 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot combination of enzyme and Pd nanoparticle catalysis for the synthesis of enantiomerically pure 1,2-amino alcohols†
Joerg H.
Schrittwieser
*a,
Francesca
Coccia
b,
Selin
Kara
a,
Barbara
Grischek
c,
Wolfgang
Kroutil
c,
Nicola
d'Alessandro
d and
Frank
Hollmann
*a
aDepartment of Biotechnology, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: j.schrittwieser@tudelft.nl; f.hollmann@tudelft.nl; Fax: (+31) 152 781415; Tel: (+31) 152 781957
bSchool of Advanced Studies in Science, University “G. d'Annunzio” of Chieti-Pescara, Via dei Vestini 31, 66013 Chieti, Italy
cDepartment of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
dDepartment of Engineering and Geology (INGEO), University “G. d'Annunzio” of Chieti-Pescara, Viale Pindaro 42, 65127 Pescara, Italy
First published on 11th September 2013
Abstract
One-pot combinations of sequential catalytic reactions can offer practical and ecological advantages over classical multi-step synthesis schemes. In this context, the integration of enzymatic and chemo-catalytic transformations holds particular potential for efficient and selective reaction sequences that would not be possible using either method alone. Here, we report the one-pot combination of alcohol dehydrogenase-catalysed asymmetric reduction of 2-azido ketones and Pd nanoparticle-catalysed hydrogenation of the resulting azido alcohols, which gives access to both enantiomers of aromatic 1,2-amino alcohols in high yields and excellent optical purity (ee >99%). Furthermore, we demonstrate the incorporation of an upstream azidolysis and a downstream acylation step into the one-pot system, thus establishing a highly integrated synthesis of the antiviral natural product (S)-tembamide in 73% yield (ee >99%) over 4 steps. Avoiding the purification and isolation of intermediates in this synthetic sequence leads to an unprecedentedly low ecological footprint, as quantified by the E-factor and solvent demand.
Introduction
The integration of several chemical transformations into one-pot processes (often referred to as ‘domino’, ‘tandem’, or ‘cascade’ systems) offers advantages with respect to operational simplicity, operating time and costs, safety, and the consumption of energy and materials.1 From an ecological perspective, the one-pot combination of catalytic methods2 is particularly appealing, and recent years have brought about remarkable developments in the use of heterogeneous, homogeneous, organo- and biocatalysts in cascade systems.3 However, these approaches typically remain within one individual ‘subfield’ of catalysis, while the one-pot combination of different types of catalysts is less explored. This is particularly true for combinations of chemical catalysts and enzymes, which are often complicated by divergent reaction conditions and detrimental interactions of the bio- and chemo-catalysts.4 Nevertheless, several excellent recent studies aimed at bringing chemo- and biocatalysis closer together clearly demonstrate the potential of chemo-enzymatic one-pot systems.5 In this context, true cascades – in which the biocatalytic and chemical reactions proceed concurrently – definitely represent the most elegant examples, but they are also most prone to the above-mentioned difficulties.6 Sequential chemo-enzymatic one-pot reactions are more easily realised and offer essentially the same environmental advantages (e.g. reduction of solvent use due to elimination of work-up and purification steps).4a,c,7The 2-amino-1-aryl alcohol moiety is a common structural motif in biologically active compounds,8 as it forms the basic scaffold of adrenergics (e.g. the hormones adrenaline and nor-adrenaline, β-adrenergic blockers, and anti-asthma drugs), amphenicol antibiotics, and several bioactive natural products (Fig. 1). In addition, 1,2-amino alcohols have found broad use as chiral ligands and auxiliaries in asymmetric synthesis.9 For both pharmaceutical and synthetic applications, a high optical purity of chiral amino alcohols is desired, causing the need for highly selective asymmetric syntheses of these compounds. Chemical methods – such as asymmetric hydrogenation10 or transfer hydrogenation11 of 2-amino ketones or their synthetic equivalents – have been intensively studied in recent years, yet they often fail to afford enantiomerically pure products. As a result, classical resolution is still a widely used technique in the preparation of chiral 1,2-amino alcohols, either for upgrading the ee of an optically enriched product or for resolving the racemate.10a
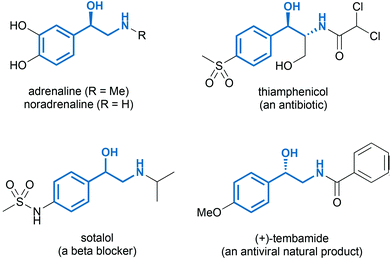 | ||
| Fig. 1 Examples of biologically active 1,2-amino alcohols (2-amino-1-aryl alcohol motif highlighted in bold). | ||
Biocatalysis, on the other hand, offers several options for the preparation of optically pure 1,2-amino alcohols in general, such as kinetic resolution or dynamic kinetic resolution of the racemate using lipases,12 oxidative kinetic resolution of N-protected amino ketones by Baeyer–Villiger monooxygenases,13 the combination of aldolase-catalysed C–C bond formation and enzymatic decarboxylation,14 or the combination of lyase-catalysed C–C bond formation with reductive amination catalysed by ω-transaminases.15 However, biocatalytic asymmetric methods for the preparation of 2-amino-1-aryl alcohols in particular are comparably scarce.12e,14,15a,16a
Herein, we describe a chemo-enzymatic approach for the asymmetric synthesis of this important substance class: a sequential one-pot combination of stereoselective azido ketone reduction by alcohol dehydrogenases and subsequent azide hydrogenation by palladium nanoparticles (Scheme 1), which provides access to both enantiomers of 2-amino-1-aryl alcohols in high yields and with excellent optical purities. The integration of an upstream azidolysis step into the one-pot process and in situ benzoylation of the crude amino alcohol have also been achieved, enabling the one-pot asymmetric synthesis of the natural product tembamide (Fig. 1) in four steps from the corresponding commercially available bromo ketone 1e. The ecological benefits of this one-pot concept are demonstrated by a basic environmental impact assessment.
