36, 1709–1715 CrossRef CAS.
- R. Cortesi, E. Esposito, E. Menegatti, R. Gambari and C. Nastruzzi, Effect of cationic liposome composition on in vitro cytotoxicity and protective effect on carried DNA, Int. J. Pharm., 1996, 139, 69–78 CrossRef CAS.
- A. P. Leonov, J. W. Zheng, J. D. Clogston, S. T. Stern, A. K. Patri and A. Wei, Detoxification of gold nanorods by treatment with polystyrenesulfonate, ACS Nano, 2008, 2, 2481–2488 CrossRef CAS.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, Controlled release of biologically active silver from nanosilver surfaces, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS.
- E. Oberdorster, Manufactured nanomaterials (fullerenes, C60) induce oxidative stress in the brain of juvenile largemouth bass, Environ. Health Perspect., 2004, 112, 1058–1062 CrossRef CAS.
- S. Zhu, E. Oberdorster and M. L. Haasch, Toxicity of an engineered nanoparticle (fullerene, C60) in two aquatic species, Daphnia and fathead minnow, Mar. Environ. Res., 2006, 62(Suppl), S5–S9 CrossRef CAS.
- A. Au, J. Ha, M. Hernandez, A. Polotsky, D. S. Hungerford and C. G. Frondoza, Nickel and vanadium metal ions induce apoptosis of T-lymphocyte Jurkat cells, J. Biomed. Mater. Res. A, 2006, 79, 512–521 CrossRef.
- A. Barchowsky, N. V. Soucy, K. A. O'Hara, J. Hwa, T. L. Noreault and A. S. Andrew, A novel pathway for nickel-induced interleukin-8 expression, J. Biol. Chem., 2002, 277, 24225–24231 CrossRef CAS.
- A. Jaramillo and G. Sonnenfeld, Effect of nickel sulfide on induction of interleukin-1 and phagocytic activity, Environ. Res., 1993, 63, 16–25 CrossRef CAS.
- N. V. Soucy, A. S. Andrew and A. Barchowsky, Nickel induces IL-8 expression in human airway epithlial cells, The Toxicologist, 2001, 60, 317 Search PubMed.
- F. W. Sunderman Jr, S. M. Hopfer, S. M. Lin, M. C. Plowman, T. Stojanovic, S. H. Wong, O. Zaharia and L. Ziebka, Toxicity to alveolar macrophages in rats following parenteral injection of nickel chloride, Toxicol. Appl. Pharmacol., 1989, 100, 107–118 CrossRef CAS.
- M. Waseem, R. Bajpai and J. L. Kaw, Reaction of pulmonary macrophages exposed to nickel and cadmium in vitro, J. Environ. Pathol. Toxicol. Oncol., 1993, 12, 47–54 CAS.
- A. W. Richter and E. Akerblom, Antibodies against polyethylene glycol produced in animals by immunization with monomethoxy polyethylene glycol modified proteins, Int. Arch. Allergy Immunol., 1983, 70, 124–131 CrossRef CAS.
- Y. Arima, M. Toda and H. Iwata, Complement activation on surfaces modified with ethylene glycol units, Biomaterials, 2008, 29, 551–560 CrossRef CAS.
- C. Boeckler, B. Frisch, S. Muller and F. Schuber, Immunogenicity of new heterobifunctional cross-linking reagents used in the conjugation of synthetic peptides to liposomes, J. Immunol. Methods, 1996, 191, 1–10 CrossRef CAS.
- P. Adiseshaiah, J. B. Hall and S. McNeil, Nanomaterial standards for efficacy and toxicity assessment, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 99–112 CrossRef CAS.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Protein nanoparticles as drug carriers in clinical medicine, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS.
- T. E. Stinchcombe, Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel, Nanomedicine, 2007, 2, 415–423 CrossRef CAS.
- T. Safra, F. Muggia, S. Jeffers, D. D. Tsao-Wei, S. Groshen, O. Lyass, R. Henderson, G. Berry and A. Gabizon, Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2, Ann. Oncol., 2000, 11, 1029–1033 CrossRef CAS.
- S. T. Stern, J. B. Hall, L. L. Yu, L. J. Wood, G. F. Paciotti, L. Tamarkin, S. E. Long and S. E. McNeil, Translational considerations for cancer nanomedicine, J. Controlled Release, 2010, 146, 164–174 CrossRef CAS.
- H. M. Weiss, M. Fresneau, G. P. Camenisch, O. Kretz and G. Gross, In vitro blood distribution and plasma protein binding of the iron chelator deferasirox (ICL670) and its iron complex Fe-[ICL670]2 for rat, marmoset, rabbit, mouse, dog, and human, Drug metabolism and disposition: the biological fate of chemicals, 2006, 34, 971–975 CAS.
| This journal is © The Royal Society of Chemistry 2013 |