In situ click chemistry: from small molecule discovery to synthetic antibodies
Steven W.
Millward
*a,
Heather D.
Agnew
b,
Bert
Lai
b,
Su Seong
Lee
c,
Jaehong
Lim
c,
Arundhati
Nag
d,
Suresh
Pitram
b,
Rosemary
Rohde
b and
James R.
Heath
d
aDepartment of Experimental Diagnostic Imaging, University of Texas MD Anderson Cancer Center, Experimental Diagnostic Imaging, Unit 603, P.O. Box 301402, Houston, TX 77230-1402, USA. E-mail: smillward@mdanderson.org
bIntegrated Diagnostics, 6162 Bristol Parkway, Culver City, California 90230, USA
cInstitute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669, Singapore
dNanosystems Biology Cancer Center, Division of Chemistry and Chemical Engineering, MC-127-72, California Institute of Technology, Pasadena, California 90230, USA
First published on 26th July 2012
Abstract
Advances in the fields of proteomics, molecular imaging, and therapeutics are closely linked to the availability of affinity reagents that selectively recognize their biological targets. Here we present a review of Iterative Peptide In Situ Click Chemistry (IPISC), a novel screening technology for designing peptide multiligands with high affinity and specificity. This technology builds upon in situ click chemistry, a kinetic target-guided synthesis approach where the protein target catalyzes the conjugation of two small molecules, typically through the azide–alkyne Huisgen cycloaddition. Integrating this methodology with solid phase peptide libraries enables the assembly of linear and branched peptide multiligands we refer to as Protein Catalyzed Capture Agents (PCC Agents). The resulting structures can be thought of as analogous to the antigen recognition site of antibodies and serve as antibody replacements in biochemical and cell-based applications. In this review, we discuss the recent progress in ligand design through IPISC and related approaches, focusing on the improvements in affinity and specificity as multiligands are assembled by target-catalyzed peptide conjugation. We compare the IPISC process to small molecule in situ click chemistry with particular emphasis on the advantages and technical challenges of constructing antibody-like PCC Agents.
 Steven W. Millward | Dr Steven Millward grew up just south of Pittsburgh, Pennsylvania and received his Bachelors of Arts in Biology in 2000 from The Johns Hopkins University. His graduate work in molecular evolution at The California Institute of Technology was carried out under the supervision of Richard W. Roberts. He continued his studies at Caltech as a postdoc with James Heath where he employed in situ click chemistry to design branched peptide kinase inhibitors. He is currently developing new experimental compounds for targeted molecular imaging of cancer as an assistant professor at the University of Texas MD Anderson Cancer Center. |
 Heather D. Agnew | Dr Heather Agnew earned bachelor's degrees in chemistry and BMB (2003) from Penn State University. As a Gates Scholar, she received an MPhil in chemistry (2005) from University of Cambridge. Heather went on to study under Professor James R. Heath at Caltech, where she received the Lemelson-MIT Caltech Student Prize for her role in developing techniques to create synthetic, site-specific binding molecules as alternatives to antibodies. After receiving her PhD (2010), Heather joined Integrated Diagnostics, where she leads a program focused on commercialization of PCC Agents. She holds an adjunct appointment in the David Geffen School of Medicine at UCLA. |
Insight, innovation, integrationLigands for high-specificity protein recognition are of considerable interest to the biomedical community. This review provides a critical analysis of the emerging field of Iterative Peptide In Situ Click Chemistry (IPISC) as a means of developing highly selective ligands that bind to proteins with antibody-like affinity. In situ click chemistry is a target-guided synthesis approach where the target protein is used as a scaffold upon which binding ligands are covalently assembled by the azide–alkyne cycloaddition. Drawing inspiration from the antigen-binding site of monoclonal antibodies, IPISC has the potential to direct the assembly of artificial multi-peptide structures that recognize protein surfaces with high-affinity and specificity while retaining the stability and chemical accessibility of synthetic compounds. |
Introduction
Molecular recognition underlies all aspects of biology and is a critical component of therapeutic design, molecular imaging, and molecular diagnostics. The simplicity and robustness of nucleic acid recognition though specific base pairing has enabled tremendous technological advances in genomics and transcriptomics. A similarly deep understanding of protein recognition has yet to emerge despite considerable study. As a result, molecules developed for specific protein recognition are usually identified through combinatorial screening processes, rather than through rational design.Antibodies are the primary molecular tool for protein recognition, and find almost universal use in the biomedical community for basic research, immunohistochemistry, diagnostic imaging, and therapeutics. A key feature of antibodies is that they can often be developed to exhibit high specificity for their target protein antigen (although high specificity is not guaranteed1). However, they are prone to proteolytic, chemical, and thermal degradation, which can limit their utility in non-laboratory diagnostic environments. In addition, as biological compounds, they are subject to batch-to-batch variability and chemical modifications with dyes and affinity tags can detrimentally influence their properties. While antibodies have found extensive use as therapeutics against extracellular protein targets, their utility in imaging applications can be compromised by long serum half-lives, leading to increased background signal in all perfused tissue.