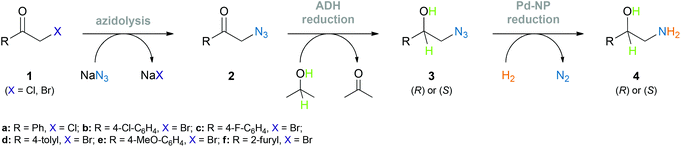 | ||
| Scheme 1 Chemo-enzymatic approach towards optically pure 1,2-amino alcohols via azidolysis, alcohol dehydrogenase (ADH) catalysed asymmetric reduction, and Pd nanoparticle (Pd-NP) catalysed azide hydrogenation. | ||
Results and discussion
Azido alcohol hydrogenation catalysed by metal nanoparticles
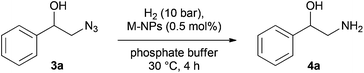
In our one-pot approach, we envisioned combining the asymmetric reduction of 2-azido ketones catalysed by alcohol dehydrogenases (ADHs) with azide hydrogenation catalysed by recently reported17 lignin-stabilised metal nanoparticles (NPs). Since the latter reaction had not been investigated before, we began our studies by screening six different metal nanoparticle preparations (Pd or Pt, stabilised by three different lignin varieties) in the hydrogenation of 2-azido-1-phenylethanol 3a. Phosphate buffer containing 5% (v/v) of 2-propanol as organic co-solvent was chosen as a reaction medium to ensure compatibility with the conditions for the enzyme-catalysed step. As shown in Table 1, Pd-LC and Pd-LK nanoparticles (for explanation of nanoparticle types, see Table 1 footnotes) gave the best results, leading to full conversion within 4 h (entries 1 and 3). Minor amounts of unidentified side products were formed in all reactions, but subsequent experiments showed that by carrying out the reduction under basic conditions (pH 9) the chemoselectivity can be raised further, such that 2-amino-1-phenyl-ethanol 4a was the only product detectable by GC–FID and NMR analysis (Table 1, entry 7). The hydrogenation of 3b–f under identical conditions proceeded with the same high level of selectivity, affording the corresponding 1,2-amino alcohols 4b–f in essentially pure form.18|
|
|||
|---|---|---|---|
| Entry | NP typeb | Conversionc [%] | Selectivityc [%] |
| a Conditions: 100 mM 3a, 0.5 mM NPs, 10 bar H2, 30 °C, 4 h. For details, see the Experimental section. b Lignin varieties: LC = sulfonated lignin with Ca2+ counterions, LA = sulfonated lignin with NH4+ counterions, LK = low-sulfonate Kraft lignin. c Determined by GC–FID analysis. d Reaction under basic conditions (pH 9). | |||
| 1 | Pd–LC | >99 | 86 |
| 2 | Pd–LA | 95 | 89 |
| 3 | Pd–LK | >99 | 85 |
| 4 | Pt–LC | 52 | 89 |
| 5 | Pt–LA | 97 | 85 |
| 6 | Pt–LK | 97 | 87 |
| 7 | Pd–LKd | >99 | >99 |
Asymmetric azido ketone reduction catalysed by ADHs
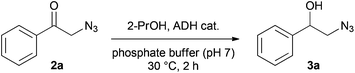
Next, we turned our attention to the ADH-catalysed asymmetric reduction of aromatic 2-azido ketones. Although there is literature precedence for this transformation,19 a broad survey of suitable enzymes has not yet been reported. Therefore, we carried out an extensive screening of 79 commercial ADHs of unspecified origin and four bacterial ADHs (ADH-A from Rhodococcus ruber DSM 44541, TbADH from Thermoanaerobium brockii, LkADH from Lactobacillus kefir, LbADH from Lactobacillus brevis)20 in the reduction of 2a to 3a. The screening was performed at 50 mM concentration of 2a and in the presence of 5% (v/v) 2-propanol, the latter serving both as co-solvent and co-substrate. Table 2 shows selected results of the ADH screening (for complete results, see ESI†). As a general trend, we identified more anti-Prelog-selective21,22 ADHs (18) with activity on 2a than Prelog-selective ones (9), and on average, the former showed about 5–10 times higher activity than the latter. Only eleven enzymes afforded optically pure 3a, whereby four gave the (R)-enantiomer and seven the (S)-enantiomer.23 Based on the observed activities and stereoselectivities, we considered the commercial ADHs listed in Table 2 (entries 5–8) as well as ADH-A (entry 1), the most promising biocatalysts for the reaction under study.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Enzyme | Source/supplier | Cofactorb | Conv.c [%] | Act.d [U mg−1] | eee [%] |
| a Conditions: 50 mM 2a, 0.1 mM NAD+, 0.1 mg mL−1 ADH preparation, 5% (v/v) 2-PrOH, 30 °C, 2 h. For details, see the Experimental section. b Preferred cofactor according to literature or supplier's information. c Determined by GC–FID analysis. d Apparent activity [μmol product/(min mg enzyme preparation)] calculated from conversion. e Enantiomeric excess of 3a determined by chiral-phase GC–FID analysis. f nd = not determined. | ||||||
| 1 | ADH-A | R. ruber | NADH | 12 | 0.5 | >99 (R) |
| 2 | TbADH | T. brockii | NADPH | <1 | <0.1 | nd |
| 3 | LkADH | L. kefir | NADPH | 2 | 0.1 | 98 (S) |
| 4 | LbADH | L. brevis | NADPH | 2 | 0.1 | >99 (S) |
| 5 | KRED-NADH-110 | Codexis | NADH | 58 | 2.4 | >99 (S) |
| 6 | KRED-P3-B03 | Codexis | NADPH | 5 | 0.2 | >99 (R) |
| 7 | evo-1.1.030 | evocatal | NADH | 7 | 0.3 | >99 (R) |
| 8 | evo-1.1.200 | evocatal | NADH | 73 | 3.1 | >99 (S) |
A further selection was made according to the enzymes’ tolerance for higher substrate concentrations and their operational stability. We identified ADH-A and KRED-NADH-110 as the most robust of the investigated Prelog- and anti-Prelog-selective enzymes, respectively, showing reasonable activity at 100 mM concentration of 2a (Suppl. Fig. 1 and 2, ESI†), and a good performance over 24 h of reaction time (Suppl. Fig. 3 and 4, ESI†). The substrate scope of ADH-A and KRED-NADH-110 was investigated using the representative 2-azido-1-arylethanones 2a–f as substrates. Both biocatalysts converted all six ketones tested with excellent stereoselectivity and with similar relative rates; only the reduction of 2f by KRED-NADH-110 was surprisingly slow in comparison to the other 2-azido ketones (Fig. 2). Test transformations with 100 mM substrate concentration and optimised enzyme loadings (ADH-A: 1.5–2.5 mg mL−1, KRED-NADH-110: 0.2–1.0 mg mL−1; for details see ESI†) proceeded to complete conversion within 20 h in all cases.
