Antigen-loaded pH-sensitive hydrogel microparticles are taken up by dendritic cells with no requirement for targeting antibodies†
Laura E. Ruffab, Enas A. Mahmoudc, Jagadis Sankaranarayananc, José M. Morachisc, Carol D. Katayamaa, Maripat Corrd, Stephen M. Hedricka and Adah Almutairi*c
aDivision of Biological Sciences and the Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla CA 92093, USA
bBiomedical Sciences Graduate Program, University of California, San Diego, La Jolla CA 92093, USA
cSkaggs School of Pharmacy and Pharmaceutical Sciences and Departments of Materials Science and Engineering and NanoEngineering, University of California, San Diego, La Jolla CA 92093, USA. E-mail: aalmutairi@ucsd.edu
dDepartment of Medicine, University of California, San Diego, La Jolla CA 92093, USA
First published on 18th September 2012
Abstract
Particle-based delivery of encapsulated antigens has great potential for improving vaccine constructs. In this study, we show that antigen-loaded, pH-sensitive hydrogel microparticles are taken up and presented by bone marrow-derived dendritic cells (BMDCs) in vitro and are taken up by dendritic cells (DCs) and monocytes in vivo. This uptake is irrespective of targeting antibodies. BMDCs in vitro and DCs in vivo also display upregulation of activation markers CD80 and CD86 when treated with microparticles, again with no difference in conjugated antibodies, even the agonistic CD40 antibody. We further show that these particles induce enhanced expansion of cytokine-producing CD8 T cells in response to challenge with ovalbumin-expressing vesicular stomatitis virus, in both an accelerated vaccination strategy using pre-loaded BMDCs and a traditional mouse immunization setting.
Insight, innovation, integrationIn this work, we utilize particle engineering and immunology to develop a novel vaccine carrier and explore the cellular mechanism by which it triggers immunity. We adapt pH-sensitive hydrogel microparticles for this purpose, as they should allow escape from endosomes and therefore lead to presentation of encapsulated antigen via the MHC Class I pathway, necessary to induce a cytotoxic T cell response. Not only do we show that the encapsulating antigen in these microparticles generates a stronger T cell response than the free antigen, but we made the intriguing observation that the microparticles are mostly taken up by monocytes rather than dendritic cells, generally considered the target cell type for vaccines. |
1. Introduction
Vaccine strategies based on protein antigens are sought after based on their low toxicity and broad applications.1 They show promise in inducing CD4 and CD8 T cell responses, which are required for effective vaccines against certain pathogens and cancers. However, suitable methods of delivery are required for protein antigens as they can be degraded due to their small size or taken up by non-beneficial cell subsets, and proteins presented in the absence of an adjuvant are frequently tolerogenic.2,3 Particles have been made from lipids, proteins, silica, and polymeric systems to enhance delivery of protein antigens.4 Biodegradable polymeric nano- or microparticles, such as poly(lactic-co-glycolic acid) (PLGA) and pH-sensitive hydrogels, are particularly attractive delivery vehicles, as they can encapsulate cargo, present targeting moieties on their surface, and are easily formulated to control for their size and intracellular release kinetics.5,6We have chosen to use pH-sensitive hydrogel particles; these particles are stable at pH 7.4, but upon phagocytosis and entry into endosomes, they degrade into linear polymer chains and small molecules due to incorporation of an acid-sensitive crosslinker in their matrix.7 These particles have been proposed to produce a colloid osmotic disruption of the endosome, and this releases their cargo into the cytoplasm allowing for presentation via the MHC class I pathway.7 This lysosomal disruption may activate dendritic cells; bone marrow-derived dendritic cells (BMDCs) that take up degradable hydrogel nanoparticles display a dendritic (mature) cell surface, while BMDCs that take up non-degradable hydrogel nanoparticles display a stellate (immature) surface.8 Leakage into the cytoplasm also circumvents possible degradation by hydrolytic enzymes in late endosomes or lysosomes.7,9 Additionally, these particles remain neutral upon degradation, limiting potential toxicity and charge interactions with proteins.10 In contrast, degradation of PLGA produces glycolic and lactic acid, which creates an acidic microenvironment that can denature and inactivate encapsulated proteins.11 pH-sensitive hydrogel particles are also more readily modified than PLGA. The simple formulation procedure for these particles allows further investigation of variables such as size, charge, and surface decoration. pH-sensitive hydrogel particles can also be tailored to encapsulate different types of cargos including proteins, DNA, and siRNA.
pH-sensitive hydrogel particles have been used to deliver the model antigen ovalbumin (OVA) in several studies. In one study, particles induced cell surface receptors indicative of activation on BMDCs even in the absence of an adjuvant.12 Particles have been made that incorporate CpG to provide increased activation,8,12 and other particles have incorporated DEC205-specific antibodies to attempt to increase targeting to DCs.13 However, even without activating or targeting components, an OVA-encapsulating particle enhanced survival rates for mice injected with OVA-expressing EG7 tumor cells.9 It should be noted that only phagocytic cells, such as macrophages and dendritic cells, are able to engulf particles of ∼0.5–3 μm in diameter to an appreciable extent; the particles in the above-mentioned studies ranged in size from 217 nm–1 μm, meaning some of them may be passively targeted to antigen presenting cells (APCs) simply based on their size.