These shortcomings have prompted the development of numerous chemical and biological display technologies for designing “antibody-like” ligands.6 The goal is typically to optimize desirable features such as reduced size, increased stability, and ease of synthesis and labeling while achieving antibody-like affinity and specificity. These approaches include aptamer technology,8 phage display,9 ribosome display,10 mRNA display,11 yeast display,12 and one-bead-one-compound (OBOC) solid phase libraries.13 These techniques typically yield linear or cyclic biopolymer ligands that bind to a single site, or “hot spot”, on the surface of the protein target with high affinity. We review here the recently developed technique of Iterative Peptide In Situ Click Chemistry (IPISC) for producing protein capture agents. This technique draws from the above-mentioned methodologies, but with a few critical differences which are described below. The advantages are briefly listed here. First, the protein target itself provides a highly selective catalytic scaffold for assembling its own capture agent. Through the application of novel screening approaches, the resultant capture agent can be developed to exhibit high selectivity for the target. Because of the protein-catalyzed process, we have named these types of ligands Protein Catalyzed Capture Agents, or PCC Agents. Second, PCC Agents are assembled stepwise from comprehensive, chemically synthesized OBOC libraries allowing stability-enhancing functionalities (e.g. unnatural amino acids) to be incorporated at the start, biasing the final products toward bio-stability. Third, the approach permits the development of a wide variety of capture agent architectures – linear, branched, cyclic or combinations thereof, opening a regime of chemical space that is not easily accessible with alternative approaches. Finally, PCC Agents are defined chemical structures that can be scaled up by automated chemical synthesis, avoiding the problem of batch-to-batch reproducibility.
This review will discuss the use of Iterative Peptide In Situ Click Chemistry to create minimized protein-binding surfaces through the templated assembly of unique peptide sequences. We will begin by touching upon the enabling technology of small molecule in situ click chemistry (SISC), which provided the initial foundation for IPISC. We will then consider the architecture of the antigen-binding site of antibodies as a model for protein recognition and biological inspiration for IPISC. Finally, we will review the recent developments in IPISC and related topics, comparing the two in situ click methodologies and discussing the advantages and challenges of designing multi-peptide PCC Agents.
Small molecule in situ click chemistry (SISC)
Small molecule in situ click chemistry (SISC) was originally described by Sharpless and co-workers in 2002 to design potent small molecule inhibitors of acetylcholinesterase (AChE).14 The principle behind this methodology is that the thermodynamically favorable 1,3-dipolar Huisgen cycloaddition reaction can be catalyzed in the absence of metal catalyst provided that the two reactants are brought into close proximity in the correct spatial orientation by a protein target (Fig. 1). Thus, for SISC, a known inhibitor is split into two molecules, one presenting an azide, and one presenting an acetylene group. One or both of those halves is expanded into a library, and the enzyme provides the scaffold for coupling those library elements together. Only those elements that are mutually compatible with the protein scaffold are clicked together. The resulting triazole-linked compound will have a binding energy as high as the sum of the free energies of the two reactants.15 Acetylcholinesterase was chosen as the target because of its deep, well-characterized binding pocket which was known to have two independent binding sites. The most potent of the final compounds had a dissociation constant of less than 40 fM against eel acetylcholinesterase representing almost the full sum of the component ligand binding energies. Highly potent inhibitors of nicotinic acetylcholine receptor,16 carbonic anhydrase II (CA II),17 HIV-1 protease,18 and bacterial chitinases19 have also been discovered by these methods. For additional discussion of this methodology, we refer the reader to a recent review of small molecule in situ click chemistry.20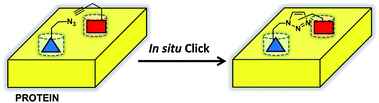 | ||
| Fig. 1 Schematic of small molecule in situ click chemistry (SISC). An azide-bearing compound (blue) is brought into close proximity to an acetylene-bearing compound (red) within the active site of a protein (yellow). Proximity and orientation drive 1,2,3-triazole formation and conjugation of the two compounds. | ||
SISC is well suited when there exists a known inhibitor structure that can be deconstructed into two or more independent chemical functionalities. The small molecules that are built for SISC represent a small, focused sampling of chemical space and their design draws from knowledge of the original inhibitor and the structure of the binding pocket. In most cases, it is the linker length, rigidity, and composition being optimized by the in situ screen, effectively determining the optimal spatial orientation between independent ligands. This is a powerful molecular design feature, which has been employed to optimize the orientation of larger peptide functionalities in Iterative Peptide In Situ Click Chemistry. The rationale for this approach is provided, in part, by the structure of the antigen-binding site used by monoclonal antibodies.
Biological insight: the structural basis of antibody–protein interactions
Antibodies recognize a wide range of ligands and surfaces including nucleic acids,21 peptides,22 proteins,23 small molecules,24 complex carbohydrates,25 lipids,26 inorganic surfaces,27 and pathogen surfaces.28 The adaptability of antibodies to these disparate structures and chemistries lies in the surface available for antigen recognition. The antigen binding site is composed of six peptide loops, three from the heavy chain and three from the light chain, forming a binding surface of approximately 1400–1900 Å2 for protein targets.29 The amino acid sequence of each loop is determined by the outcome of VDJ recombination at the genetic level, resulting in a diverse repertoire of surfaces with each loop contributing to the chemistry and shape of the antigen recognition surface. However, not all six loops contribute equally to the affinity and specificity of the final interaction. The CDR-H3 loop on the heavy chain is the most variable of the six, with a typical length ranging from 10 to 22 amino acids30 and significantly higher sequence diversity relative to the other five loops.31 This sequence variability translates to increased conformational variability across known antibody structures relative to the other loops that define the binding surface.32 The CDR-H3 has been shown in many cases to define the specificity of the antibody–antigen recognition with the other loops contributing stabilizing interactions to increase affinity and define the shape of the binding site.31a In one report, the CDR3 loop alone was sufficient for low-nanomolar binding of a camel single-domain antibody to carbonic anhydrase.33 This phenomenon has been exploited to design peptide ligands based on the CDR-H3 loop sequence to bind the target antigen with high nanomolar affinity.34Crystal structures of antibodies and Fab fragments bound to their cognate protein antigens reveal a diverse repertoire of antigen-binding site architectures. In some cases, only a subset of the six CDR loops are utilized in the high-affinity antigen recognition surface (Fig. 2). For example, antibody binding of HIV p24 protein, Lysozyme, and VEGF, is mediated mainly through the CDR-H3, CDR-L3, CDR-H1, and CDR-L3 loops, with the CDR-H3 loop contributing significant binding interactions in all three structures.