![Activity of ADH-A and KRED-NADH-110 in the reduction of azido ketones 2a–f. Apparent activity [μmol product/(min mg enzyme preparation)] determined from conversion after 2 h; conditions: 50 mM 2, 0.1 mM NAD+, 0.1 mg mL−1 ADH preparation, 5% (v/v) 2-PrOH, 30 °C. For details, see the Experimental section.](/image/article/2013/GC/c3gc41666f/c3gc41666f-f2.gif) | ||
| Fig. 2 Activity of ADH-A and KRED-NADH-110 in the reduction of azido ketones 2a–f. Apparent activity [μmol product/(min mg enzyme preparation)] determined from conversion after 2 h; conditions: 50 mM 2, 0.1 mM NAD+, 0.1 mg mL−1 ADH preparation, 5% (v/v) 2-PrOH, 30 °C. For details, see the Experimental section. | ||
Chemo-enzymatic one-pot transformations
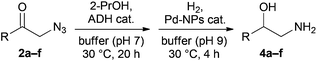
After suitable ADHs had been identified, we proceeded with combining the enzymatic azido ketone reduction and Pd-NP-catalysed azide hydrogenation in a one-pot sequence. The biocatalytic reduction was run to completion (20 h) before adjusting the pH of the reaction mixture to 9 and adding a Pd-NP stock solution. First experiments showed that the activity of the nanoparticles was not impaired by the presence of the enzymes, and complete reduction of the intermediate azido alcohols 3a–f was achieved in 4 h. However, in addition to the expected products 4, the corresponding 2,2-dimethyloxazolidines 5 were also formed in minor amounts (4–10%), apparently by the reaction of 4 with acetone generated as by-product in the ADH-catalysed reduction (Scheme 2).24 When the bioreduction was carried out under reduced pressure, so as to remove the acetone from the solution, the one-pot sequence afforded essentially pure 1,2-amino alcohols 4a–f. In semi-preparative-scale experiments (5 mL volume, 0.5 mmol substrate converted), the desired products could be isolated in good yield and excellent enantiomeric excess (Table 3). Only the isolated yields of 4f fall behind compared to the other amino alcohols, which we attribute to the high aqueous solubility and the resulting difficult extraction of 4f.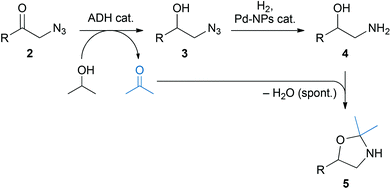 | ||
| Scheme 2 Rationalisation of the formation of 2,2-dimethyloxazolidines 5 in the ADH/Pd-NP one-pot sequence. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Subst. | ADH | GC yield (4)b [%] | Isol. yield (4)c [%] | ee (4)d [%] |
| a Conditions: 100 mM 2, 0.1 mM NAD+, 0.2–2.5 mg mL−1 ADH preparation, 0.5 mM Pd-NPs, 5% (v/v) 2-PrOH, 30 °C, 24 h. For details, see the Experimental section. b Relative amount of 4 in the crude product determined by GC–FID analysis. Rest to 100% is the corresponding 2,2-dimethyloxazolidine 5. c Isolated yield of pure 4 after column chromatography; semi-preparative scale (5 mL; 0.5 mmol substrate). d Enantiomeric excess of 4 as determined by chiral GC–FID analysis after conversion into the corresponding 2,2-dimethyloxazolidine 5. e Switch in substituent priorities according to Cahn–Ingold–Prelog rules. | |||||
| 1 | 2a | ADH-A | 97 | 87 (60 mg) | >99 (R) |
| 2 | 2b | ADH-A | 98 | 89 (76 mg) | >99 (R) |
| 3 | 2c | ADH-A | 99 | 86 (67 mg) | >99 (R) |
| 4 | 2d | ADH-A | 99 | 85 (64 mg) | >99 (R) |
| 5 | 2e | ADH-A | 98 | 85 (71 mg) | >99 (R) |
| 6 | 2f | ADH-A | >99 | 55 (35 mg) | >99 (S)e |
| 7 | 2a | KRED-NADH-110 | 98 | 86 (59 mg) | >99 (S) |
| 8 | 2b | KRED-NADH-110 | 98 | 78 (67 mg) | >99 (S) |
| 9 | 2c | KRED-NADH-110 | 98 | 88 (68 mg) | >99 (S) |
| 10 | 2d | KRED-NADH-110 | 98 | 86 (65 mg) | >99 (S) |
| 11 | 2e | KRED-NADH-110 | 99 | 87 (73 mg) | >99 (S) |
| 12 | 2f | KRED-NADH-110 | >99 | 38 (24 mg) | >99 (R)e |
To further prove the preparative value and the scalability of the one-pot two-step reaction system, we performed the conversion of azidoketone 2b into 1,2-amino alcohol (R)-4b on gram scale (75 mL, 7.5 mmol substrate). ADH-A (1.5 mg mL−1) was used as a biocatalyst, and the target compound was isolated in 84% yield (1.08 g) and >99% ee.
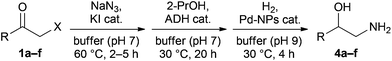
Encouraged by the positive results of the ADH/Pd-NP combination, we sought to integrate the in situ formation of azido ketones 2 from the corresponding halo ketones 1 into the one-pot system. As a first test, 1a was reacted at 60 °C with 1.2 equiv. of NaN3 in buffer containing 5% (v/v) 2-propanol and varied amounts of potassium iodide as a nucleophilic substitution catalyst. With 5 and 10 mol% of iodide, the reaction proceeded almost equally fast, and 90% conversion was achieved within 4 h (Suppl. Fig. 5, ESI†). Transferring the same conditions (10 mol% KI) to the autoclave setup used for the one-pot sequence, the azidolysis reaction of 1a–e proceeded to completion within 5 h, while the furan derivative 1f only required 2 h for full conversion. In addition, we found that the ADH-catalysed reduction could be performed on the crude reaction mixture of the azidolysis step, as a minor decrease in enzyme activity was easily compensated by slightly raising the enzyme loading. Hence, the sequential combination of all three reactions (azidolysis, ADH reduction, and hydrogenation) proved feasible. All steps of the one-pot process could be run to complete conversion, and no accumulation of any side products was observed. Consequently, the 1,2-amino alcohols 4a–f were obtained with the same level of chemical and enantiomeric purity as in the two-step process (see Table 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Subst. | ADH | GC yield (4)b [%] | Isol. yield (4)c [%] | ee (4)d [%] |
| a Conditions: 100 mM 1, 120 mM NaN3, 10 mM KI, 0.1 mM NAD+, 0.3–4.0 mg mL−1 ADH preparation, 0.5 mM Pd-NPs, 5% (v/v) 2-PrOH, 60–30 °C, 26–29 h. For details, see the Experimental section. b Relative amount of 4 in the crude product determined by GC–FID analysis. Rest to 100% is the corresponding 2,2-dimethyloxazolidine 5. c Isolated yield of pure 4 after column chromatography; semi-preparative scale (5 mL; 0.5 mmol substrate). d Enantiomeric excess of 4 as determined by chiral GC–FID analysis after conversion into the corresponding 2,2-dimethyloxazolidine 5. e Switch in substituent priorities according to Cahn–Ingold–Prelog rules. | |||||
| 1 | 1a | ADH-A | >99 | 83 (57 mg) | >99 (R) |
| 2 | 1b | ADH-A | >99 | 75 (63 mg) | >99 (R) |
| 3 | 1c | ADH-A | >99 | 89 (69 mg) | >99 (R) |
| 4 | 1d | ADH-A | >99 | 81 (61 mg) | >99 (R) |
| 5 | 1e | ADH-A | >99 | 75 (63 mg) | >99 (R) |
| 6 | 1f | ADH-A | >99 | 39 (25 mg) | >99 (S)e |
| 7 | 1a | KRED-NADH-110 | 99 | 76 (52 mg) | >99 (S) |
| 8 | 1b | KRED-NADH-110 | >99 | 80 (69 mg) | >99 (S) |
| 9 | 1c | KRED-NADH-110 | >99 | 79 (61 mg) | >99 (S) |
| 10 | 1d | KRED-NADH-110 | >99 | 82 (62 mg) | >99 (S) |
| 11 | 1e | KRED-NADH-110 | >99 | 86 (72 mg) | >99 (S) |
| 12 | 1f | KRED-NADH-110 | >99 | 36 (23 mg) | >99 (R)e |
An exemplary gram-scale conversion (75 mL, 7.5 mmol substrate) of 1c into (R)-4c using ADH-A (2.0 mg mL−1) as a biocatalyst provided the 1,2-amino alcohol in 84% isolated yield (0.97 g) and optically pure form (ee >99%).