We produced 1.5 μm, pH-sensitive hydrogel microparticles encapsulating OVA with various antibodies decorating the surface to determine if targeting and/or activating components could enhance APC-microparticle interactions. We selected a DEC205-specific antibody (anti-DEC205) as the targeting antibody, agonistic CD40 antibody (anti-CD40) as an activating component, and hemagglutinin antibody (anti-HA) as a negative control. DEC205 is an endocytic receptor, thought to be involved in recognizing ligands expressed by apoptotic or necrotic cells and found primarily on CD8+ DCs.14 Anti-DEC205 has been conjugated with antigen and utilized to target that antigen to DCs. However, without concomitant delivery of an adjuvant, such as anti-CD40, DEC205 targeting induces tolerance to the antigen.15 Anti-HA recognizes the nonapeptide sequence YPYDVPDYA derived from the human influenza virus hemagglutinin (HA) protein, and as such, will not react with mouse cells. Thus, we engineered OVA-encapsulating particles displaying anti-DEC205 or anti-HA with or without anti-CD40 to determine if receptor-mediated endocytic targeting is observed with our formulation of particles, and to determine if anti-CD40 enhances activation of APCs by the particles. As a control, OVA-encapsulating particles without antibodies were also produced.
2. Materials and methods
2.1. Mice
OT-I (CD8 TCR transgenic OVA-specific mice), OT-II (CD4 TCR transgenic OVA-specific mice), and C57BL/6 mice were purchased from the Jackson Laboratories and maintained in specific pathogen-free facilities at UCSD. Animal protocols were approved by the Institutional Animal Care and Use Committee.2.2. Antibody preparation
Anti-HA (12CA5), anti-DEC205 (NLDC-145), and anti-CD40 (FGK45.5) hybridomas were obtained from American Type Culture Collection. Antibodies were then produced by BioNexus, purified using a Montage Prosep G kit (Millipore), and used in production of microparticles.2.3. Preparation of microparticles
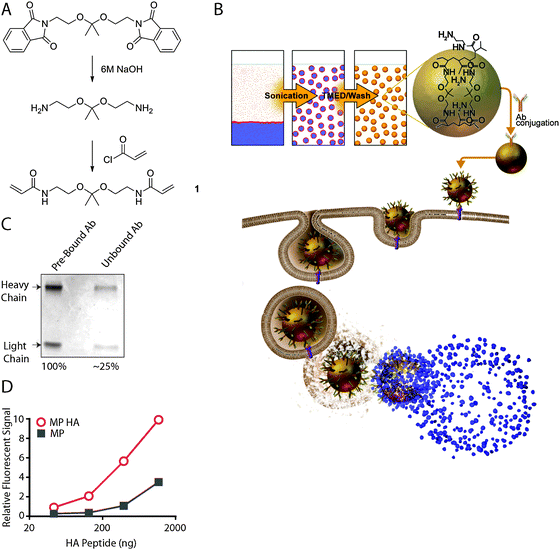 | ||
| Fig. 1 Antibodies conjugated to pH-sensitive hydrogel microparticles maintain binding activity. (A) Synthesis of acid-sensitive crosslinker. (B) Schematic of microparticle production, uptake by a cell, and release of cargo in the endosome. (C) Conjugation efficiency of antibodies to particles. (D) Fluorescein-HA peptide binding activity of MP HA compared to MP. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Span 80:Tween 80 in hexanes were added to the aqueous phase followed by 30 seconds of sonication at an amplitude of 40 (1/8 in tip, Misonix 4000s). The polymerization was initiated by the addition of 25 μL TMED and the particles were left to stir for 10 min at room temperature (RT). The particles were collected by centrifugation at 4000×g for 5 min and washed with hexanes (10 mL, two times) then with acetone (10 mL, three times), and then in PBS, pH 8. The particles were dispersed in PBS, pH 8, passed through a 35 μm filter, and lyophilized after the addition of 5% mannitol. OVA Alexa Fluor 488 was used to prepare particles for in vivo uptake studies.
:1 Span 80:Tween 80 in hexanes were added to the aqueous phase followed by 30 seconds of sonication at an amplitude of 40 (1/8 in tip, Misonix 4000s). The polymerization was initiated by the addition of 25 μL TMED and the particles were left to stir for 10 min at room temperature (RT). The particles were collected by centrifugation at 4000×g for 5 min and washed with hexanes (10 mL, two times) then with acetone (10 mL, three times), and then in PBS, pH 8. The particles were dispersed in PBS, pH 8, passed through a 35 μm filter, and lyophilized after the addition of 5% mannitol. OVA Alexa Fluor 488 was used to prepare particles for in vivo uptake studies.To allow for direct comparison to free OVA, the amount of microparticles is listed as the absolute amount of encapsulated OVA (μg) or as OVA concentrations in a certain volume (μg mL−1) for all experiments. Encapsulated OVA was determined to be 2.4 μg OVA per mg particle powder.
100×g for 30 seconds and washing with PBS, pH 8. Then, antibody solution corresponding to 0.1 mg (0.1 mg for one antibody, or 0.1 mg each for two antibodies) was added and the microparticles were mixed by placing on a shaker at RT for another 30 min. The unbound antibody was separated by washing the particles, centrifuging, and resuspending in 10 mM Tris-HCl, pH 8. Finally, the particles were lyophilized in the presence of 5% mannitol.2.4. Antibody conjugation efficiency and binding activity
The conjugation efficiency was measured by comparing the initial antibody concentration to the concentration of unbound antibodies after the conjugation reaction. The antibody samples were boiled for 5 min in 1x SDS loading buffer (Bio-Rad) and loaded on a 10% SDS poly-acrylamide gel (Bio-Rad). The gel was stained with Coomassie blue (Bio-Rad) for 3 hours followed by 3 destaining washes.The binding activity of anti-HA-conjugated particles encapsulating OVA (MP HA) was compared to particles without antibody encapsulating OVA (MP). A sample of particles containing 15 μg of MP HA or MP was mixed with 3-fold dilutions of fluorescein-labeled HA peptide [YPYDVPDYA; highest dose: 1170 ng (900 pmol); Synthetic Biomolecules]. The unbound peptide was washed away using BioSpin Columns (Bio-Rad) and the quantity of HA peptide bound on particles was measured using fluorescence intensity.