 | ||
| Fig. 2 Antibody/Fab Recognition of Protein Surfaces: using a subset of CDR loops. Binding of the protein antigen (yellow) to the antibody/Fab fragment (grey) is mediated by six CDR loops, three from the light chain and three from the heavy chain. The CDR3 loops are shown in red and the CDR1 and CDR2 loops are shown in blue. (A) Lysozyme–antibody complex (PDB code 1MLC).2 (B) HIVp24–Fab complex (1E6J).4 (C) VEGF–antibody complex (1BJ1).7 Protein structure images were generated in PyMol. | ||
These observations suggest (1) 2–4 peptides in the proper spatial arrangement can potentially recognize folded protein antigens with high affinity and specificity and (2) a single loop can provide the majority of the interactions to mediate the binding event. While existing library display methodologies can determine the optimal sequence of the individual peptides, their ideal relative orientation to one another, particularly in a non-linear configuration, can be challenging to optimize. In situ click chemistry, in combination with one-bead-one-compound library technology, can provide the spatial information to assemble a multi-peptide ligand with the affinity and specificity of a monoclonal antibody.
PCC Agents through Iterative Peptide In Situ Click Chemistry (IPISC)
The application of in situ click chemistry to the design of multimeric PCC Agent peptide ligands was first demonstrated by Agnew and co-workers in 2009,3 who developed a triligand capture agent against bovine carbonic anhydrase II (bCAII). PCC Agent development through IPISC proceeds stepwise, beginning with a 1°, or anchor ligand. That anchor ligand is then used in an in situ screen to identify a 2° ligand. The 1° and 2° ligands are clicked together to form a biligand. The biligand can be converted into an anchor for use in an in situ screen to identify a 3° ligand, from which a triligand is formed, and so on. A generalized schematic of Iterative Peptide In Situ Click Chemistry shown in Fig. 3.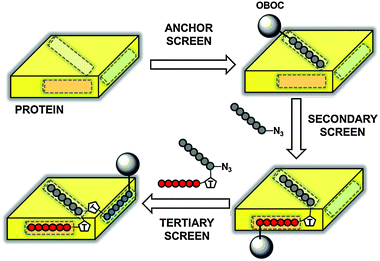 | ||
| Fig. 3 Generalized Schematic of Iterative Peptide In Situ Click Chemistry (IPISC). An anchor peptide (black circles) is selected for binding to a site on the protein surface (yellow) from a one-bead-one-compound peptide library (grey sphere). The anchor peptide is appended with an azide and incubated with the protein and an OBOC library containing acetylene-functionalized peptides (secondary screen). In situ triazole formation links the secondary peptide (red circles) to the anchor to form the biligand. The biligand is then appended with an azide and incubated with the acetylene-containing OBOC library to direct triazole formation between the tertiary peptide (blue circles) and the biligand to form the triligand (tertiary screen). | ||
For the development of the bCAII PCC Agent, the 1° ligand was identified through a standard OBOC target screen for protein binding. The OBOC library was a random hexameric library appended with D-propargylglycine. The library was comprised of D-amino acids to yield protease resistance and improved bio-stability. Hit elements were selected for binding to bCAII via a two-generation screen. The consensus anchor ligand exhibited a weak affinity (KD ∼ 500 μM) against bCAII. The anchor compound was then incubated with bCAII and a second D-amino acid peptide library bearing azido-amino acids of variable side-chain length. After two screens, a consensus 2° peptide was identified and conjugated to the anchor peptide by the copper-catalyzed azide–alkyne cycloaddition (CuAAC) to yield a biligand with significantly improved affinity (KD = 3 μM). The process was repeated to generate a high affinity triligand (KD ∼ 45 nM) that could function as a drop-in antibody replacement in immunoblotting experiments. Importantly, the triligand was formed on-bead only in the presence of bCAII or human carbonic anhydrase II (hCAII) but not in the presence of the unrelated proteins BSA or transferrin.