Finally, we wanted to apply our newly developed chemo-enzymatic one-pot reaction system to the asymmetric synthesis of a biologically active molecule. As a target compound, we chose (S)-tembamide, a naturally occurring benzamide derivative, for which antiviral (HIV) activity has been reported.25 The synthesis required benzoylation of amino alcohol (S)-4e as the final step, which was achieved by simply adding a solution of benzoyl chloride (1.2 eq.) in MTBE to the alkaline, aqueous reaction mixture obtained after the Pd-NP-catalysed hydrogenation step (Scheme 3). A gram-scale reaction (50 mL, 5.0 mmol substrate, 0.5 mg mL−1 KRED-NADH-110 as biocatalyst) yielded 0.98 g (73% from 1e) of (+)-(S)-tembamide, thus providing access to this natural product in a four-step one-pot operation.
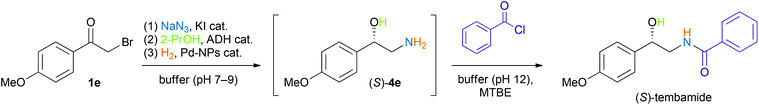 | ||
| Scheme 3 Asymmetric synthesis of (S)-tembamide in a chemo-enzymatic four-step one-pot sequence. | ||
Environmental impact assessment
Because of its highly ‘integrative’ nature we considered the tembamide synthesis a suitable test case for assessing the ecological benefits of the multi-step one-pot concept. Therefore, we performed a basic environmental impact analysis, in which we compared our four-step one-pot preparation of tembamide to previously reported asymmetric syntheses of this compound. We chose Sheldon's E-factor26 (mass of waste produced per mass of desired product) as a simple metric. We also decided to exclude solvents from the E-factor analysis and calculate the solvent demand as a second, independent indicator, since solvent waste and non-volatile waste (particularly salts) require very different processing. Both metrics can only provide a rough estimation of environmental impact, as they do not take into account the chemical composition (and hence the toxicity) of the waste, the energy demand of the involved processes, or the waste generated in the preparation of starting materials and catalysts. On the other hand, such a basic analysis is easily performed, and can thus serve as a quick eco-assessment of several synthetic options.Table 5 provides an overview of the environmental performance of the six synthetic sequences under investigation. The chemo-enzymatic four-step one-pot system presented herein achieves the second-highest yield and has clear environmental advantages over the previously published procedures. Only the synthesis developed by Brown et al. reaches comparable values for yield, E-factor and solvent demand; however, it requires highly toxic hydrogen cyanide as a reagent in the asymmetric key step.
| Article | Stepsa | Asymmetric key step | Yieldb [%] | E-factorc | Solventd [mL g−1] | Ref. |
|---|---|---|---|---|---|---|
| a Total number of chemical transformations. The number of steps carried out individually (with product isolation) is given in parentheses. b Overall yield. c Overall E-factor (excluding solvents). d Overall solvent demand. e Please note that this synthesis starts from 2-azido ketone 2e, which is not commercially available, and therefore also needs to be synthesised. | ||||||
| Present work | 4 (1) | Asymmetric ketone reduction (ADH) | 73 | 11.1 | 309 | — |
| Lee et al. 2007 | 5 (5) | Asymmetric ketone reduction (Rh catalyst) | 62 | 57.8 | 1600 | 27 |
| Baeza et al. 2005 | 3 (2) | Asymmetric cyano-O-phosphorylation (Lewis acid/Brønsted base catalyst) | 65 | 23.3 | 1031 | 28 |
| Kamal et al. 2004 | 5 (4) | Enantioselective transesterification (lipase) | 42 | 114.9 | 1801 | 29 |
| Yadav et al. 2001 | 3 (2)e | Asymmetric ketone reduction (carrot root) | 85 | 97.5 | 826 | 16b |
| Brown et al. 1993, 1994 | 3 (3) | Asymmetric hydrocyanation (peptide catalyst) | 72 | 14.6 | 483 | 30 |
Differentiation of the E-factor into the contributions of the reaction itself (excess of reagents, coupled products, by-products, catalysts) and the down-stream processing reveals the main advantage of the one-pot concept: the elimination of the isolation and purification steps leads to significant reductions in waste generation, which for all syntheses except the one reported by Yadav et al. (which uses large quantities of carrot root as a catalyst), is mainly linked to work-up and purification rather than loss of material in the reaction itself (Fig. 3). Nevertheless, our synthesis also features the lowest reaction-linked E-factor contribution (3.2) of the six procedures analysed.
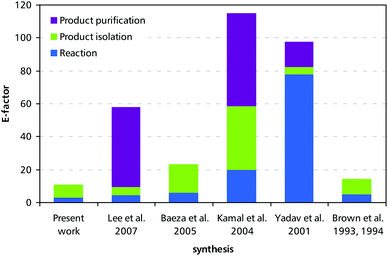 | ||
| Fig. 3 Contributions of reaction steps, product isolation steps, and product purification steps to the overall E-factor (excluding solvents) of catalytic asymmetric syntheses of tembamide. | ||
Differentiation by the type of waste (see Suppl. Fig. 10, ESI†) shows that silica gel contributes substantially to the E-factor of the syntheses that make use of it. Those sequences that do not require chromatographic purifications (Baeza et al., Brown et al., and the present work), mainly produce inorganic salts (NaBr, NaCl, NaOH, Na2SO4, MgSO4, and others) as waste, which are arguably less problematic.
Finally, a more detailed analysis of the different types of solvents used (see Suppl. Table 7, ESI†) shows that our chemo-enzymatic sequence generally employs more environmentally acceptable solvents31 (mostly water, ethyl acetate, and ethanol) than the other processes, especially because it avoids the use of chlorinated solvents and does not require eluents for chromatography, which often contain large amounts of hexane.