2.5. BMDC cultures
BMDCs were prepared as described previously18 and harvested on day 8.2.6. T cell isolation and purification
For T cell purification, spleens and lymph nodes (LNs) were taken from OT-I or OT-II mice. Splenocytes were depleted of red blood cells (RBCs) using ACK lysis buffer (0.15 M NH4Cl, 1 M KHCO3, 0.1 mM Na2EDTA). Cells were then incubated with a mixture of biotinylated anti-B220 (RA3-6B2, eBioscience—all antibodies are from eBioscience unless otherwise noted), anti-Gr-1 (RB6-8C5), anti-MHC class II (M5/114.15.2), anti-DX5 (DX5), anti-CD11b (M1/70), anti-Ter119 (TER119), and either anti-CD4 (OT-I purification; GK1.5) or anti-CD8α (OT-II purification; 53-6.7). LNs were incubated with a mixture of biotinylated anti-B220 and anti-CD4 (OT-I purification) or biotinylated anti-B220 and anti-CD8α (OT-II purification). Cells were then negatively selected with streptavidin-coupled microbeads (Miltenyi Biotec).2.7. 3[H]-thymidine proliferation assay
20000 BMDCs were cultured for 1 day at 37 °C with 3-fold dilutions of microparticles or EndoGrade OVA (highest dose 1 μg mL−1), then washed twice and incubated with 150000–200000 magnetic bead-sorted OT-I or OT-II T cells for 3 days at 37 °C. 1 μCi per well 3[H]-thymidine (Perkin Elmer Life Sciences) was added for the last 8 hours of culture.2.8. In vitro activation
BMDCs were cultured for 12 hours (in vitro cytokine assay), or 2 days (cell surface receptor assay and ELISA) at 37 °C with 3-fold dilutions of microparticles or EndoGrade OVA (highest dose 1 μg mL−1). 10 μg mL−1 lipopolysaccharide (LPS, Sigma) and PBS were used as controls. Cells used for in vitro cytokine assay were given Brefeldin A (1 μg mL−1; BD Biosciences) 3 hours prior to collection. For flow cytometry experiments, cells were resuspended in FACS buffer (PBS with 1 mM EDTA, 1% FCS, and 0.1% sodium azide), and incubated with anti-mouse FcχRII-III (supernatant from hybridoma 2.4G2 cultures) for 15 min at 4 °C. Cells for in vitro cytokine assay were stained with anti-CD86 APC-Cy7 (Biolegend), anti-B220 PerCP-Cy5.5, and anti-CD11c PE-Cy7. Antibodies for cell surface receptor assay were anti-CD80 FITC (16-10A1, Biolegend), anti-CD86 PE (GL1), anti-B220 PerCP (Biolegend), and anti-CD11c allophycocyanin (N418). In both cases, cells were stained for 20 min at 4 °C.For the in vitro cytokine assay, BMDCs were then fixed and permeabilized to detect intracellular cytokines with anti-IL-10 FITC (JES5-16E3), anti-IL-6 PE (MP5-20F3), anti-IL-12/IL-23 p40 eFluor660 (C17.8), and anti-TNFα Pacific Blue (MP6-XT22; Biolegend).
Supernatants from BMDC used in ELISA were collected after 2 days and IL-1β was detected with the ELISA Ready-SET-Go! kit (eBioscience).
2.9. In vivo DC activation and fluorescent particle uptake
10 μg microparticles were injected subcutaneously (s.c.) in both hind footpads of mice (20 μg per mouse, 3–4 mice per group). Mice were sacrificed at day 1 for fluorescent particle uptake and day 2 for in vivo DC activation. Popliteal and inguinal LNs were taken and analyzed by flow cytometry.2.10. BMDC vaccinations for VSV-OVA challenge
BMDCs were incubated overnight with 0.85 μg mL−1 MP HA, 0.85 μg mL−1 MP HA + 0.42 μg mL−1 LPS, 0.42 μg mL−1 LPS + 5 μM SIINFEKL peptide positive control (OT-I peptide; derived from OVA257-264, the epitope to which the T cells of TCR transgenic OT-I mice react; Genemed Synthesis) or PBS control. 250000 of these BMDCs were injected intravenously (i.v.) into mice (4 mice per group). One week after BMDC vaccination, mice were challenged i.v. with 1 × 105 PFU vesicular stomatitis virus-OVA (VSV-OVA; Dr Leo Lefrançois, University of Connecticut Health Center).2.11. Vaccination for VSV-OVA challenge
10 μg MP HA, 10 μg MP HA + 10 μg LPS, 10 μg LPS + 10 μg EndoGrade OVA, 10 μg EndoGrade OVA, or PBS were injected s.c. in both hind footpads of mice (4 mice per group). 45 days after vaccination, mice were challenged i.v. with 105 PFU VSV-OVA.2.12. Flow cytometry
Peripheral blood lymphocytes (PBL), LNs, or spleen was isolated from mice at various time points. For DC analysis, LNs were first incubated for 20 min at 37 °C with collagenase D (1 mg mL−1; Roche). Single-cell suspensions were prepared and cells from PBL and spleen were subjected to RBC lysis using ACK. Cell staining was performed as for BMDC staining (2.8 In vitro activation). Antibodies used for: in vivo DC activation—CD80 FITC, CD86 PE, CD11c PE-Cy7, B220 APC-Cy7, and CD3 PerCP-Cy5.5 (145-2C11); in vivo fluorescent microparticle uptake—Ly6G PE (1A8, BD Pharmingen), B220 APC-Cy7, TCRβ APC-Cy7 (H57-597), Ly6C allophycocyanin (HK1.4), CD11c PE-Cy7, CD11b eFluor450, CD8 PerCP (Biolegend), CD4 biotin, and streptavidin Qdot525 (Invitrogen); and VSV-OVA challenge experiments—B220 FITC, CD11b FITC, Ter119 FITC (used in the BMDC accelerated vaccination experiment only), CD4 PE-Cy7, CD8 Pacific Blue (Biolegend), and CD44 APC-Cy7 (IM7).2.13. Intracellular cytokine staining
For detection of intracellular cytokines, PBL, LNs, or spleen was isolated and cultured with 5 μM OT-I peptide. After 1 hour, Brefeldin A was added and the cultures were incubated at 37 °C overnight. Cells were stained as above, then fixed and permeabilized to detect intracellular IFNγ allophycocyanin (XMG1.2).3. Results
3.1. Production of pH-sensitive hydrogel microparticles