The architectural flexibility afforded by IPISC is illustrated in the structures, shown in Fig. 4, of four of the PCC Agent triligands we have developed over the past couple of years. These structures are color-coded so that the 1°, 2°, and 3° ligands are red, black, and blue, respectively, and each triligand exhibits a mid- to low nM affinity for their cognate proteins. Fig. 4a is the linear anti-bCAII triligand discussed above, while Fig. 4b is a branched variant on that structure, in which a D-tryptophan at the 3-position in the 6-mer 2° ligand is replaced with an azide-containing amino acid for the development of the triligand branch. Fig. 4c is an anti-PSA PCC Agent triligand that utilizes a cyclic peptide from the literature35 as the initial 1° ligand and anchor. Note that the triazole linkage connecting the 2° and 3° ligands is a 1,5-triazole, as compared to the 1,4-triazoles that bridge all of the other ligands. The in situ click reaction can generate either isoform and, if both variants are tested, there can be a preference for one or the other, depending on the flexibility of the linkage connecting the ligands. The branched triligand shown in Fig. 4d is discussed below.
 | ||
| Fig. 4 Structures of Triligand PCC Agents Obtained by IPISC. (a) Linear anti-bCAII triligand.3 (b) Branched anti-bCAII triligand (unpublished). (c) Anti-PSA triligand with cyclic anchor peptide (unpublished). (d) Branched anti-Akt triligand.5 | ||
In 2011, Millward and co-workers applied this technique to design a triligand capture agent with mid-nanomolar affinity for Protein Kinase B (Akt1).5 The resulting branched triligand was found to inhibit the catalytic activity of Akt1 without directly binding to the ATP or peptide substrate pockets, suggesting an allosteric mode of inhibition. The triligand immunoprecipitated Akt from OVCAR3 ovarian cancer cell lysates and a fluorescent variant was used to image membrane-localized Akt in cell culture after stimulation with insulin and EGF. In both cases, the triligand compared favorably with commercial monoclonal antibodies, particularly in immunoprecipitation experiments where the efficiency of the antibody was found to be very poor.
An innovation of this work was the use of “product screens” to select hit beads not on the basis of target binding (the standard screening approach), but on the basis of on-bead formation of the triazole linkage between the bead-immobilized peptide and the soluble anchor peptide in the presence of the protein target. For product screens, the soluble biotinylated anchor peptide is incubated with the on-bead library in the presence of the target biomolecule, and the beads are then probed for the presence of biotin. Only the beads whose sequences participate in the in situ click reaction will have a covalently-linked biotin functionality. When carried out in the absence of a target screen, the product screen resulted in modest affinity gains but significant gain in selectivity for Akt over the GSK3β, the primary off-target binding interaction, implying that the product screen can serve as a selectivity screen. It also served as a powerful approach to removing false-positive hits, which can account for 90% of the hit beads in the target screen. The authors went on to use the principle of the product screen to quantitate the efficiency of the on-bead in situ click reaction through a novel quantitative PCR (QPCR)-based technique. These experiments demonstrated the efficiency of the in situ reaction was dependent on the target protein as well the orientation of the peptide ligands. The yield of the on-bead target-catalyzed reaction was found to be approximately 1/1000th of the copper-catalyzed process. Table 1 summarizes the screening conditions and results of selected SISC and IPISC screens.
| Target | [Target] (nM) | Screen | d | Anchor KD (nM) | Biligand KD (nM) | Triligand KD (nM) | ΔΔG/Heavy atom (kcal mol−1 atom−1) | NROT/kDa (bonds/kDa) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Target concentration denotes the concentration of target protein present in the screen. The parameter d is number of unique compounds present in the combinatorial library from which the ligand components were selected.a Denotes an IC50 value.b Denotes a concentration in mUnit per mL. NROT/kDa is a measure of ligand conformational flexibility and denotes the number of rotatable bonds per kDa of molecular weight of the final biligand or triligand. ΔΔG/Heavy atom is a measure of the change in affinity per addition of nonhydrogen atoms to the anchor ligand. This was obtained by converting affinity data for the ligands into ΔG (ΔG = −RT ln KD) and dividing the change in ΔG by the change in nonhydrogen atom composition. When unavailable, IC50 values were substituted for KD values without additional correction. For SISC screens the ΔΔG/heavy atom of the biligand was obtained relative to the anchor. For IPISC screens, the anchor was used as the reference compound for the biligand and the biligand was used as the reference compound for the triligand. | |||||||||
| AChE | 1000 | SISC | 98 | 10–100 | 0.000077 | n/a | 0.25 | 18 | 14 |
| AChE | 1000 | SISC | 104 | 10–100 | 0.000100 | n/a | 0.20–0.25 | 17 | 45 |
| AChE | 1000 | SISC | 23 | 10–100 | 0.000033 | n/a | 0.29 | 21 | 46 |
| bCAII | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
SISC | 24 | 31–43 | 0.2–7.1 | n/a | 0.05–0.15 | 14–19 | 17 |
| bCAII | 10–50 | IPISC | 2 × 107 | 500000 |
3000 | 45 | 0.037 (Biligand) | 3 | |
| 0.038 (Triligand) | 37 | ||||||||
| HIV protease | 15000 |
SISC | 5 | 3600–4800a | 6a | n/a | 0.23 | 21 | 18 |
| Chitinase B | 9.6b | SISC | 71 | 536–624a | 20–24a | n/a | 0.12 | 24 | 19 |
| Akt1 | 9–110 | IPISC | 5.7 × 106 | 25000 |
300 | 200 | 0.038 (Biligand) | 5 | |
| 0.003 (Triligand) | 36 | ||||||||
Analysis of IPISC screens; affinity and specificity