Conclusions
In summary, we have developed chemo-enzymatic one-pot reaction sequences that provide access to enantiomerically pure 1,2-amino alcohols either in two steps from the corresponding 2-azido ketones or in three steps from 2-halo ketones. The biocatalytic reduction of 2-azido ketones using alcohol dehydrogenases (ADHs) is the asymmetric key step in these processes, and by selecting suitable ADHs, both enantiomers of the target compounds can be obtained in excellent enantiomeric excess (ee >99%). The 2-amino-1-arylethanol derivatives 4a–f thus prepared are important building blocks in pharmaceutical research, for instance in the synthesis of anti-inflammatory, antiviral, or antitumour agents.32Furthermore, the one-pot concept has been applied to the asymmetric synthesis of the antiviral natural product (S)-tembamide, obtained in 73% yield over four steps and >99% ee from commercially available bromo ketone 1e. This synthesis reaches high catalyst turnover (TON = 200 for Pd, 1000 for NAD+, several 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for the ADH),33 uses only a small excess of reagents (1.2 eq. of NaN3 and BzCl), and affords a chemically pure product after a final recrystallisation as the sole purification step. Due to these features, our method compares favourably with previous syntheses of tembamide not only in terms of yield, but especially regarding its ecological impact, as quantified by E-factor and solvent demand. Hence, our study highlights the advantages of chemo-enzymatic one-pot processes in the multi-step synthesis of chiral compounds, and since it uses catalysts that are either commercially available or easily prepared, we believe that it will also be of practical value to synthetic chemists.
000 for the ADH),33 uses only a small excess of reagents (1.2 eq. of NaN3 and BzCl), and affords a chemically pure product after a final recrystallisation as the sole purification step. Due to these features, our method compares favourably with previous syntheses of tembamide not only in terms of yield, but especially regarding its ecological impact, as quantified by E-factor and solvent demand. Hence, our study highlights the advantages of chemo-enzymatic one-pot processes in the multi-step synthesis of chiral compounds, and since it uses catalysts that are either commercially available or easily prepared, we believe that it will also be of practical value to synthetic chemists.
Experimental
General materials and methods
Unless otherwise noted, reagents and organic solvents were obtained from chemical suppliers in reagent grade quality and used without further purification. Petroleum ether (boiling range 40–60 °C) and ethyl acetate used for extraction and column chromatography were purchased in technical grade quality and were distilled prior to use. Pro analysi (p.a., >99% purity) grade solvents were used for handling the 1,2-amino alcohols 4a–f to avoid any undesired oxazolidine formation due to contaminating acetone. Sulfonated lignin with either calcium or ammonium counterions was obtained from Burgo Group S.p.A., Tolmezzo, Italy. Low-sulfonate Kraft lignin was obtained from Sigma-Aldrich. The halo ketones 1a–e as well as both enantiomers of amino alcohol 4a were obtained commercially; all other substrates and reference compounds were synthesised as described in the ESI.†The proprietary enzymes used in this study are part of the Codexis Codex KRED screening kit, the Almac selectAZyme CRED screening kit, and the evocatal ADH screening kit. The ADHs from Lactobacillus kefir and Thermoanaerobium brockii were obtained from Sigma-Aldrich. ADH-A from Rhodococcus ruber DSM 44541 and LbADH from Lactobacillus brevis were heterologously expressed in E. coli as described in the ESI.†
Hydrogenation reactions, as well as analytical-scale (2 mL) and semi-preparative scale (5 mL) chemo-enzymatic one-pot transformations were carried out in magnetically stirred stainless steel autoclaves (16 mL total volume) that are part of a HEL PolyBlock8 parallel reactor system, and reactor temperature as well as stirring speed were controlled using the associated HEL WinISO software (v. 2.3.85.1). Gram-scale chemo-enzymatic one-pot transformations were carried out in a mechanically stirred Parr 4560 series stainless steel autoclave (452HC2 bomb cylinder, 160 mL total volume), and the reactor temperature was controlled using a Parr 4841 heater/controller. Non-enzymatic reactions were generally stirred at 500 rpm, while biotransformations were stirred at 300 rpm (to minimise mechanical stress).
Thin layer chromatography was carried out on silica gel 60 F254 plates (Merck) and compounds were visualized either by UV or by dipping into cerium ammonium molybdate stain [50 g L−1 (NH4)6Mo7O24·4H2O, 2 g L−1 Ce(SO4)2·4H2O in 10% (v/v) sulfuric acid] or basic permanganate stain (50 g L−1 Na2CO3, 10 g L−1 KMnO4, 0.85 g L−1 NaOH in demineralised water). Melting points were determined in open capillary tubes on a Büchi B-540 apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded in CDCl3 solution on a Bruker Avance 400 instrument at 400 and 100 MHz, respectively. Chemical shifts are given in parts per million (ppm) relative to the residual CHCl3 peak (1H: δ = 7.26 ppm, 13C: δ = 77.2 ppm) and coupling constants (J) are reported in Hertz (Hz). Specific optical rotation values [α]20D were determined on a Perkin-Elmer Model 343 Polarimeter at 20 °C and a wavelength of 589 nm (sodium D-line) using a cuvette of 1 dm path length.
Azido alcohol hydrogenation catalysed by metal nanoparticles
Biotransformations
Test transformations to ensure complete conversion under the conditions of the one-pot sequence were carried out in small-scale autoclave reactors (16 mL): azido ketone 2a–f (200 μmol; final conc. 100 mM) was dissolved/dispersed in 2-propanol (100 μL; final conc. 5% v/v, approx. 650 mM). Potassium phosphate buffer (1.7 mL; 100 mM, pH 7.0, 1 mM MgSO4) and a stock solution of ADH (2–25 mg mL−1; final conc. 0.2–2.5 mg mL−1, see Table 6) and NAD(P)+ (0.7 mg mL−1, 1 mM; final conc. 100 μM) in potassium phosphate buffer (200 μL) were added, and the mixture was stirred at 30 °C and 300 rpm for 20 h. The reaction mixture was transferred to microcentrifuge tubes, the product was extracted into EtOAc (2 × 1 mL), and the extract was dried over MgSO4. Conversion was determined by GC–FID analysis, while product ee was determined by GC–FID analysis (3a, 3f) or HPLC analysis (3b–e).
Chemo-enzymatic one-pot transformations
The ADH concentrations used for the one-pot, two-step transformation of azido ketones 2a–f into amino alcohols 4a–f are summarised in Table 6.
The ADH concentrations used for the one-pot, three-step transformation of halo ketones 1a–f into amino alcohols 4a–f are summarised in Table 7.
Environmental impact assessment
E-factor calculations were performed using the EATOS (v. 1.1) software tool,43 while solvent demand was calculated using Microsoft Excel (determining the solvent use of each step separately and carrying forward the solvent demand of all intermediates). All reactions were treated as proceeding to complete conversion, hence all losses in yield are accounted for as ‘unknown by-products’. In cases where the exact quantities of reagents or auxiliary materials were not given in the literature, the following estimations were used: solvents for diluting solutions: 3 times the initial volume, Celite for filtration: 0.1 g mL−1 of filtrate, solvents for extraction: 50 mL g−1 of crude product, aqueous solutions for washing extracts: 20% of extract volume, except brine: 10% of extract volume, anhydrous salts for drying extracts: 0.02 g mL−1 of extract volume, solvents for recrystallisation: 10 mL g−1 of crude product, silica gel for chromatography: 20 g g−1 of crude product, eluents for chromatography: 500 mL g−1 of crude product (total eluent volume). The EATOS and Excel files used for the calculations are available as ESI.†Acknowledgements
This study has been financed by the Austrian Science Fund (FWF project J3244-N17). The authors would like to thank Florian Mayer for assistance with the NMR measurements, Maarten Gorseling and Remco van Oosten for general technical assistance, and Caroline Paul for proof-reading the manuscript.Notes and references
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS.
- (a) L. M. Ambrosini and T. H. Lambert, ChemCatChem, 2010, 2, 1373–1380 CrossRef CAS; (b) A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477–1489 CAS; (c) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed; (d) J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–312 RSC.
- For recent reviews, see: (a) H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS PubMed; (b) I. Oroz-Guinea and E. García-Junceda, Curr. Opin. Chem. Biol., 2013, 17, 236–249 CrossRef CAS PubMed; (c) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC; (d) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237–294 CrossRef CAS; (e) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821–1849 RSC; (f) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef PubMed; (g) E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS; (h) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (i) M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2009, 2, 500–506 CrossRef CAS PubMed.
- (a) I. Schnapperelle, W. Hummel and H. Gröger, Chem.–Eur. J., 2012, 18, 1073–1076 CrossRef CAS PubMed; (b) M. Poizat, I. W. C. E. Arends and F. Hollmann, J. Mol. Catal. B: Enzym., 2010, 63, 149–156 CrossRef CAS PubMed; (c) E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551–9554 CrossRef CAS PubMed.
- For recent reviews, see: (a) A. C. Marr and S. Liu, Trends Biotechnol., 2011, 29, 199–204 CrossRef CAS PubMed; (b) P. N. R. Vennestrøm, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 249–258 CrossRef; (c) H. C. Hailes, P. A. Dalby and J. M. Woodley, J. Chem. Technol. Biotechnol., 2007, 82, 1063–1066 CrossRef CAS.
- For recent reviews on the combination of chemo- and biocatalysis for dynamic kinetic resolution in organic media, see: (a) P. Hoyos, V. Pace and A. R. Alcántara, Adv. Synth. Catal., 2012, 354, 2585–2611 CrossRef CAS; (b) J. H. Lee, K. Han, M. J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999–1015 CrossRef CAS; (c) B. Martín-Matute and J. E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226–232 CrossRef PubMed; (d) H. Pellissier, Tetrahedron, 2008, 64, 1563–1601 CrossRef CAS PubMed . For recent examples of the concurrent combination of chemo- and biocatalysis in aqueous or bi-phasic media, see: ; (e) Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS PubMed; (f) V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed; (g) F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC.
- For selected recent examples of the sequential combination of chemo- and biocatalysis in aqueous medium, see: ref. 4a and 4c; (a) A. Cuetos, F. R. Bisogno, I. Lavandera and V. Gotor, Chem. Commun., 2013, 49, 2625–2627 RSC; (b) J. S. Willemsen, J. C. M. van Hest and F. P. J. T. Rutjes, Chem. Commun., 2013, 49, 3143–3145 RSC; (c) J. M. Foulkes, K. J. Malone, V. S. Coker, N. J. Turner and J. R. Lloyd, ACS Catal., 2011, 1, 1589–1594 CrossRef CAS; (d) H. Maid, P. Böhm, S. M. Huber, W. Bauer, W. Hummel, N. Jux and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 2397–2400 CrossRef CAS PubMed; (e) M. Kraußer, T. Winkler, N. Richter, S. Dommer, A. Fingerhut, W. Hummel and H. Gröger, ChemCatChem, 2011, 3, 293–296 CrossRef; (f) K. Tenbrink, M. Seßler, J. Schatz and H. Gröger, Adv. Synth. Catal., 2011, 353, 2363–2367 CrossRef CAS; (g) G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944–7947 CrossRef CAS PubMed; (h) P. N. R. Vennestrøm, E. Taarning, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 943–945 CrossRef; (i) E. Burda, W. Bauer, W. Hummel and H. Gröger, ChemCatChem, 2010, 2, 67–72 CrossRef CAS; (j) V. Gauchot, W. Kroutil and A. R. Schmitzer, Chem.–Eur. J., 2010, 16, 6748–6751 CrossRef CAS PubMed; (k) W. Szymanski, C. P. Postema, C. Tarabiono, F. Berthiol, L. Campbell-Verduyn, S. De Wildeman, J. G. De Vries, B. L. Feringa and D. B. Janssen, Adv. Synth. Catal., 2010, 352, 2111–2115 CrossRef CAS; (l) K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2009, 48, 9355–9358 CrossRef CAS PubMed; (m) A. Prastaro, P. Ceci, E. Chiancone, A. Boffi, R. Cirilli, M. Colone, G. Fabrizi, A. Stringaro and S. Cacchi, Green Chem., 2009, 11, 1929–1932 RSC; (n) R. M. Haak, F. Berthiol, T. Jerphagnon, A. J. A. Gayet, C. Tarabiono, C. P. Postema, V. Ritleng, M. Pfeffer, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2008, 130, 13508–13509 CrossRef CAS PubMed.
- S. C. Bergmeier, Tetrahedron, 2000, 56, 2561–2576 CrossRef CAS.
- For reviews, see: (a) J. L. Vicario, D. Badía, L. Carrillo, E. Reyes and J. Etxebarria, Curr. Org. Chem., 2005, 9, 219–235 CrossRef CAS; (b) K. Everaere, A. Mortreux and J. F. Carpentier, Adv. Synth. Catal., 2003, 345, 67–77 CrossRef CAS; (c) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–876 CrossRef CAS PubMed.
- For a review, see: (a) F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed . For recent examples, see: ; (b) R. Kuwano, N. Kameyama and R. Ikeda, J. Am. Chem. Soc., 2011, 133, 7312–7315 CrossRef CAS PubMed; (c) J. F. McGarrity and A. Zanotti-Gerosa, Tetrahedron: Asymmetry, 2010, 21, 2479–2486 CrossRef CAS PubMed; (d) G. Shang, D. Liu, S. E. Allen, Q. Yang and X. Zhang, Chem.–Eur. J., 2007, 13, 7780–7784 CrossRef CAS PubMed; (e) A. Hu and W. Lin, Org. Lett., 2005, 7, 455–458 CrossRef CAS PubMed; (f) A. Lei, S. Wu, M. He and X. Zhang, J. Am. Chem. Soc., 2004, 126, 1626–1627 CrossRef CAS PubMed; (g) T. Morimoto, K. Yoshikawa, M. Murata, N. Yamamoto and K. Achiwa, Chem. Pharm. Bull., 2004, 52, 1445–1450 CrossRef CAS; (h) T. Ohkuma, D. Ishii, H. Takeno and R. Noyori, J. Am. Chem. Soc., 2000, 122, 6510–6511 CrossRef CAS.
- For recent examples, see: (a) H. Vázquez-Villa, S. Reber, M. A. Ariger and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979–8981 CrossRef PubMed; (b) O. Soltani, M. A. Ariger, H. Vázquez-Villa and E. M. Carreira, Org. Lett., 2010, 12, 2893–2895 CrossRef CAS PubMed; (c) Z. Xu, S. Zhu, Y. Liu, L. He, Z. Geng and Y. Zhang, Synthesis, 2010, 811–817 CrossRef CAS PubMed; (d) M. Watanabe, K. Murata and T. Ikariya, J. Org. Chem., 2002, 67, 1712–1715 CrossRef CAS PubMed; (e) A. M. Kawamoto and M. Wills, J. Chem. Soc., Perkin Trans. 1, 2001, 1916–1928 RSC.
- (a) A. Prechter, H. Gröger and M. R. Heinrich, Org. Biomol. Chem., 2012, 10, 3384–3387 RSC; (b) F. J. Quijada, F. Rebolledo and V. Gotor, Tetrahedron, 2012, 68, 7670–7674 CrossRef CAS PubMed; (c) R. Lihammar, R. Millet and J. E. Bäckvall, Adv. Synth. Catal., 2011, 353, 2321–2327 CrossRef CAS; (d) A. Rouf, P. Gupta, M. A. Aga, B. Kumar, R. Parshad and S. C. Taneja, Tetrahedron: Asymmetry, 2011, 22, 2134–2143 CrossRef CAS PubMed; (e) A. Chaubey, R. Parshad, P. Gupta, S. C. Taneja, G. N. Qazi, C. R. Rajan and S. Ponrathnam, Bioorg. Med. Chem., 2009, 17, 29–34 CrossRef CAS PubMed.
- (a) J. Rehdorf, M. D. Mihovilovic and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2010, 49, 4506–4508 CrossRef CAS PubMed; (b) J. Rehdorf, M. D. Mihovilovic, M. W. Fraaije and U. T. Bornscheuer, Chem.–Eur. J., 2010, 16, 9525–9535 CrossRef CAS PubMed.
- (a) K. Baer, N. Dückers, W. Hummel and H. Gröger, ChemCatChem, 2010, 2, 939–942 CrossRef CAS; (b) J. Steinreiber, M. Schürmann, F. Van Assema, M. Wolberg, K. Fesko, C. Reisinger, D. Mink and H. Griengl, Adv. Synth. Catal., 2007, 349, 1379–1386 CrossRef CAS; (c) J. Steinreiber, M. Schürmann, M. Wolberg, F. Van Assema, C. Reisinger, K. Fesko, D. Mink and H. Griengl, Angew. Chem., Int. Ed., 2007, 46, 1624–1626 CrossRef CAS PubMed.
- (a) T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed; (b) S. Matosevic, G. J. Lye and F. Baganz, J. Biotechnol., 2011, 155, 320–329 CrossRef CAS PubMed; (c) C. U. Ingram, M. Bommer, M. E. B. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. J. Lye, Biotechnol. Bioeng., 2007, 96, 559–569 CrossRef CAS PubMed.
- (a) G. Sello, F. Orsini, S. Bernasconi and P. D. Gennaro, Tetrahedron: Asymmetry, 2006, 17, 372–376 CrossRef CAS PubMed; (b) J. S. Yadav, P. T. Reddy, S. Nanda and A. B. Rao, Tetrahedron: Asymmetry, 2001, 12, 3381–3385 CrossRef; (c) P. A. Procopiou, G. E. Morton, M. Todd and G. Webb, Tetrahedron: Asymmetry, 2001, 12, 2005–2008 CrossRef CAS.
- (a) F. Coccia, L. Tonucci, D. Bosco, M. Bressan and N. d'Alessandro, Green Chem., 2012, 14, 1073–1078 RSC; (b) F. Coccia, L. Tonucci, N. d'Alessandro, P. d'Ambrosio and M. Bressan, Inorg. Chim. Acta, 2013, 399, 12–18 CrossRef CAS PubMed.
- Notably, this high chemoselectivity is in stark contrast to the hydrogenation of 3b over Pd/C in methanol, where reductive dehalogenation was observed and 4a·HCl was formed as the sole product in quantitative yield.
- For studies using isolated enzymes, see: ref. 7a; (a) F. R. Bisogno, E. Garcia-Urdiales, H. Valdes, I. Lavandera, W. Kroutil, D. Suarez and V. Gotor, Chem.–Eur. J., 2010, 16, 11012–11019 CrossRef CAS PubMed; (b) F. R. Bisogno, I. Lavandera, W. Kroutil and V. Gotor, J. Org. Chem., 2009, 74, 1730–1732 CrossRef CAS PubMed; (c) H. Ankati, Y. Yang, D. Zhu, E. R. Biehl and L. Hua, J. Org. Chem., 2008, 73, 6433–6436 CrossRef CAS PubMed; (d) K. Edegger, C. C. Gruber, T. M. Poessl, S. R. Wallner, I. Lavandera, K. Faber, F. Niehaus, J. Eck, R. Oehrlein, A. Hafner and W. Kroutil, Chem. Commun., 2006, 2402–2404 RSC . For studies using whole microbial cells, see: ref. 16c;; (e) L. C. Fardelone, J. A. R. Rodrigues and P. J. S. Moran, J. Mol. Catal. B: Enzym., 2006, 39, 9–12 CrossRef CAS PubMed; (f) H. Antunes, L. d. C. Fardelone, J. A. R. Rodrigues and P. J. S. Moran, Tetrahedron: Asymmetry, 2004, 15, 2615–2620 CrossRef CAS PubMed; (g) J. S. Yadav, P. T. Reddy, S. Nanda and A. B. Rao, Tetrahedron: Asymmetry, 2001, 12, 63–67 CrossRef CAS; (h) A. E. P. M. Sorillha, M. Marques, I. Joekes, P. José, S. Moran, J. Augusto and R. Rodrigues, Bioorg. Med. Chem. Lett., 1992, 2, 191–196 CrossRef . For studies using carrot root as biocatalyst, see: ref. 16b;; (i) J. S. Yadav, S. Nanda, P. Thirupathi Reddy and A. Bhaskar Rao, J. Org. Chem., 2002, 67, 3900–3903 CrossRef CAS PubMed.
- (a) B. Kosjek, W. Stampfer, M. Pogorevc, W. Goessler, K. Faber and W. Kroutil, Biotechnol. Bioeng., 2004, 86, 55–62 CrossRef CAS PubMed; (b) W. Stampfer, B. Kosjek, C. Moitzi, W. Kroutil and K. Faber, Angew. Chem., Int. Ed., 2002, 41, 1014–1017 CrossRef CAS; (c) W. Hummel, Trends Biotechnol., 1999, 17, 487–492 CrossRef CAS; (d) W. Hummel, Adv. Biochem. Eng. Biotechnol., 1997, 58, 145–184 CrossRef CAS; (e) R. J. Lamed and J. G. Zeikus, Biochem. J., 1981, 195, 183–190 CAS.