To produce pH-sensitive hydrogel microparticles, an acid-sensitive crosslinker (Fig. 1A) was produced as described previously16,17 and copolymerized with acrylamide by inverse emulsion polymerization (Fig. 1B). This crosslinker is cleaved by acid-catalyzed hydrolysis; thus, once a particle is taken up, the acidic environment of the endosome will initiate particle degradation and release of its contents, resulting in release of antigen into the cytoplasm. Presumably some endocytic vesicles are shunted into the MHC class II presentation pathway, whereas, access to the cytoplasm also promotes presentation in association with MHC class I (Fig. 1B). We chose ovalbumin (OVA) as the cargo to load in the particles, since the experimental tools for studying the immune response to OVA are extensive. Specifically, to study T cell responses, we used CD8 and CD4 OVA-specific TCR transgenic mice (OT-I and OT-II, respectively) and VSV-OVA, a pathogen engineered to express OVA. The amount of OVA loaded was 2.4 μg per mg of particles, with an encapsulation efficiency of 52.6% ± 14.7%. To allow for direct comparison of particles to free OVA, we list particle concentration as the concentration of OVA and not the concentration of particles. Following particle formulation, anti-DEC205, anti-HA, and/or anti-CD40 were added via BS3 ligation of amine groups on the antibodies (MP DEC, MP HA, MP CD40, MP DEC CD40, and MP HA CD40; particles without antibodies are referred to as MP). To estimate conjugation efficiency of the antibodies, a protein gel was run, comparing total antibody loaded and unbound antibody following BS3 conjugation (Fig. 1C). Approximately 75% of antibody starting material was conjugated to the particles, with only minimal non-specific binding. The particles average diameter based on volume was 1.5 ± 0.8 μm; the measurement was done by laser diffraction particle sizing using a Mastersizer 2000 instrument (Malvern), and confirmed using Multisizer 4 (Beckman) and Qnano (Izone). Finally, endotoxin testing (PyroGene® Recombinant Factor C Endotoxin Detection Assay; Lonza) revealed that the final particles were indeed endotoxin-free, as they contained <0.01 EU per mg of particles.3.2. Antibody binding is preserved following production of microparticles
Antibody activity following conjugation to particles is generally not assessed, even though a particle formulation that destroys antibody activity or blocks antibody binding sites effectively nullifies addition of that component. We took advantage of the well-established anti-HA–HA peptide chemistry to develop an assay utilizing fluorescein-tagged HA peptide to determine whether the binding ability of anti-HA was still active following BS3 conjugation. Peptide ligands for anti-DEC and anti-CD40 are not known, and thus could not be tested. MP HA and MP were incubated with increasing amounts of the labeled HA peptide, excess peptide was washed out, and fluorescence intensity was measured and compared between microparticles (Fig. 1D). Greater than 5-fold binding activity was observed for MP HA versus MP, indicating that anti-HA is still capable of binding peptide. DEC-205 and CD40 antibody conjugates are expected to behave similarly, as BS3 will randomly couple any primary amines on the antibodies to the primary amines on the particles.3.3. CD8 and CD4 T cells incorporate 3[H] in an in vitro proliferation assay in the presence or absence of targeting antibodies on microparticles
To determine whether targeting antibodies could influence antigen delivery to DCs and subsequent proliferation by T cells, we incubated microparticles with BMDCs. Following overnight incubation, BMDCs were washed to remove free microparticles and incubated with magnetic-bead sorted OT-I (CD8) and OT-II (CD4) T cells for three days. 3[H]-thymidine uptake was used to quantitate DNA synthesis induced in culture. We found that OVA was presented by BMDCs on both MHC class I and II (Fig. 2A) for all microparticles, regardless of conjugated antibodies, whereas, at the concentrations tested, free OVA was ineffective. 3[H]-thymidine incorporation was statistically significant for all microparticles by two-way ANOVA, compared to OVA alone. Fig. 2B shows post hoc testing at the 1 μg mL−1 OVA dose.![Co-culture with microparticle-injected BMDCs produces CD8 and CD4 proliferation in vitro. BMDCs were incubated with 3-fold dilutions of microparticles or OVA (highest dose: 1 μg mL−1) for 1 day. Cells were then incubated with OT-I (CD8) or OT-II (CD4) T cells for 3 days. 1 μCi per well 3[H]-thymidine was added for the final 8 hours of culture. (A) Graph of CPM vs. dose (amount of OVA in microparticles). Data shown are mean ± SD. (B) CPM graphs for microparticles at 1 μg mL−1 OVA with two-way ANOVA Dunnett's post hoc test. Microparticles are compared to OVA. ***p < 0.001, two-way ANOVA.](/image/article/2013/IB/c2ib20109g/c2ib20109g-f2.gif) | ||
| Fig. 2 Co-culture with microparticle-injected BMDCs produces CD8 and CD4 proliferation in vitro. BMDCs were incubated with 3-fold dilutions of microparticles or OVA (highest dose: 1 μg mL−1) for 1 day. Cells were then incubated with OT-I (CD8) or OT-II (CD4) T cells for 3 days. 1 μCi per well 3[H]-thymidine was added for the final 8 hours of culture. (A) Graph of CPM vs. dose (amount of OVA in microparticles). Data shown are mean ± SD. (B) CPM graphs for microparticles at 1 μg mL−1 OVA with two-way ANOVA Dunnett's post hoc test. Microparticles are compared to OVA. ***p < 0.001, two-way ANOVA. | ||
3.4. Microparticles induce upregulation of activation markers on DCs in vitro and in vivo
To examine whether microparticles could activate DCs in vitro, we incubated BMDCs with the different microparticles for two days, then performed FACS analysis for upregulation of the activation markers CD80 and CD86. All particles similarly induced both molecules, compared with the same dose of OVA (Fig. 3A–C). All microparticles conjugated with anti-CD40 upregulated CD80 and CD86 to the same levels as MP DEC, MP HA, or MP, despite the latter three not having an activating moiety. | ||