The improvement of ligand affinity as a function of molecular weight is a key metric for the efficiency of IPISC screens. To quantify this, we have calculated the change in binding energy (ΔΔG) per additional heavy (nonhydrogen) atom as anchors are translated to biligands and as biligands are translated to triligands. This approach was originally described by Kuntz and co-workers36 and later adapted by Hajduk to analyze the efficiency of fragment-based drug discovery efforts.37 We found that the average change in binding energy for SISC screens was approximately 0.2 kcal mol−1 per heavy atom which is in excellent agreement with previous studies (0.27 kcal mol−1 per heavy atom).37 Similar analysis of the two IPISC screens reveal an average change of approximately 0.03 kcal mol−1 per heavy atom, almost an order of magnitude lower than for SISC-derived biligands. However, in the previous work mentioned above, molecules in this size regime (>50 heavy atoms) are predicted to show almost no change in binding energy as the molecular weight increases (ΔΔG ∼ −0.0002 kcal mol−1 per heavy atom). This suggests the possibility that peptide multiligands are making additional energetic contacts with the protein surface outside the anchor-binding site, resulting in modest improvements in affinity as size increases. This is in contrast to SISC and other small molecule fragment-based approaches where the energetic benefits of increasing the molecular weight are essentially limited by the size and composition of the binding pocket.The initial IPISC experiments suggest that screening for triligand formation rather than target binding can lead to increased multiligand specificity rather than affinity. While the tertiary peptide in the Akt triligand appears to contribute very little to the binding energy for Akt, it partially destabilizes binding to GSK3β, the primary off-target interaction.5 This is consistent with a pure product screen which does not select for affinity for the target, but rather compatibility with target surface proximal to the anchor peptide binding site. In this case, specificity can be achieved by excluding non-target molecules whose surfaces are incompatible with one or more multi-ligand components. This concept of “negative design” has been exploited to design highly specific coiled-coil systems38 and a recent study of SH3 domains demonstrates the significant gains in specificity from destabilizing off-target interactions.39 Previous work with Herceptin, a therapeutic anti-Her2 antibody, demonstrated that the specificity of two antibodies sharing common CDR3 loops (analogous to anchor peptides in IPISC) can be dramatically altered by a handful of mutations in the CDR-L1 loop.40 Here specificity is controlled, in part, by what is excluded from the binding site. While it is difficult to draw broad conclusions from two screening experiments, the product screen shows promise as a design tool for improving ligand specificity without the need for complex and costly counter-screening strategies.
Design of artificial receptors using in situ click chemistry
The ligands obtained from the IPISC screens described above target full-length folded proteins. However, a recent report from Tanaka and co-workers describes the use of in situ click chemistry to assemble an SH2 domain mimic which targets a short phosphorylated peptide.41 A resin-bound bis-lysine known to bind to phosphate was employed as the anchor ligand and a phosphorylated cyclic peptide specific for the Grb2 SH2 domain was used as the target. Two azides were appended to the anchor which could form independent triazole linkages with members of an alkyne-modified, solution phase, tetrapeptide library. In the presence of the target cyclic peptide and low concentrations of copper catalyst, the authors demonstrated that the each azide on the bis-lysine anchor formed a triazole linkage with the same tetrapeptide sequence. The resulting, branched structure bound to the Grb2-specific phospho-peptide with low micromolar affinity and showed in vivo tumor volume reduction in C6-glioma mouse xenografts. The use of copper to drive the reaction to completion suggests a potential route to improving the yield of the reaction, albeit with a corresponding increase in the background (non-templated) click reaction. This approach differs from IPISC screen notably in its use of copper, rather than the protein target, to drive triazole formation. In addition, it also utilizes an immobilized anchor ligand, as well as a low diversity solution-phase library. However, the use of target-guided click chemistry to assemble a branched peptide further demonstrates that selective molecular recognition can arise from the correct spatial orientation of short peptides.Optimizing IPISC screening strategies
The Akt1 experiments demonstrated that the in situ click screen was specific, but low yielding relative to the copper-catalyzed process. While there is no explicit data for the product yield in SISC screens, the observation of the product by liquid chromatography indicates that the yield is significantly higher than the peptide-based process where the on-bead product can only be visualized after PCR-based amplification. The low concentration of on-bead product leads to low signal-to-noise ratios and selection of false positive hit beads. The yield of the on-bead reaction is the most significant hurdle to the rapid and efficient design of ligands through Iterative Peptide In Situ Click Chemistry. There are various explanations for this inefficiency, however we will focus on the linkage between the IPISC ligand components and the poor catalytic efficiency of the target protein.The number of rotatable bonds (normalized for molecular weight) in the final product is significantly higher in iterative peptide screens than in small molecule screens (Table 1). This arises from use of conformationally flexible component peptides linked to each other through even more flexible hydrocarbon chains. One potential consequence is a reduced probability of azide and alkyne residing in the correct orientation necessary for the in situ reaction to occur, resulting in a low observed yield of triazole formation. A long, conformationally flexible linker would also be predicted to prevent optimal energetic coupling between the component peptides in the multiligand, lowering the affinity of the final product. This could be addressed in two ways, either by reducing the length of the linkage between peptide components or decreasing its conformational flexibility. The results from the Akt1 study suggest that the IPISC screen selects for the best peptide component in the context of the linker length used in the screening experiment; arbitrarily shortening the linker was found to significantly attenuate the affinity of the designed biligand.5 The use of shorter linkers would also reduce the amount of protein surface that can be sampled in an IPISC screen. Indeed, a recent study by Mack and co-workers using a series of bivalent bCAII inhibitors with long, flexible linkers demonstrated only modest differences in binding affinity to engineered bCAII dimers, even when the linker lengths were significantly longer than required (∼2 kcal mol−1 variation in binding energy over 40 Å of extended linker length).42 This observation suggests that long linkers can be used to search for additional binding sites on the protein surface while still retaining high affinity in the multiligand. Given the advantage of a long linker, it may be useful to focus on increasing its conformational rigidity though introduction of non-rotatable bonds.