- Due to the effect of the azide group on the Cahn–Ingold–Prelog priorities, enzymes with a stereoselectivity according to Prelog's rule will produce the (R)-enantiomer of 3a. However, in the suppliers’ catalogs these enzymes are categorised as (S)-selective, in accordance with the stereopreference observed with ‘standard substrates’ such as acetophenone or 2-octanone.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- Interestingly, the ADHs from T. brockii and L. brevis, which have been reported to reduce 2a, did not give substantial conversion in our screening, despite proving active in a photometric assay on 2-propanol oxidation (for details, see ESI†).
- The oxazolidine 5 can also be turned into the main product by adding additional acetone after extraction of the crude product from the aqueous phase, and preliminary experiments have shown that more complex oxazolidines (which may contain a second chiral centre) can also be accessed by this method.
- M.-J. Cheng, K.-H. Lee, I.-L. Tsai and I.-S. Chen, Bioorg. Med. Chem., 2005, 13, 5915–5920 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC; (c) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- D.-M. Lee, J.-C. Lee, N. Jeong and K.-I. Lee, Tetrahedron: Asymmetry, 2007, 18, 2662–2667 CrossRef CAS PubMed.
- A. Baeza, C. Nájera, J. M. Sansano and J. M. Saá, Chem.–Eur. J., 2005, 11, 3849–3862 CrossRef CAS PubMed.
- A. Kamal, A. A. Shaik, M. Sandbhor and M. S. Malik, Tetrahedron: Asymmetry, 2004, 15, 3939–3944 CrossRef CAS PubMed.
- (a) R. F. C. Brown, A. C. Donohue, W. R. Jackson and T. D. McCarthy, Tetrahedron, 1994, 50, 13739–13752 CrossRef CAS; (b) R. F. C. Brown, W. R. Jackson and T. D. McCarthy, Tetrahedron: Asymmetry, 1993, 4, 205–206 CrossRef CAS.
- For ecological perspectives of solvent use, see: (a) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC; (b) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- For recent articles, see: (a) K. P. Bhabak and C. Arenz, Bioorg. Med. Chem., 2012, 20, 6162–6170 CrossRef CAS PubMed; (b) A. S. Aboraia, S. W. Yee, M. S. Gomaa, N. Shah, A. C. Robotham, B. Makowski, D. Prosser, A. Brancale, G. Jones and C. Simons, Bioorg. Med. Chem., 2010, 18, 4939–4946 CrossRef CAS PubMed; (c) M. D. Wittman, B. Balasubramanian, K. Stoffan, U. Velaparthi, P. Liu, S. Krishnanathan, J. Carboni, A. Li, A. Greer, R. Attar, M. Gottardis, C. Chang, B. Jacobson, Y. Sun, S. Hansel, M. Zoeckler and D. M. Vyas, Bioorg. Med. Chem. Lett., 2007, 17, 974–977 CrossRef CAS PubMed; (d) V. Nesterenko, K. S. Putt and P. J. Hergenrother, J. Am. Chem. Soc., 2003, 125, 14672–14673 CrossRef CAS PubMed . For recent patents, see: ; (e) K. Eis, F. Puehler, L. Zorn, A. Scholz, P. Lienau, M. J. Gnoth, U. Boemer, J. Guenther, J. Fanghaenel and D. Knorr (Bayer GmbH), Internat. Pat., WO 2012/156367, 2012 Search PubMed; (f) J. A. Linn, T. Longstaff and K. L. Stevens (Smithkline Beecham Corp.), Internat. Pat., WO 2008/024634, 2008 Search PubMed; (g) A. Slassi, B. Joseph, F. Ma, I. Egle, J. Clayton, M. Isaac and K. Swierczek (Astrazeneca AB; NPS Pharmaceuticals, Inc.), Internat. Pat., WO 2007/078523, 2007 Search PubMed; (h) A. Ali, J. M. Napolitano, Q. Deng, Z. Lu, P. J. Sinclair, G. E. Taylor, C. F. Thompson, N. Quraishi, C. J. Smith and J. A. Hunt (Merck & Co., Inc.), Internat. Pat., WO 2006/014357, 2006 Search PubMed; (i) P. J. Hergenrother, V. Nesterenko, K. Putt, B. J. Allen, R. S. Dothager and B. J. Leslie (Univ. Illinois), Internat. Pat., WO 2005/044191, 2005 Search PubMed; (j) M. E. Schnute, M. M. Cudahy, M. Eggen, D. J. Anderson, T. M. Judge, E. J. Kim and S. A. Collier (Pharmacia & Upjohn, Co.), Internat. Pat., WO 2004/106345, 2004 Search PubMed.
- Since the ADH preparation is a crude cell-free extract, the exact concentration of the productive enzyme is not known. An approximation assuming 20% target protein content and a molecular weight of 30 kDa yields a concentration of 3.33 μM, corresponding to a TON of 30000 at full conversion (100 mM substrate).
- A. Kamal, G. B. R. Khanna, T. Krishnaji and R. Ramu, Bioorg. Med. Chem. Lett., 2005, 15, 613–615 CrossRef CAS PubMed.
- A. V. Malkov, A. J. P. S. Liddon, P. Ramírez-López, L. Bendová, D. Haigh and P. Kočovský, Angew. Chem., Int. Ed., 2006, 45, 1432–1435 CrossRef CAS PubMed.
- M. J. Bosiak and M. M. Pakulski, Synthesis, 2011, 316–324 CrossRef CAS PubMed.
- L. Kniežo, P. Kristian and S. Velebný, Collect. Czech. Chem. Commun., 1978, 43, 1917–1923 CrossRef.
- B. T. Cho, S. K. Kang and S. H. Shin, Tetrahedron: Asymmetry, 2002, 13, 1209–1217 CrossRef CAS.
- W. Becker, H. Freund and E. Pfeil, Angew. Chem., 1965, 77, 1139–1139 CrossRef.
- C. Paolucci and G. Rosini, Tetrahedron: Asymmetry, 2007, 18, 2923–2946 CrossRef CAS PubMed.
- A. S. Demir, C. Tanyeli, H. Aksoy, V. Gulbeyaz and A. S. Mahasneh, Synthesis, 1995, 1071–1073 CrossRef CAS PubMed.
- R. Somanathan, H. R. Aguilar, R. Ventura and K. M. Smith, Synth. Commun., 1983, 13, 273–280 CrossRef CAS.
- (a) M. Eissen and J. O. Metzger, Chem.–Eur. J., 2002, 8, 3580–3585 CrossRef CAS; (b) EATOS: Environmental Assessment Tool for Organic Syntheses, http://www.metzger.chemie.uni-oldenburg.de/eatos/english.htm, Accessed 13.06.2013.
Footnote |
| † Electronic supplementary information (ESI) available: Complete results of ADH screening, additional optimisation studies, characterisation data of Pd-NPs, additional details on the environmental assessment of tembamide syntheses, additional experimental procedures, full characterisation data of compounds isolated from the chemo-enzymatic transformations, EATOS and Excel files used in the environmental impact assessment. See DOI: 10.1039/c3gc41666f |
| This journal is © The Royal Society of Chemistry 2013 |