| Fig. 3 All microparticles, regardless of antibody conjugation, induce upregulation of CD80 and CD86 but not cytokine production by BMDCs. (A–C) BMDCs were incubated with 3-fold dilutions of microparticles or OVA (highest dose: 1 μg mL−1). Controls were PBS or LPS. After 2 days, cells were stained for CD80, CD86, B220 and CD11c and analyzed by flow cytometry. (A) Maximum fluorescence intensity (MFI) of CD80 or CD86 for CD11c + B220-cells. Two-way ANOVA for CD80 and CD86 *** p < 0.001. (B) CD80 and CD86 expression induced by microparticles at 1 μg mL−1 OVA with two-way ANOVA Dunnett's post hoc test versus OVA. PBS and LPS were not included in ANOVA analysis. (C) Representative histograms of CD80 and CD86 staining. Shown: OVA, MP HA, both 1 μg mL−1 OVA, and PBS. (D, E) Activation of DCs in vivo. Mice were given 10 μg microparticles s.c. in both hind footpads. After 2 days, mice were sacrificed and popliteal and inguinal LNs were taken. Cells were stained for CD11c, B220, CD3, CD80, and CD86. Graphs are MFI of (D) CD86 or (E) CD80 for CD3-B220-CD11c + cells. 4 mice per group. Statistics are one-way ANOVA with Dunnett's post hoc test (compared to OVA). *p < 0.05, **p < 0.01, ***p < 0.001. (F, G) Cytokine production by BMDC. BMDC were cultured as in A–C, and harvested after 12 hours (F) or 2 days (G). (F) Cells were stained with CD86, CD11c, B220, IL-12/IL-23p40, IL-10, IL-6, and TNFα and analyzed by flow cytometry. Graph is MFI of cytokines and CD86 for CD11c + B220-cells. (G) ELISA for IL-1β on supernatants from BMDC cultures. | ||
To measure in vivo activation of DCs, we injected microparticles s.c. into both hind footpads of mice. Two days later, the mice were sacrificed and popliteal (draining) and inguinal LNs were taken and stained for CD80 and CD86. The microparticles induced modest upregulation of both markers in popliteal LNs (Fig. 3D), and CD86 was upregulated in inguinal LNs (Fig. 3E). This induction was similar to LPS activation.
To further examine whether microparticles induced DC activation, we measured production of the cytokines IL-6, IL-12/23p40, IL-10, and TNFα by flow cytometry, following a 12 hour culture of BMDCs with MP, MP HA, MP CD40, or OVA (Fig. 3F). No cytokine production was observed in response to any of the particles or OVA. To test for inflammasome induction, we measured IL-1β production in BMDC supernatants by ELISA (Fig. 3G). No cytokine response was observed by this measure either. Combined, these results indicate that the particles provide suboptimal activation in vitro, and CD40 conjugation does not enhance activation.
3.5. Fluorescent microparticles interact with monocytes and DCs in vivo
To determine which cells were interacting with (and potentially taking up) microparticles in vivo, we made MP, MP HA, and MP DEC using Alexa Fluor 488-tagged OVA. We injected the microparticles s.c. in both hind footpads. Free OVA-Alexa Fluor 488 and PBS were used as controls. Mice were sacrificed 1 day later and popliteal LNs were examined. Cells were phenotyped as T cells (TCRβ+), B cells (B220+), plasmacytoid DCs (pDCs, CD11c+B220+), DCs (CD11c+B220−), monocytes (CD11bhi Ly6C+Ly6G−), and neutrophils (CD11bhi Ly6C+Ly6G+). Interestingly, a substantial CD11bhi CD11clo population emerged in all microparticle-injected mice that was almost completely absent from OVA-injected or PBS-injected mice (Fig. 4A, B). This population consisted of both monocytes and neutrophils, based on Ly6G and Ly6C expression (Fig. 4A, bottom row). The percentage of each cell subset (B, T, monocyte, neutrophil, DC, and pDC) present in the popliteal LNs is shown in Fig. 4B. Monocyte and neutrophil cell subsets were dramatically increased in these LNs for all microparticle-injected mice versus OVA-injected mice. No increase was observed for the other four subsets. | ||
| Fig. 4 Fluorescent particles recruit neutrophils and monocytes to the draining LN. 10 μg microparticles encapsulating OVA Alexa Fluor 488 were injected s.c. in both hind footpads. Mice were sacrificed after 1 day, and popliteal LNs were taken. Cells were stained for CD11c, CD11b, B220, TCRβ, CD4, CD8, Ly6G and Ly6C. (A) Flow cytometry of cells previously gated to include only live cells (FSC vs. SSC). Top row: representative FACS plots from MP HA-, OVA-, and PBS-injected mice. Bottom row: CD11bhi CD11clo cells were both neutrophils and monocytes. (B, C) Data were combined from two experiments, three mice each. (B) Percentage of monocytes and neutrophils in microparticle- versus OVA-injected popliteal LNs. (C) The efficiency of particle or OVA uptake for each cell subset. | ||
Popliteal LN cells were analyzed for Alexa Fluor 488 staining to identify the populations that had taken up the microparticles. Monocytes and DCs were most effective at taking up antigen in microparticle-injected mice, while neutrophils were comparatively less efficient (Fig. 4C). There was no difference in the efficiency of microparticle uptake compared with OVA in the monocyte and DC subsets; however, as there were many more monocytes in the popliteal LNs of the microparticle-injected mice there was a much larger total number of Alexa Fluor 488+ monocytes in these groups. DC uptake of particles versus OVA was not significantly different (Fig. 4C), and no difference was seen in uptake amongst DC subsets (CD4+, CD8+, CD4–CD8- or DEC205+; data not shown). Finally, no differences in uptake were observed between the different microparticles (Fig. 4C).
3.6. Vaccination with microparticles increases IFNγ + CD8 T cells in response to VSV-OVA challenge
To assess memory-inducing potential of the microparticles, two vaccination strategies were used: accelerated vaccination using BMDCs pre-incubated with microparticles, and injection of mice directly with microparticles. As no differences had been observed between the particles in the previous experiments, only MP HA was selected for these experiments.For accelerated vaccination using BMDCs, we incubated cells overnight with MP HA, MP HA + LPS, PBS, or as a positive control, LPS plus the 8-amino acid antigenic peptide (SIINFEKL, OT-I peptide). Cells were then collected and washed, and 250000 were injected per mouse i.v. (4 mice per group). After 1 week, mice were challenged with VSV-OVA. Mice were bled over the following 2 months and sacrificed at day 55, when spleens were taken. To track immune response to OVA, PBL or spleen cells were restimulated overnight with SIINFEKL in the presence of Brefeldin A, and then analyzed for intracellular IFNγ. Mice given MP HA or MP HA + LPS BMDCs had a large percentage of SIINFEKL-specific IFNγ + CD8 T cells (15–20% out of CD8+), as tracked in blood at the peak of the response (Fig. 5A). This response was significantly greater than that of the PBS control group. MP HA + LPS was also significantly greater than LPS + SIINFEKL peptide and MP HA. At sacrifice, spleens from the MP HA + LPS group had a significantly greater percentage of IFNγ + CD8 T cells than PBS; LPS + peptide and MP HA groups were also elevated, though not statistically significant (Fig. 5B).
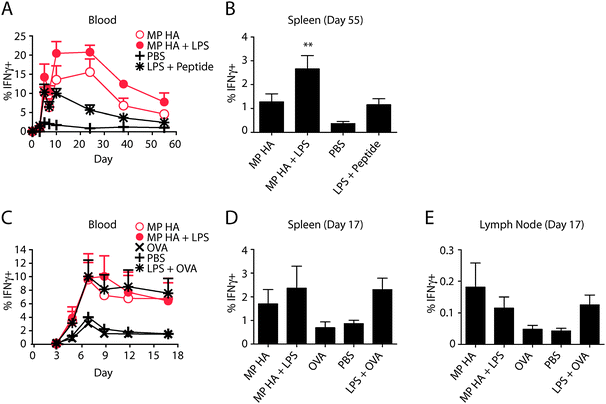 | ||
| Fig. 5 Vaccination with microparticles increases IFNγ + CD8 T cells in response to VSV-OVA challenge. BMDCs were incubated overnight with 0.85 μg mL−1 MP HA, 0.85 μg mL−1 MP HA + 0.42 μg mL−1 LPS, 0.42 μg mL−1 LPS + 5 μM SIINFEKL (OT-I peptide), or PBS control. 250000 of these BMDCs were given to mice i.v. 7 days post-BMDC injection, mice were challenged with 105 PFU VSV-OVA i.v. and (A) the T cell response was tracked via blood on days 0, 3, 5, 7, 10, 24, 38, and 55 post-challenge. Lymphocytes from blood were purified, restimulated with OT-I peptide, and stained for CD4, CD8, IFNγ, CD44, B220, Ter119, and CD11b. Antigen-specific cells were detected by IFNγ + CD44+ staining. Data shown are previously gated on CD8+ T cells. (B) At day 55, mice were sacrificed, spleens were taken, and antigen-specific cells were detected as in blood. One-way ANOVA with Bonferroni post hoc testing. (C–E) Direct immunogenicity. Mice were given 10 μg MP HA, 10 μg MP HA + 10 μg LPS, 10 μg LPS + 10 μg EndoGrade OVA, 10 μg EndoGrade OVA, or PBS s.c. in both hind footpads. At day 45, mice were challenged with 105 PFU VSV-OVA i.v., (C) and subsequently bled at days 3, 5, 7, 9, 12, and 17. Lymphocytes from blood were purified, restimulated with OT-I peptide and stained for CD4, CD8, IFNγ, CD44, B220, and CD11b. Antigen-specific cells were detected by IFNγ + CD44+ staining. Data shown are previously gated on CD8+ T cells. (D, E) At day 17, mice were sacrificed, spleens and LNs were taken, and antigen-specific cells were detected as in blood. | ||
The direct immunogenicity of microparticles was also tested. Mice were given MP HA, MP HA + LPS, LPS + OVA, OVA, or PBS s.c. in both hind footpads, and at day 45 they were challenged with VSV-OVA. Immune responses were then tracked in blood, as for the BMDC accelerated vaccination (Fig. 5C). At day 17, mice were sacrificed and spleens and LNs were taken. Immune responses of MP HA- and MP HA + LPS-injected mice were similar to the LPS + OVA-injected mice (a secondary immune response to the challenge), while OVA-injected mice displayed an immune response similar to PBS (a primary immune response). No additive effect was observed for MP HA + LPS compared to MP HA. IFNγ + CD8 responses in spleen and LNs were elevated in MP HA and MP HA + LPS to similar levels as LPS + OVA (Fig. 5D–E). OVA-specific CD4 responses were too small to be tracked in blood (data not shown).
4. Discussion
We have produced 1.5 μm, OVA-encapsulating, pH-sensitive hydrogel microparticles, with or without targeting and activating antibodies. These particles were all effective at generating CD4 and CD8 T cell responses in vitro and upregulating cell surface receptors on DCs in vitro and in vivo. No preferential uptake was observed with MP DEC versus MP HA. No increased activation was observed with microparticles displaying an antibody that binds CD40 and stimulates an activation program in DCs and B cells.19 Testing of the MP HA construct revealed that antibody binding sites were still active, indicating that particle production did not damage the antibodies. Cytokine responses by BMDC were not observed in vitro; however, MP HA immunization and BMDC-MP HA transfer both induced an expanded CD8 T cell response to VSV-OVA challenge, more comparable to a secondary than a primary immune response. Furthermore, immunization of LPS with MP HA did not induce greater CD8 T cell responses, as may have been expected if the APCs taking up the particles were suboptimally primed.Previous studies have shown that the efficacy of targeting antibodies on particles depends on antibody density, analysis timepoint, size of the particles, and route of injection.13,20–22 It has recently been shown that the density of DEC205 antibody on the surface of PLGA nanoparticles dictates increases in IL-10 production by DCs, with no change in CD80/86 upregulation. DEC205 on these particles also had only a small targeting effect, observed at very early timepoints.20 Our targeted particles may have effected similar changes. Cruz et al. compared targeting by DC-SIGN antibodies between PLGA nano- and microparticles in vitro, and only observed a targeting effect with nanoparticles.21 Kwon et al. showed a non-significant trend towards increased targeting of DEC205 on hydrogel microparticles.13 The route of injection for in vivo experiments also differed in these papers from ours; the DEC205 PLGA nanoparticles were injected intraperitoneally, Kwon et al., injected s.c. in the tail,13,20 and we injected s.c. in the footpad.
We did not observe cytokines indicative of BMDC activation or inflammasomes in vitro. However, upon MP HA immunization and subsequent challenge with VSV-OVA, we observed a CD8 T cell response comparable to a secondary immune response. This response was not increased by including LPS in the immunization. It is possible that the particles can synergize with endogenous factors in vivo, as Sharp et al. observed with PLG and PS microparticles. In this paper, IL-1β was only observed in vitro when TLR agonists were included in the assay, but TLR agonists were not required to induce IL-1β in vivo. They further showed that high-mobility group box 1 (HMGB1), which is released from necrotic cells, could synergize with their particles.23
Interestingly, the in vivo uptake of our microparticles is dominated by monocytes. DCs also take up the particles, though at the same level as fluorescent OVA. Monocytes and neutrophils are both recruited to the draining LN in large numbers, with ∼10-fold more monocytes and ∼40–50-fold more neutrophils in microparticle-injected versus OVA-injected counterparts. However, monocytes are the primary cell type that interacts with the particles; in the draining LN, mice immunized with any of the microparticles have ∼10-fold more particle-positive monocytes compared to OVA+ monocytes in mice immunized with OVA. Monocytes can differentiate into monocyte-derived DCs (moDCs) in cases of inflammation, infection, or even at the steady state.24–26 We hypothesize that monocytes may pick up the particle in the footpad and then move to the draining LN, where they may further differentiate into these APCs. In fact, 0.5–1 μm fluorescent microspheres injected s.c. are also taken up by monocytes and carried to the LN.24 Our observation is also similar to that in mice treated with LPS or gram-negative bacteria, where peripheral monocytes migrate to LNs, differentiate into moDCs, and become the predominant APC.27 The large neutrophil recruitment we observe may be due to their phagocytic nature, an acute inflammation occurring in response to the particles, or perhaps even to modulate the DC or moDC response. While neutrophils are typically thought of as short-lived cells that are recruited to phagocytose cells and release effector molecules, they have also been shown to compete for the antigen in the LN28 and induce maturation of DCs or moDCs.29–32
This study shows that pH-sensitive hydrogel microparticles are self-targeting and have the potential to induce both CD8 and CD4 T cell immune responses. When injected s.c., these particles are predominantly taken up by monocytes, and both monocytes and neutrophils are recruited to the draining lymph node in large numbers, two findings that have not been shown previously for hydrogel microparticles. Activation of APCs appears to be suboptimal in vitro, but no such defect is observed in vivo, where particles may synergize with endogenous factors. The properties of these particles cannot be improved by addition of targeting/activating antibodies or concurrent administration of LPS as an adjuvant. The results illustrate a pathway to improved vaccination by focusing on the facilitated uptake of microparticles and on their ability to induce DC and myeloid activation.
Competing interests statement
The authors declare no competing financial interests.Acknowledgements
We acknowledge Osayi Eidgin and Jamie Moser for their help in preparing the hydrogel particles. L.E.R. was supported by University of California, San Diego (UCSD) Immunology training grant NIH T32AI060536. J.M.M. was supported by UCSD IRACDA Fellowship Grant GM06852. We thank NIH Directors New Innovator Award (1 DP2 OD006499-01) and King Abdul Aziz City of Science and Technology (KACST) for financially supporting this study.References
- J. A. Berzofsky, J. D. Ahlers and I. M. Belyakov, Nat. Rev. Immunol., 2001, 1, 209–219 CrossRef CAS.
- C. A. Janeway, Cold Spring Harbor Symp. Quant. Biol., 1989, 54 Pt 1, 1–13 CrossRef.
- L. S. K. Walker and A. K. Abbas, Nat. Rev. Immunol., 2002, 2, 11–19 CrossRef CAS.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther. Ser., 2008, 83, 761–769 CrossRef CAS.
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Control Release, 2008, 125, 193–209 CrossRef CAS.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- N. Murthy, M. Xu, S. Schuck, J. Kunisawa, N. Shastri and J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4995–5000 CrossRef CAS.
- Y. J. Kwon, S. M. Standley, S. L. Goh and J. M. J. Fréchet, J. Control Release, 2005, 105, 199–212 CrossRef CAS.
- S. M. Standley, Y. J. Kwon, N. Murthy, J. Kunisawa, N. Shastri, S. J. Guillaudeu, L. Lau and J. M. J. Fréchet, Bioconjucate Chem., 2004, 15, 1281–1288 CrossRef CAS.
- A. P. Sassi, A. J. Shaw, H. W. Blanch and J. M. Prausnitz, Polymer, 1996, 37, 2151–2164 CrossRef CAS.
- G. Zhu, S. R. Mallery and S. P. Schwendeman, Nat. Biotechnol., 2000, 18, 52–57 CrossRef CAS.
- S. M. Standley, I. Mende, S. L. Goh, Y. J. Kwon, T. T. Beaudette, E. G. Engleman and J. M. J. Fréchet, Bioconjucate Chem., 2007, 18, 77–83 CrossRef CAS.
- Y. J. Kwon, E. James, N. Shastri and J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18264–18268 CrossRef CAS.
- R. E. Shrimpton, M. Butler, A.-S. S. Morel, E. Eren, S. S. Hue and M. A. Ritter, Mol. Immunol., 2009, 46, 1229–1239 CrossRef CAS.
- L. Bonifaz, D. Bonnyay, K. Mahnke, M. Rivera, M. C. Nussenzweig and R. M. Steinman, J. Exp. Med., 2002, 196, 1627–1638 CrossRef CAS.
- R. Jain, S. M. Standley and J. M. J. Fréchet, Macromolecules, 2007, 40, 452–457 CrossRef CAS.
- S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. J. Fréchet, Bioconjucate Chem., 2008, 19, 911–919 CrossRef CAS.
- A. S. Dejean, D. R. Beisner, I. L. Ch'en, Y. M. Kerdiles, A. Babour, K. C. Arden, D. H. Castrillon, R. A. DePinho and S. M. Hedrick, Nat. Immunol., 2009, 10, 504–513 CrossRef CAS.
- A. Rolink, F. Melchers and J. Andersson, Immunity, 1996, 5, 319–330 CrossRef CAS.
- A. Bandyopadhyay, R. L. Fine, S. Demento, L. K. Bockenstedt and T. M. Fahmy, Biomaterials, 2011, 32, 3094–3105 CrossRef CAS.
- L. J. Cruz, P. J. Tacken, R. Fokkink, B. Joosten, M. C. Stuart, F. Albericio, R. Torensma and C. G. Figdor, J. Control Release, 2010, 144, 118–126 CrossRef CAS.
- J. M. Morachis, E. A. Mahmoud and A. Almutairi, Pharmacol. Rev., 2012, 505–519 CrossRef.
- F. A. Sharp, D. Ruane, B. Claass, E. Creagh, J. Harris, P. Malyala, M. Singh, D. T. O'Hagan, V. Pétrilli, J. Tschopp, L. A. J. O'Neill and E. C. Lavelle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 870–875 CrossRef CAS.
- G. J. Randolph, K. Inaba, D. F. Robbiani, R. M. Steinman and W. A. Muller, Immunity, 1999, 11, 753–761 CrossRef CAS.
- B. León, M. López-Bravo and C. Ardavín, Immunity, 2007, 26, 519–531 CrossRef.
- C. Varol, L. Landsman, D. K. Fogg, L. Greenshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann and S. Jung, J. Exp. Med., 2007, 204, 171–180 CrossRef CAS.
- C. Cheong, I. Matos, J.-H. Choi, D. B. Dandamudi, E. Shrestha, M. P. Longhi, K. L. Jeffrey, R. M. Anthony, C. Kluger, G. Nchinda, H. Koh, A. Rodriguez, J. Idoyaga, M. Pack, K. Velinzon, C. G. Park and R. M. Steinman, Cell, 2010, 143, 416–429 CrossRef CAS.
- C.-W. Yang, B. S. I. Strong, M. J. Miller and E. R. Unanue, J. Immunol., 2010, 185, 2927–2934 CrossRef CAS.
- K. P. J. M. van Gisbergen, M. Sanchez-Hernandez, T. B. H. Geijtenbeek and Y. van Kooyk, J. Exp. Med., 2005, 201, 1281–1292 CrossRef CAS.
- K. P. J. M. van Gisbergen, I. S. Ludwig, T. B. H. Geijtenbeek and Y. van Kooyk, FEBS Lett., 2005, 579, 6159–6168 CrossRef CAS.
- A. M. Megiovanni, F. Sanchez, M. Robledo-Sarmiento, C. Morel, J. C. Gluckman and S. Boudaly, J. Leukocyte Biol., 2006, 79, 977–988 CrossRef CAS.
- S. Bennouna and E. Y. Denkers, J. Immunol., 2005, 174, 4845–4851 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR of the acid-sensitive crosslinker. See DOI: 10.1039/c2ib20109g |
| This journal is © The Royal Society of Chemistry 2013 |