The target protein also plays a critical role in the efficiency of the in situ reaction. Ideally, the protein target will undergo multiple turnover events and catalyze the formation of more than one multiligand from the component ligands. However, if the protein target templates the formation of a high affinity multiligand, this could lead to a very slow off rate (koff) for the product, effectively preventing dissociation and rebinding of a new pair of component ligands. This phenomenon has been observed in DNA-templated synthesis due to the high-affinity DNA duplexes that are formed after covalent bond formation.43 A recent report by Sharma and co-workers illustrates the high target concentrations required to drive a low-turnover in situ click reaction to completion.44 In this study, cocaine was used to template the ligation of two pieces of a cocaine-binding split aptamer by in situ click chemistry. While the reported yield was as high as 74%, this yield was obtained with a 500-fold excess of cocaine. The low turnover of triazole product in in situ click screens highlights the need for high concentrations of target to generate adequate signal-to-noise ratios in IPISC screens. SISC screens are generally carried out with micromolar concentrations of target and we have found that these concentrations result in more efficient product screens and fewer false-positive hits in IPISC.
Conclusions and future directions
As more potential protein biomarkers for disease are discovered, the demand for compounds that can capture, detect, and inhibit these proteins will commensurately increase. The IPISC design process allows access to novel ligand architecture and composition, opening the possibility for specific recognition of biomolecules that are difficult to target through traditional approaches. As demonstrated by Tanaka and co-workers, branched peptides have enormous potential to recognize phosphorylated peptide epitopes. The IPISC design process can readily applied to other this and other modifications including glycosylation, a post-translational modification that has proven difficult to target with high affinity. Small peptide epitope recognition, currently the province of antibodies and their derivatives, represents another exciting application for branched PCC Agents. Our groups are actively refining the IPISC screening process to decrease the time from anchor to final compound while concurrently improving the affinity and specificity of the ligand at each step. We envision these compounds finding use as antibody alternatives in immunohistochemistry, probes for molecular imaging, and novel therapeutics that couple the most desirable properties of antibodies with the stability and economy of synthetic compounds.Acknowledgements
This work was supported by the Institute for Collaborative Biotechnologies through grant W911NF-09-0001 from the U.S. Army Research Office and by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore) and by the National Cancer Institute Grant No. 5U54 CA119347 (JRH). SWM acknowledges support from an NRSF postdoctoral fellowship 1F32CA13615001.References
- (a) G. Kijanka, S. IpCho, S. Baars, H. Chen, K. Hadley, A. Beveridge, E. Gould and D. Murphy, Rapid characterization of binding specificity and cross-reactivity of antibodies using recombinant human protein arrays, J. Immunol. Methods, 2009, 340(2), 132–137 CrossRef CAS; (b) G. A. Michaud, M. Salcius, F. Zhou, R. Bangham, J. Bonin, H. Guo, M. Snyder, P. F. Predki and B. I. Schweitzer, Analyzing antibody specificity with whole proteome microarrays, Nat. Biotechnol., 2003, 21(12), 1509–1512 CrossRef CAS.
- B. C. Braden, H. Souchon, J.-L. Eiselé, G. A. Bentley, T. N. Bhat, J. Navaza and R. J. Poljak, Three-dimensional structures of the free and the antigen-complexed Fab from monoclonal anti-lysozyme antibody D44.1, J. Mol. Biol., 1994, 243(4), 767–781 CrossRef CAS.
- H. D. Agnew, R. D. Rohde, S. W. Millward, A. Nag, W. S. Yeo, J. E. Hein, S. M. Pitram, A. A. Tariq, V. M. Burns, R. J. Krom, V. V. Fokin, K. B. Sharpless and J. R. Heath, Iterative in situ click chemistry creates antibody-like protein-capture agents, Angew. Chem., Int. Ed., 2009, 48(27), 4944–4948 CrossRef CAS.
- S. Monaco-Malbet, C. Berthet-Colominas, A. Novelli, N. Battaï, N. Piga, V. Cheynet, F. Mallet and S. Cusack, Mutual Conformational Adaptations in Antigen and Antibody upon Complex Formation between an Fab and HIV-1 Capsid Protein p24, Structure, 2000, 8(10), 1069–1077 CrossRef CAS.
- S. W. Millward, R. K. Henning, G. A. Kwong, S. Pitram, H. D. Agnew, K. M. Deyle, A. Nag, J. Hein, S. S. Lee, J. Lim, J. A. Pfeilsticker, K. B. Sharpless and J. R. Heath, Iterative in situ click chemistry assembles a branched capture agent and allosteric inhibitor for Akt1, J. Am. Chem. Soc., 2011, 133(45), 18280–18288 CrossRef CAS.
- T. Kodadek, Synthetic receptors with antibody-like binding affinities, Curr. Opin. Chem. Biol., 2010, 14(6), 713–720 CrossRef CAS.
- Y. A. Muller, Y. Chen, H. W. Christinger, B. Li, B. C. Cunningham, H. B. Lowman and A. M. de Vos, VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 Å resolution and mutational analysis of the interface, Structure (London), 1998, 6(9), 1153–1167 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, In vitro selection of RNA molecules that bind specific ligands, Nature, 1990, 346(6287), 818–822 CrossRef CAS.
- J. K. Scott and G. P. Smith, Searching for peptide ligands with an epitope library, Science, 1990, 249(4967), 386–390 CAS.
- J. Hanes and A. Plückthun, In vitro selection and evolution of functional proteins by using ribosome display, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(10), 4937–4942 CrossRef CAS.
- R. W. Roberts and J. W. Szostak, RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(23), 12297–12302 CrossRef CAS.
- E. T. Boder and K. D. Wittrup, Yeast surface display for screening combinatorial polypeptide libraries, Nat. Biotechnol., 1997, 15(6), 553–557 CrossRef CAS.
- K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, A new type of synthetic peptide library for identifying ligand-binding activity, Nature, 1991, 354(6348), 82–84 CrossRef CAS.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radic, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks, Angew. Chem., Int. Ed., 2002, 41(6), 1053–1057 CAS.
- W. P. Jencks, On the attribution and additivity of binding energies, Proc. Natl. Acad. Sci. U. S. A., 1981, 78(7), 4046–4050 CrossRef CAS.
- N. P. Grimster, B. Stump, J. R. Fotsing, T. Weide, T. T. Talley, J. G. Yamauchi, A. Nemecz, C. Kim, K. Y. Ho, K. B. Sharpless, P. Taylor and V. V. Fokin, Generation of candidate ligands for nicotinic acetylcholine receptors via in situ click chemistry with a soluble acetylcholine binding protein template, J. Am. Chem. Soc., 2012, 134(15), 6732–6740 CrossRef CAS.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Roper, K. B. Sharpless, C. H. Wong and H. C. Kolb, In situ click chemistry: enzyme-generated inhibitors of carbonic anhydrase II, Angew. Chem., Int. Ed., 2004, 44(1), 116–120 CrossRef.
- M. Whiting, J. Muldoon, Y. C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Inhibitors of HIV-1 protease by using in situ click chemistry, Angew. Chem., Int. Ed., 2006, 45(9), 1435–1439 CrossRef CAS.
- T. Hirose, T. Sunazuka, A. Sugawara, A. Endo, K. Iguchi, T. Yamamoto, H. Ui, K. Shiomi, T. Watanabe, K. B. Sharpless and S. Omura, Chitinase inhibitors: extraction of the active framework from natural argifin and use of in situ click chemistry, J. Antibiot., 2009, 62(5), 277–282 CrossRef CAS.
- S. K. Mamidyala and M. G. Finn, In situ click chemistry: probing the binding landscapes of biological molecules, Chem. Soc. Rev., 2010, 39(4), 1252–1261 RSC.
- M. L. Cerutti, J. M. Centeno, F. A. Goldbaum and G. de Prat-Gay, Generation of sequence-specific, high affinity anti-DNA antibodies, J. Biol. Chem., 2001, 276(16), 12769–12773 CrossRef CAS.
- R. L. Stanfield, T. M. Fieser, R. A. Lerner and I. A. Wilson, Crystal structures of an antibody to a peptide and its complex with peptide antigen at 2.8 Å, Science, 1990, 248(4956), 712–719 CAS.
- P. J. Carter, Potent antibody therapeutics by design, Nat. Rev. Immunol., 2006, 6(5), 343–357 CrossRef CAS.
- M. H. Niemi, K. Takkinen, L. K. Amundsen, H. Soderlund, J. Rouvinen and M. Hoyhtya, The testosterone binding mechanism of an antibody derived from a naive human scFv library, J. Mol. Recognit., 2011, 24(2), 209–219 CrossRef CAS.
- J. Hirakawa, K. Tsuboi, K. Sato, M. Kobayashi, S. Watanabe, A. Takakura, Y. Imai, Y. Ito, M. Fukuda and H. Kawashima, Novel anti-carbohydrate antibodies reveal the cooperative function of sulfated N- and O-glycans in lymphocyte homing, J. Biol. Chem., 2010, 285(52), 40864–40878 CrossRef CAS.
- B. G. Schuster, M. Neidig, B. M. Alving and C. R. Alving, Production of antibodies against phosphocholine, phosphatidylcholine, sphingomyelin, and lipid A by injection of liposomes containing lipid A., J. Immunol., 1979, 122(3), 900–905 CAS.
- T. Hattori, M. Umetsu, T. Nakanishi, T. Togashi, N. Yokoo, H. Abe, S. Ohara, T. Adschiri and I. Kumagai, High affinity anti-inorganic material antibody generation by integrating graft and evolution technologies: potential of antibodies as biointerface molecules, J. Biol. Chem., 2010, 285(10), 7784–7793 CrossRef CAS.
- R. A. Williamson, R. Burioni, P. P. Sanna, L. J. Partridge, C. F. Barbas, 3rd and D. R. Burton, Human monoclonal antibodies against a plethora of viral pathogens from single combinatorial libraries, Proc. Natl. Acad. Sci. U. S. A., 1993, 90(9), 4141–4145 CrossRef CAS.
- L. Lo Conte, C. Chothia and J. Janin, The atomic structure of protein–protein recognition sites, J. Mol. Biol., 1999, 285(5), 2177–2198 CrossRef CAS.
- V. Morea, A. Tramontano, M. Rustici, C. Chothia and A. M. Lesk, Conformations of the third hypervariable region in the VH domain of immunoglobulins, J. Mol. Biol., 1998, 275(2), 269–294 CrossRef CAS.
- (a) J. L. Xu and M. M. Davis, Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities, Immunity, 2000, 13(1), 37–45 CrossRef CAS; (b) M. Zemlin, M. Klinger, J. Link, C. Zemlin, K. Bauer, J. A. Engler, H. W. Schroeder, Jr. and P. M. Kirkham, Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures, J. Mol. Biol., 2003, 334(4), 733–749 CrossRef CAS.
- C. Chothia, A. M. Lesk, A. Tramontano, M. Levitt, S. J. Smith-Gill, G. Air, S. Sheriff, E. A. Padlan, D. Davies and W. R. Tulip, et al., Conformations of immunoglobulin hypervariable regions, Nature, 1989, 342(6252), 877–883 CrossRef CAS.
- A. Desmyter, K. Decanniere, S. Muyldermans and L. Wyns, Antigen Specificity and High Affinity Binding Provided by One Single Loop of a Camel Single-domain Antibody, J. Biol. Chem., 2001, 276(28), 26285–26290 CrossRef CAS.
- B. W. Park, H. T. Zhang, C. Wu, A. Berezov, X. Zhang, R. Dua, Q. Wang, G. Kao, D. M. O'Rourke, M. I. Greene and R. Murali, Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo., Nat. Biotechnol., 2000, 18(2), 194–198 CrossRef CAS.
- P. Wu, J. Leinonen, E. Koivunen, H. Lankinen and U. H. Stenman, Identification of novel prostate-specific antigen-binding peptides modulating its enzyme activity, Eur. J. Biochem., 2000, 267(20), 6212–6220 CrossRef CAS.
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The maximal affinity of ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002 CrossRef CAS.
- P. J. Hajduk, Fragment-based drug design: how big is too big?, J. Med. Chem., 2006, 49(24), 6972–6976 CrossRef CAS.
- (a) J. J. Havranek and P. B. Harbury, Automated design of specificity in molecular recognition, Nat. Struct. Biol., 2003, 10(1), 45–52 CrossRef CAS; (b) J. M. Mason, K. M. Muller and K. M. Arndt, Positive aspects of negative design: simultaneous selection of specificity and interaction stability, Biochemistry, 2007, 46(16), 4804–4814 CrossRef CAS.
- M. Gorelik, K. Stanger and A. R. Davidson, A Conserved residue in the yeast Bem1p SH3 domain maintains the high level of binding specificity required for function, J. Biol. Chem., 2011, 286(22), 19470–19477 CrossRef CAS.
- J. Bostrom, S. F. Yu, D. Kan, B. A. Appleton, C. V. Lee, K. Billeci, W. Man, F. Peale, S. Ross, C. Wiesmann and G. Fuh, Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site, Science, 2009, 323(5921), 1610–1614 CrossRef CAS.
- K. Tanaka, S. Shirotsuki, T. Iwata, C. Kageyama, T. Tahara, S. Nozaki, E. R. Siwu, S. Tamura, S. Douke, N. Murakami, H. Onoe, Y. Watanabe and K. Fukase, Template-Assisted and Self-Activating Clicked Peptide as a Synthetic Mimic of the SH2 Domain, ACS Chem. Biol., 2012, 7(4), 637–645 CrossRef CAS.
- E. T. Mack, P. W. Snyder, R. Perez-Castillejos, B. Bilgicer, D. T. Moustakas, M. J. Butte and G. M. Whitesides, Dependence of avidity on linker length for a bivalent ligand–bivalent receptor model system, J. Am. Chem. Soc., 2012, 134(1), 333–345 CrossRef CAS.
- (a) H. Abe and E. T. Kool, Destabilizing Universal Linkers for Signal Amplification in Self-Ligating Probes for RNA, J. Am. Chem. Soc., 2004, 126(43), 13980–13986 CrossRef CAS; (b) C. Dose, S. Ficht and O. Seitz, Reducing Product Inhibition in DNA-Template-Controlled Ligation Reactions, Angew. Chem., Int. Ed., 2006, 45(32), 5369–5373 CrossRef CAS; (c) T. N. Grossmann, A. Strohbach and O. Seitz, Achieving Turnover in DNA-Templated Reactions, ChemBioChem, 2008, 9(14), 2185–2192 CrossRef CAS.
- A. K. Sharma and J. M. Heemstra, Small-molecule-dependent split aptamer ligation, J. Am. Chem. Soc., 2011, 133(32), 12426–12429 CrossRef CAS.
- R. Manetsch, A. Krasinski, Z. Radic, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, In situ click chemistry: enzyme inhibitors made to their own specifications, J. Am. Chem. Soc., 2004, 126(40), 12809–12818 CrossRef CAS.
- A. Krasinski, Z. Radic, R. Manetsch, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors, J. Am. Chem. Soc., 2005, 127(18), 6686–6692 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |