 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOlefin metathesis in aqueous media
Jasmine
Tomasek
and
Jürgen
Schatz
*
Organic Chemistry 1 and ICMM (Interdisciplinary Center for Molecular Materials), University Erlangen-Nuremberg, Henkestr. 42, D-91054 Erlangen, Germany. E-mail: juergen.schatz@fau.de; Fax: +49 9131 85-24707; Tel: +49 9131 85-25766
First published on 15th July 2013
Abstract
The worldwide undisputable and unattainable chemist is nature, using water as a solvent of choice in biosynthesis. Water as a solvent not only indicates “green chemistry” but is also inevitable in biochemical reactions as well as syntheses of several pharmaceutical products. In the last few decades, several organic reactions were successfully carried out under aqueous conditions, a powerful and attractive tool in organic synthesis metathesis reaction. This review summarises advances made in metathesis reaction in aqueous media. Two main strategies can be distinguished: the design of water soluble catalysts to obtain homogeneous conditions and using commercially available catalysts to utilize the advantages of heterogeneous conditions.
 Jasmine Tomasek | Jasmine Tomasek studied chemistry at the Friedrich-Alexander University (FAU) of Erlangen-Nuremberg and obtained an M.Sc. in Chemistry in 2011. After this, she joined the research group of J. Schatz and is perusing her Ph.D. research related to the application of supramolecular chemistry in aqueous olefin metathesis reactions. |
 Jürgen Schatz | In 1994 Jürgen Schatz completed his Ph.D. in Organic Chemistry under the guidance of Jürgen Sauer at the University of Regensburg. After a postdoctoral stay with C. W. Rees, CBE FRS, at the Imperial College, London, he returned to Germany to start his independent career at the University of Ulm. After one semester at LMU Munich 2007, he moved to his current position at the Friedrich-Alexander University (FAU) of Erlangen-Nuremberg. His research is focused on supramolecular chemistry and its use in catalytic processes with a focus on water as a solvent. |
Introduction
In organic chemistry, C–C coupling reactions open a wide range of applications for effective synthesis, which otherwise would be difficult or even hardly feasible. The olefin metathesis reaction displays one of these atom efficient catalysis reactions under mild conditions.1 The term “olefin metathesis” was coined by Calderon and displays a catalytic reaction where “olefins undergo bond reorganization, resulting in a redistribution of alkylidene moieties”.2 This C–C double-bond transformation reaction includes not only one kind of reaction, but also a wide variety of different types of metathesis reactions, meaning coupling reactions of cyclic and acyclic alkenes or alkynes as well as polymerisation reactions (Scheme 1).1 Accordingly, the metathesis has been of great interest since its discovery in the mid-1950s3 and reveals a powerful tool for both industrial applications, especially in petro- and polymer chemistry and organic synthesis.4,5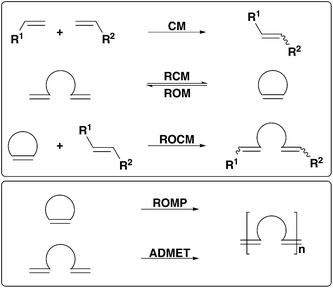 | ||
| Scheme 1 Variety of olefin metathesis reactions. Cross metathesis (CM), ring-closing metathesis (RCM), ring-opening metathesis (ROM), ring-opening cross metathesis (ROCM), ring-opening metathesis polymerisation (ROMP), acyclic dienmetathesis polymerisation (ADMET). | ||
Already in 1966, one of the first metathesis applications was carried out on an industrial scale, the Philips-triolefin-process,4a years before the mechanism and the role of the catalyst were unambiguously elucidated. While several research groups dealt with this topic and suggested a pair-wise interchange of the alkylidene moieties,6Chauvin postulated a more complex catalytic cycle, which displays the generally accepted and Nobel-Prize honoured mechanism (Scheme 2).7,8 The main difference is the assumption that there is no direct alkylidene exchange between the olefins, but a transfer via the catalyst by building a metal carbene complex III. The catalytic cycle consists of sequential [2 + 2] cycloaddition reactions followed by rearrangement of the double bonds. This mechanism includes fully reversible steps. To avoid a consequential mixture of olefins and to obtain only the desired product, a shift of the equilibrium in the desired direction is crucial.
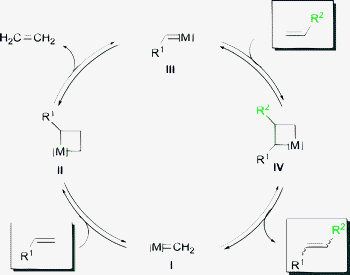 | ||
| Scheme 2 Chauvin-mechanism of a CM. | ||
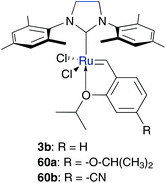
Tailor-made catalysts are crucial for an optimized chemical reaction. At the beginning of metathesis research, catalysts usually consisted not only of a single component, but commonly of two or even three different species.9 In those cases the active catalyst was generated in situ by mixing transition metal halides and main group metal alkyls as co-catalysts, such as WCl6/EtAlCl2/EtOH, WOCl4/EtAlCl2, MoCl5/Et3Al, ReCl5/EtAlCl2, to name only a few.9 In other cases catalysts based on transition metals were supported on metal–oxide surfaces, such as Al2O3 and SiO2, or were bound chemically on polymers.9 However, there are a number of factors influencing the formation of the active species and therefore influencing the efficiency of the catalytic reaction. For supported catalysts a pre-treatment step is needed, which requires harsh conditions. Otherwise, in many cases the initiation step is very slow and leads to a low concentration of the metathesis active catalyst. Additionally, in polymerisation reactions it is sometimes hard to control the propagating step. For these reasons the focus of catalyst design switched to single component catalysts, based on metal–carbene centres. First improvements of such carbene complexes are related to polymerisation experiments. Thereby carbene catalysts based on various metals showed all living ring-opening metathesis (ROMP).10 To prevent unwanted side reactions or interactions with the metal centre, which interfere with the catalytic activity, metals with high functional group tolerance are needed. Grubbs generated a reactivity table based on the results of ROMP and other organic reactions, using titanium, tungsten, molybdenum and ruthenium as the metal centre (Table 1).10 Olefins, compared to aldehydes, ketones and esters, preferentially belong to soft functional groups. Taking this into account, it is not surprising that ruthenium as the softest metal, i.e. the metal with most d-electrons, shows the highest affinity to olefins and consequently a high tolerance to functional groups as a metathesis catalyst.

The best-known well-defined (pre)catalysts were prepared in the 1990s, based on molybdenum and ruthenium as early transition metals (Fig. 1). While the Schrock catalyst exhibits high activity, the tolerance to functional groups is very limited and the sensitivity to moisture and air is high.11 Five coordinated, ruthenium based Grubbs catalysts partially overcome these disadvantages and are now an attractive tool for practical applications.12,13 Especially the insertion of N-heterocyclic carbene (NHC) ligands is conducive to pre-catalysts with high metathesis activity and high tolerance to functional groups, air and moisture.14 As a result of these accomplishments, ruthenium and NHC based pre-catalysts serve as a promising basic framework in the followed catalyst design.15,16
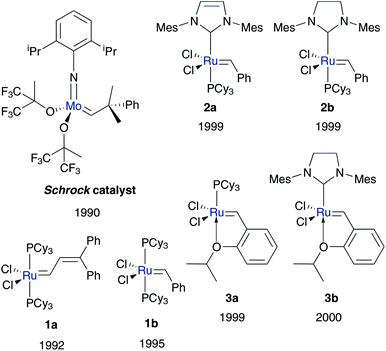 | ||
| Fig. 1 Schrock, Grubbs 1st generation 1, Grubbs 2nd generation 2 and Hoveyda–Grubbs3 (pre)catalysts. | ||
Organic reactions in aqueous media
Simultaneously with the development of metathesis reaction, a trend towards water as a solvent in organic reactions has been evolving for several decades.17 Water is termed as a “green solvent”. This includes a lot of advantages over conventionally used organic solvents. Water is economical and safe, and it is neither flammable, potentially explosive, mutagenic, nor carcinogenic. Otherwise, the term “green” also indicates a “green workup” or “green accruing waste”. To claim this adjective, high efforts and costs for purification and extraction methods have to be excluded.18 Nevertheless, owing to its beneficial properties the literature already shows several efficient organic reactions carried out in aqueous media.17,19,20This “green-ness” and environmental friendliness of water as a solvent is based on the “hydrophilicity of nature”; water is the universal solvent in nature. The largest proportion of biochemical reactions in living cells proceed in aqueous media.17 According to this, pharmaceutical and biologically relevant molecules are usually polar and only soluble in water/polar media and, thus, there is a significant need for synthetic methodologies which can be applied in such polar, aqueous and protic media. An atom efficient approach with high functional group tolerance and mild reaction conditions is displayed by Grubbs-type metathesis reaction in aqueous media.
Thinking about the first organometallic catalysts with benchmark reactions such as Grignard,21Reformatsky22 and Barbier23 primarily leads to the question of catalyst stability in water, because of their sensitivity to moisture.24 However, the last few decades show a plethora of organometallic reactions, which are viable in aqueous media and do not need inert conditions or even a glove box.25
Considering the stability of carbene–metal complexes, Ru based pre-catalysts are expected to be the most promising candidates for metathesis reactions in aqueous media. Besides decomposition studies of Grubbs-type (pre)catalysts in organic solvents and at higher temperatures,26Dinger and Moll tested the influence of alcohols, water and oxygen on Grubbs pre-catalysts of the first 1b and the second generation 2b.27 In the presence of primary alcohols and temperatures up to 60 °C or the addition of a base, both pre-catalysts decompose to a monohydride species (Scheme 3), which can now act as α-olefin isomerization catalysts.27b In contrast to alcohols, water leads to other unknown products and oxygen leads to the formation of both the monohydride species and another decomposition compound. Nevertheless, these results seemed not to interfere with metathesis reactions performed in water. All discussed decomposition reactions only occur at elevated temperatures and after a prolonged reaction time. Therefore, such processes should not be completely ignored, but can usually be neglected as potential side reactions in aqueous metathesis reactions. This is confirmed by several efficient examples, which add to metathesis reactions in organic solvent, and also opens up a new and wide research area and will be discussed in the following sections.
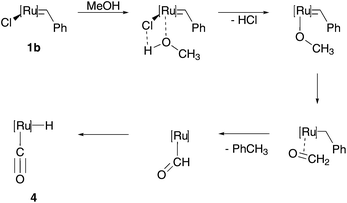 | ||
| Scheme 3 Decomposition of Grubbs precatalysts 1b to monohydride species 4 in the presence of methanol.27 | ||
Water-soluble Grubbs-type catalysts
In metathesis reactions carried out in aqueous solvent, several strategies were taken into account, which can be divided into homogeneous and heterogeneous approaches.28 The former refers to the design of water-soluble pre-catalysts and will be discussed in this section.Ruthenium salts as first water-soluble metathesis catalysts
First approaches in metathesis reactions are based on the use of simple transition metal salts as catalysts, just as in first attempts at metathesis reactions performed in water.9 According to early positive results of ROMP mediated by transition metal salts in polar solution,29Grubbs and co-workers investigated ROMP of functionalized oxanorbornenes. RuCl3(H2O)n and OsCl3(H2O)n turned out to be the most promising candidates for several solvents and solvent-mixtures, because of their high functional group tolerance.30 While these experiments were carried out in the absence of water, the results in aqueous media and even neat water negated former expectations that inert conditions are necessary. Water as a solvent exceeded the results of organic ROMP, with lower PDI values, a faster initiation rate and a higher molecular mass of the polymer.31,32 Further the complex Ru(tos)2(H2O)6 (tos = p-toluenesulfonate) shows similar improved results in ROMP of carboxyimide functionalized 7-oxanorbornene derivatives performed in pure water.33 These catalysts in fact are one of the first water-soluble ruthenium based metathesis catalysts, not only active in aqueous media, but with a beneficial influence on the initiation process. However, the active metal–carbene species is formed in situ and complexes did not polymerize in a living manner. It is assumed that RuIII is reduced in water to RuII, which forms a stable RuII–carbene species and initiates a polymerization reaction.31,34 To improve control of the active catalytic species in the reaction and therefore improve control of the entire reaction, such simple ruthenium components were replaced by well-defined water-soluble ruthenium based catalysts.Water-soluble pre-catalysts tagged with ionic groups
One possibility to enhance the water-solubility of an organic compound is to introduce ionic groups into one or more ligands or into the carbene moiety (Fig. 2). Besides, data in the literature indicate that cationic excel anionic tailored metathesis catalysts.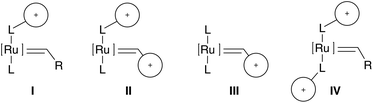 | ||
| Fig. 2 Strategies for enhanced polarity by introducing cationic groups. | ||
Ionic tagged phosphine Ru-catalysts
In the early stages of water-soluble catalyst design, cationic and anionic tagged phosphine ligands were used. The cationic pre-catalysts (6 and 7), as well as anionic ones (8 and 9) were synthesized via ligand exchange of phosphine species 5 (Fig. 3).35,36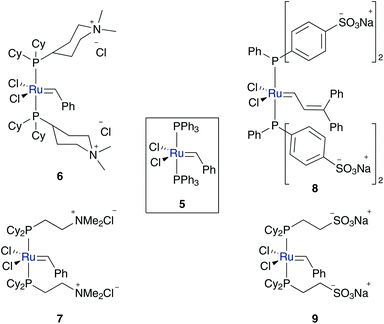
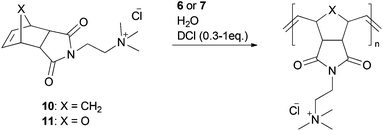
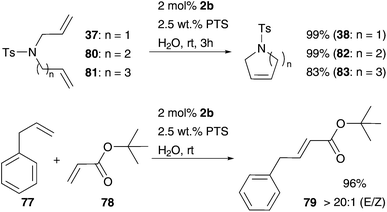
Both pre-catalysts 6 and 7 show high solubility in water and methanol, while they are insoluble in organic solvents, such as THF, ethanol, benzene or acetone. However, they are highly air-sensitive and decompose in a time interval of some weeks in methanolic solution and even in a brief span of 2 days in water.35 According to this, ROMP of water-soluble monomers 10 and 11 in pure water results in a non-quantitative conversion of 45 to 80%, because of the transient propagating species. It is suggested, based on earlier research, that generated hydroxide ions cause this decomposition.37 To overcome this problem and eliminate those stability interfering hydroxide ions, a Brønsted acid was added to the solution. Besides neutralizing the hydroxide ions, the acid also protonates the dissociated phosphine ligand, leading to a more stable phosphonium salt and thus accelerating the polymerization rate (Scheme 4).38
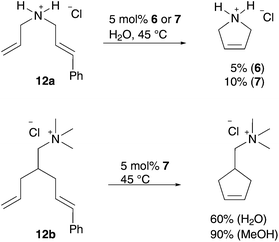
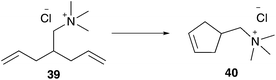
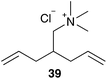
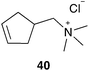
In this way, a homogeneous living polymerisation process and in addition the formation of block copolymers are feasible.38 A further application of aqueous metathesis reaction using cationic pre-catalysts 6 and 7 is RCM of water soluble acyclic dienes 12 containing one terminal and one internal olefin moiety, whereas the latter is the more active one. However, efficient conversion in RCM depends highly on the substrate (Scheme 5).39,40 Additionally, CM of terminal olefins and RCM of α,ω-olefins is almost inefficient, due to less stability of the active methylidene species, which rapidly decompose to metathesis inactive hydride species.40
In contrast to cationic phosphine bearing pre-catalysts IV (Fig. 2), also water soluble complexes possessing anionic phosphine ligands were synthesised. However, pre-catalyst 8 is water-soluble; it did not initiate aqueous ring opening polymerization, because of its triphenyl phosphine moieties. In general such ligands induce a too small cone angle and are only poor electron-donating.36,41 Anionic tricyclohexyl phosphine tagged complex 9, theoretically, should overcome these problems, but was too unstable to be isolated.36,41
Although these results seemed very unpromising, exchange of the carbene moiety to vinylidene 13 and allenylidene 14 species generates anionic, water soluble ROCM active complexes. These catalysts were tested in ring-opening metathesis of cyclopentene 15 and methyl acrylate 16 to give polyunsaturated esters 17. While vinylidene catalyst 13 is only active in homogeneous methanolic media to form 17a (yield 64%), its allenylidene 14 analogue also shows metathesis activity under biphasic diethyl ether–water conditions, to give a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 mixture of 17a to 17b (total yield 42%) (Scheme 6).42
:12 mixture of 17a to 17b (total yield 42%) (Scheme 6).42
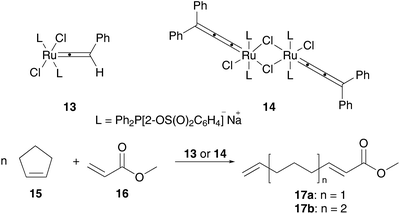 | ||
| Scheme 6 ROCM of cyclopentene 15 and methyl acrylate 16 in protic media.42 | ||
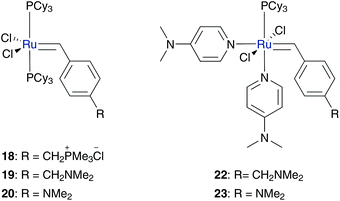
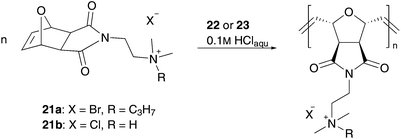
Another opportunity for water soluble catalyst design is to tag ionic functionalities to the benzylidene moiety III (Fig. 2). Schanz reported pH-responsive catalysts bearing phosphonium (18) and ammonium (19 and 20) functionalities on the benzylidene moiety (Fig. 4).43 All catalysts successfully performed controlled ROMP of cationic exo-oxanorbornene 21 in protic, acidic media. In this case, the acid protonates the dissociated phosphine ligand, which leads to a shift in the equilibrium of the initiation step with the active catalytic species. It also protonates the amine group of complexes 19 and 20 for in situ generation of the water-soluble complex. But neither of them yielded any conversion under aqueous solution, because of their limited water solubility, due to two hydrophobic PCy3 ligands. By ligand exchange of one phosphine to two basic 4-(dimethylamino)pyridine (DMAP) ligands, the resulting pre-catalysts 22 and 23 are capable of performing controlled ROMP of monomer 21 in aqueous 0.1 M HCl solution (Scheme 7).44
Ionic tagged NHC Ru-catalysts
Attributable to the synthesis of the first persistent NHC carbene complex by Arduengo,45 plenty of designed NHC bearing metathesis catalysts followed.15,16 Because of enhanced stability to moisture and air and functional group tolerance of ruthenium pre-catalysts bearing NHC ligands,14 several ionic functionalized NHC-Ru catalysts were synthesised, too.In addition to the pH-responsive catalyst reported by Schanz,43,44 also Ru-NHC catalysts were tagged on the benzylidene moiety by Grela and co-workers.46Hoveyda–Grubbs pre-catalyst 3, bearing a tertiary amine group to ensure pH-response, acts as the basic framework. Neutral complex 24 is nonpolar and demonstrates low activity in olefin metathesis in CH2Cl2. In contrast, with the addition of a strong Brønsted acid, cationic and polar in situ complex 25 is generated, which enables efficient tools for RCM and enyne metathesis reactions in organic media (Fig. 5). With this change of polarity, also electronic properties changed, from an electron donating (EDG) amine group (–NEt2) 24 to an electron withdrawing (EWG) (–NEt2H+) ammonium group 25. It has been suggested by former studies47 that electron withdrawing groups on the benzylidene moiety would weaken the O–Ru chelation, which leads to easier and faster initiation of the metathesis reaction. Although this switch enhanced the metathesis activity and initiation rate even outreaching that of Hoveyda–Grubbs3b, metathesis reactions were exclusively performed in organic media.46
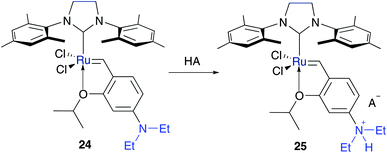 | ||
| Fig. 5 Activation of pH-responsive catalyst 24 with changing electronic properties to cationic catalyst 25.46 | ||
To ensure polar character of the metathesis catalyst, amine groups were substituted by charged ammonium groups, especially quaternary ammonium ions. This includes maintaining Hoveyda–Grubbs pre-catalyst 3b as the basis for an active metathesis catalyst with the opportunity for affecting the initiation rate by switching the electronic properties. Within this concept several water soluble and metathesis active Hoveyda–Grubbs type catalysts containing one (26–31) (Fig. 6) or two (32–34) (Fig. 7) cationic moieties were prepared.
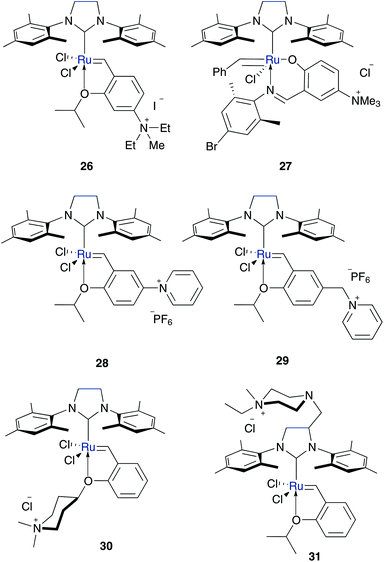
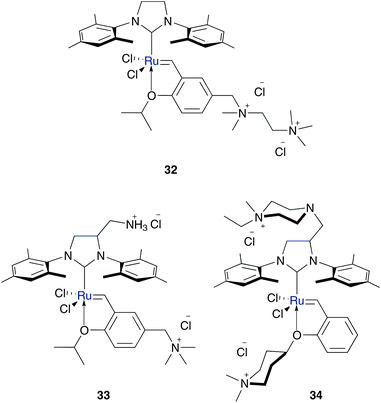 | ||
| Fig. 7 NHC ruthenium precatalysts tagged with two ammonium groups.52,53 | ||
Grela and co-workers prepared catalyst 26 with a quaternary ammonium group tagged on the benzylidene moiety. This slightly water soluble complex (<1 mg mL−1) successfully performed several RCM and CM in neat water as well as enyne metathesis in alcohol–water mixtures (Tables 2–4).48 The group of Raines combined the stabilizing and activating character of NHC ligands with a polar bidentate salicylaldimine chelating ligand, which effectively competes in former research in RCM and ROMP reactions49 to form catalyst 27.50 This catalyst is active in RCM of dienes and enynes carried out in methanol and methanol–water mixtures at a temperatures of 55 °C (Table 2). However, the stabilizing combination of both ligands also provides a slow initiation rate. Because polymers with high PDI-values would be generated, ROMP as applications is not desirable.50
| Cat. (mol%) | Solvent | t [h] | T [°C] | Conv. [%] |
|---|---|---|---|---|
|
||||
| 26 (5) | D2O | 5 | 25 | 99 |
| 30 (2.5) | D2O | 3.5 | 25 | 49 |
| 31 (2.5) | D2O | 2.5 | 25 | 96 |
| 32 (5) | D2O | 24 | 30 | >95 |
| 33 (5) | D2O | 0.5 | 30 | >95 |
| 34 (2.5) | D2O | 2.5 | 25 | 88 |
| Cat. (mol%) | Solvent | t [h] | T [°C] | Conv. [%] (43) |
|---|---|---|---|---|
|
||||
| 26 (2.5) | D2O | 3.5 | 25 | >99 |
| 28 (1) | D2O | 8 | 25 | 14 |
| 29 (1) | D2O | 8 | 25 | 19 |
| 30 (5) | D2O | 24 | 25 | 74 |
| 31 (5) | D2O | 24 | 25 | 77 |
| 32 (5) | D2O | 24 | 30 | 82 (+4) |
| 33 (5) | D2O | 6 | 30 | 69 (+12) |
| 34 (5) | D2O | 24 | 45 | 38 |
The designated aim of Mauduit and Grela in the preparation of pre-catalysts 28 and 29 was to design a recyclable catalyst in especially ionic liquids without loss of activity.51 Accordingly, they used quaternary ammonium groups as an activating EWG at the benzylidene moiety and the pyridinium species for recyclability. Nevertheless, both complexes can act as catalysts for RCM of standard dienes in alcohol–water mixtures at room temperature, while they show hardly any conversion in CM in neat water (Tables 2 and 4).51 In contrast, recently published quaternary ammonium tagged catalysts 30 and 31 mediated RCM of charged substrates and CM in neat water (Tables 2 and 3).52 While complexes 26 and 28–30 are tagged on the benzylidene moiety with an activating effect through weakening of the O–Ru bond, catalyst 31 is bearing its polar character at the NHC backbone. In this case the generated active species after the initiation step in metathesis reaction maintains its polar character.
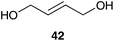
To enhance the water solubility, Grubbs and Jordan synthesized catalysts containing two cationic ammonium groups 32 and 33. Complex 33 has two ammonium groups (Fig. 2, II), one tagged on the NHC ligand and one tagged on the isopropoxy-styrene moiety.53 This ensures polar character of both, the pre-catalyst and the active species, after the catalytic initiation step. In contrast, pre-catalyst 32 with two ammonium groups stuck to the isopropoxy moiety is hardly soluble in pure water (<0.01 M). Both catalysts rapidly polymerise benchmark ROMP monomer endo-norbornene 11 in pure water. Furthermore, they mediated RCM of cationic dienes (Table 3) and CM of allylalcohol (Table 4), whereas also side-product 43 is formed.53 With regard to prospective applications in the synthesis of biologically relevant molecules which require neutral pH media, the pH dependant –NH3+ compound in complex 33 could be detrimental. Accordingly, Skowerski and Grela designed a catalyst 34 bearing a quaternary ammonium group at the benzylidene moiety as well as at the NHC backbone.52 Compared to the phosphine bearing analogue 6,35 catalyst 34 shows higher stability under air and in water and is up to ten times more soluble in water compared to catalysts 30 and 31, tagged with only one piperazine or piperidine derivative. While mediated CM of allyl alcohol gave moderate yields (Table 4), no side products were generated. Moreover, catalyst 34 also successfully performed RCM of charged dienes and even enyne in neat water (Table 3).52
Water-soluble pre-catalyst tagged with neutral, polar groups
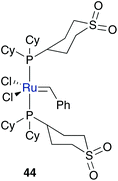
A further opportunity for enhancing water solubility of an organic compound is overcome by the addition of polar groups. Catalyst 44 represents such a neutral, electron-rich phosphine bearing complex tagged with a sulfone moiety for enhanced polarity.54 This leads to solubility in both, organic solvents as well as protic solvents. While this polar complex shows quantitative conversion of diethyl allyl (cinnamyl)malonate 45 to 46 in organic solvents such as benzene and dichloromethane, RCM in a methanol–water mixture also gave a high yield of 78% (Table 5). This discrepancy in yield arises from faster decomposition of the catalyst in protic media.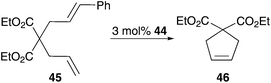
To enhance the stability of polar ruthenium complexes, again phosphine ligand was exchanged by a NHC ligand, which provides a stronger bonding to the metal centre. The resulting complex can be tagged with a polar group on the NHC ligand direct to the N-atom or at the backbone, on the benzylidene moiety or can be introduced as a further ligand (Fig. 8).
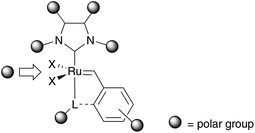 | ||
| Fig. 8 Potential positions for introducing polar groups for NHC-Ru catalyst. | ||
Ruthenium catalysts tagged with polar polymers on the benzylidene moiety
In several ruthenium catalysts as a tag with neutral polar groups, hydrophilic polymers were used. This implies a polar character and in some cases even a strategy for catalyst recycling. Because of its highly hydrophilic behaviour, in most designed polymer tagged catalysts, poly(ethylene glycol) (PEG) is the polymer of choice. Connon and Blechert published the synthesis of a phosphine free Hoveyda–Grubbs type catalyst tethered with a highly hydrophilic PEG-resin on the benzylidene moiety 47 (Fig. 9).55 This pre-catalyst shows high tolerance to oxygen and promotes several RCM and CM in methanolic solution as well as in neat water, while in benchmark RCM of 37 also side-products were formed.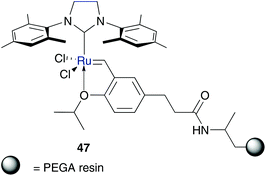
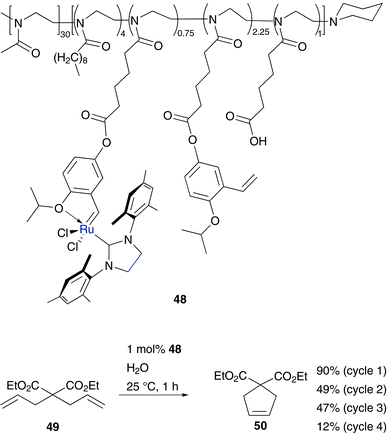
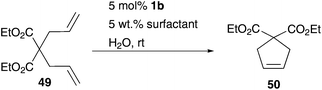
A further polymer bound amphiphilic Hoveyda–Grubbs type catalyst immobilized through benzylidene moiety was reported by Weberskirch.56 This block copolymer 48 is based on poly(2-oxazoline) and contains two isopropoxy-styrene ligands per polymer chain. This implies an advantage over polymer 47, because in this way a re-attachment of the ruthenium initiator to the solid support is feasible. Furthermore, catalyst 48 shows high conversion (90%) of benchmark RCM substrate diethyl diallylmalonate 49 in pure water with low catalyst loading of 1 mol% (Scheme 8). Owing to aqueous micellar conditions, conversion of this RCM is accelerated, while the active catalytic species is stabilized.56
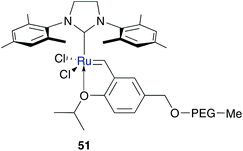
In earlier research on polar tagged ruthenium catalysts, PEG immobilized catalyst 51 turn out to be a promising tool for RCM and CM in organic solvents (Fig. 10).57,58 Considering the hydrophilic character of PEG, recently reported results demonstrate also efficient RCM of several hydrophobic substrates in acetone–water mixtures of catalyst 51 (Table 6).59
| Cat. (mol%) | Solvent | t [h] | T [°C] | Conv. [%] |
|---|---|---|---|---|
|
||||
| 51 (10) | CH2Cl2 | 0.5 | Reflux | >98 |
| 51 (5) | Me2CO–H2O (2:1) |
1 | rt | 98 |
Ruthenium catalysts tagged with polar polymers on the NHC ligand
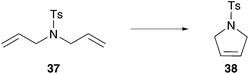
By anchoring a polar polymer group attached to the benzylidene moiety at the non-dissociating NHC ligand, the catalyst remains in solution throughout the entire metathesis reaction. Grubbs developed the first PEG labelled homogeneous aqueous olefin metathesis catalyst tagged on the N-atom of the NHC ligand 52.60 This catalyst performed efficient ROMP of exo-monomer of 10 as well as ROMP of challenging, sterically hindered endo-monomer 10 in acidified water with conversion up to 95% (Scheme 9). RCM and failed CM reactions demonstrate stability problems of 52 in methanol, due to the less stable unsaturated NHC ligand (IMes) compared to the saturated NHC ligand (SIMes).60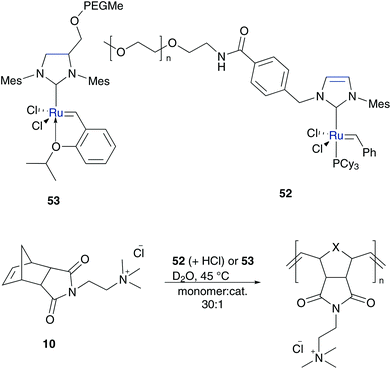 | ||
| Scheme 9 Polar ruthenium catalysts 52 and 53 tagged on the NHC ligand performed ROMP of 10 in (acidified) water.60,62 | ||
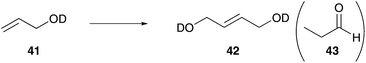
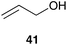
In addition to the lost stability due to an unsaturated NHC ligand, also the constitution of aryl moieties bound to the N-atoms is substantial for catalyst stability.61 To avoid these problems, Hong and Grubbs developed a PEG bearing Hoveyda–Grubbs type catalyst tagged on the saturated NHC backbone 53 (Scheme 9).62 This improved PEG containing catalyst shows unprecedented activity in ROMP of monomer 10 in RCM of cationic dienes such as 39, as well as in CM of allylalcohol 41 all performed in neat water (Table 7).
| Substrate | Product | t [h] | T [°C] | Conv. [%] |
|---|---|---|---|---|
|
|
12 | rt | >95 |
|
|
12 | 45 | >95 |
Water-soluble hexa-coordinated ruthenium pre-catalysts containing pyridine derivatives
To accelerate the initiation rate of Grubbs II type catalyst 2 it is judicious to remove the phosphine ligand. In contrast, introduced pyridine derivatives are promising candidates for fast initiation because of their labile binding to the metal centre. This was demonstrated by a 3-bromo pyridine substituted ruthenium catalyst, which shows unprecedented initiation rates in ROMP performed in dichloromethane.63Emrick and co-workers developed such a hexa-coordinated ruthenium pre-catalyst, tagged with a polar polymer throughout a pyridine derivative 54, 55, and 56 (Fig. 11).64,65 PEG catalyst 54 is not only highly soluble in both organic solvents and aqueous media, but also highly active in performed ROMP of polar oxanorbornene monomers in both solvents. However, ROMP of PEGylated monomer 57 in aqueous media only proceeds in acidified water with pH ≤ 2 and without any molecular weight control. Brønsted acids are needed to protonate the PEG-tagged pyridine derivative and diminish their ligation capability to promote initiation reaction.64 Catalyst 55 containing a PEG-triazole substituted pyridine ligand shows a similar activity and also needs acidified water for ROMP of water-soluble monomer 57. Furthermore, addition of Cu(II) salts as a pyridine scavenger facilitates ROMP even at neutral pH, but with a lower conversion rate of 70%.65 To provide water solubility and biocompatibility, PEG was replaced by phosphoryl choline (PC) groups to generate catalyst 56. In contrast to PEG tagged catalysts 54 and 55, PC containing catalyst 56 demonstrates ROMP of monomer 57 in acidified water with comparable results as under neutral pH.65 This underlines the relevant property for applications in biological systems of PC compared to PEG substituents of the pyridine derivatives (Scheme 10).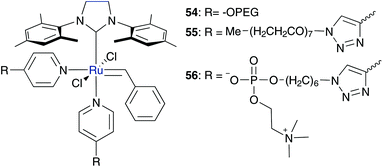 | ||
| Fig. 11 Water-soluble ruthenium precatalysts tagged with PEG 54, 55 and PC 56 substituted pyridine derivatives.64,65 | ||
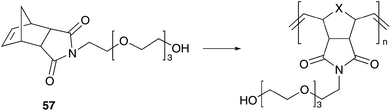 | ||
| Scheme 10 ROMP of water-soluble oxanorbornene 57. | ||
Artificial metalloenzyme for olefin metathesis
To date, the most “biocompatible” water-soluble Grubbs type catalyst is that published in the recent work of Ward and co-workers. They designed artificial enzymes containing Hoveyda–Grubbs3b catalysts via covalent 58 and non-covalent 59 binding to a protein (Fig. 12).66,67 Both catalysts are substituted on the NHC backbone with a spacer molecule. The metathesis catalyst complex of artificial enzyme 58 is bound via a spacer to a cysteine moiety of the heat shock protein from Methanocaldococcus jannaschii (MjHSP) and demonstrates metathesis activity in the performed RCM of N-tosyldiallylamine 37 in acidic buffered solution.66 However, in comparison to protein free catalysts, the protein structure shows no influence on the activity. This is due to the loss of the quaternary capsid structure of the protein in the presence of acidified media. Moreover, the catalyst sticks at the surface of the protein rather than be embedded in the pocket.66 Besides covalently bounded catalyst 58, the Hoveyda–Grubbs catalyst of artificial enzyme 59 is tagged at the NHC backbone with a biotin-anchor, which is compatible within the streptavidin and avidin pocket.67 In RCM of 37 carried out in acidified water–DMSO mixtures, conversion values up to 95% are achievable in the presence of the host–guest system of 59 and (strept)avidin. But experiments of protein-free catalysts show similar results at a lower pH value and even higher conversion values under neutral conditions.67 Nevertheless, this research area of creating artificial metalloenzymes for olefin metathesis is still at the beginnings and displays an interesting future direction of metathesis reactions in aqueous media.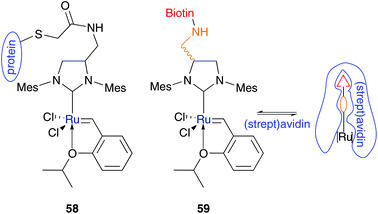 | ||
| Fig. 12 Artificial metalloenzymes covalently 58 and non-covalently bound 59.66,67 | ||
Hydrophobic Grubbs type catalysts for aqueous metathesis reactions
Besides homogeneous approaches of olefin metathesis reaction performed in water, also heterogeneous systems can be beneficial. In most cases commercially available well-defined Grubbs type catalysts 1b, 2b and 3 are used to avoid elaborate, multi-step catalyst syntheses. This strategy includes metathesis in homogeneous aqueous solvent-mixture to introduce partial solubility and mainly metathesis “on water”. The term “on water” implies heterogeneous conditions with water-insoluble components and will cause reactions that occur between the water and oil phase boundary, which can be improved by additives.68 These several attempts at the performed metathesis reaction under heterogeneous conditions are discussed in this chapter.Direct application in homogeneous aqueous solution
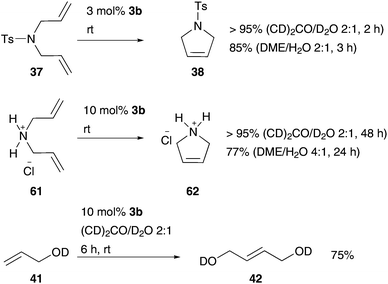
Homogeneous mixtures of water and water-miscible organic solvents belong to one of the first applications of metathesis experiments in water, using water-insoluble catalysts. Blechert and co-workers performed RCM and CM using common Grubbs type catalysts in methanol, and several mixture ratios of water and miscible organic solvents, such as DMF and MeOH (Table 8).55,69 While Hoveyda–Grubbs catalysts bearing electron-withdrawing groups at the benzylidene moiety show increased conversion in RCM in CH2Cl2,70 catalysis in protic media of more electron-deficient Hoveyda–Grubbs type pre-catalysts 3b and 60b gave poorer results compared to electron-rich pre-catalyst 60a.69 It is expected that this disagreement arises from rapid decomposition in MeOH and DMF media of fast generated catalytic active carbene species. Like complex 60a, Grubbs II precatalyst 2b demonstrates comparable conversion values in RCM of benchmark substrate 37 in pure MeOH and DMF.69 Addition of water initially leads to a decrease in conversion until the amount of water is less than 50%, while mixtures of organic solvent–water (1:3) again increase the conversion of the substrate 37.69 In contrast, Hoveyda-catalysts 3b and 60b demonstrate only low conversion in homo-CM of allylalcohols in aqueous methanol.55,69
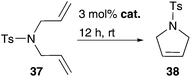
Based on these results, Raines and co-workers studied the activity of 3b in further homogeneous, aqueous solvent-mixtures with THF, dioxane, DMF, acetone, and DME.71 While Hoveyda catalyst 3b was inactive in RCM of substrate 37 in THF– and dioxane–water mixtures, conversion up to 95% was reached with acetone or DME as a co-solvent. This promising combination of pre-catalyst 3b and solvent-mixtures also demonstrates efficient RCM of further charged and neutral substrates and even conversion of 75% in CM of allylalcohol 41 (Scheme 11).71
 | ||
| Scheme 11 Selected RCM and CM reactions mediated by Hoveyda–Grubbs catalyst 3b in aqueous solution-mixtures.71 | ||
“On water” metathesis
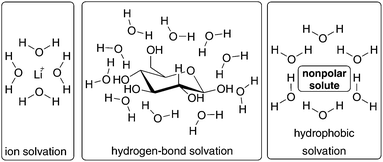
The concept of homogeneous aqueous solution is to enhance the solubility of water-insoluble or hardly water-soluble substrates and catalysts. The results of Blechert69,70 and Raines71 are classified (with respect to this review) somewhat in-between homogeneous and heterogeneous aqueous metathesis. There are limited examples in the literature dealing with this concept. In contrast, the purpose of keeping the heterogeneous character in organic reactions in water is widely used in several examples in the literature.17,24,72The “on water” approach can imply a positive impact on the rate and selectivity of the reaction. One reason for this is the “hydrophobic effect”.73 In principle, the interaction of molecules in water can be divided into three different solvation modes: ion solvation, hydrogen-bond solvation, and hydrophobic solvation (Fig. 13).73 All three modes have in common that by introducing such molecules in the hydrogen-bonding water network the orientation of the water molecules is disturbed. This is displayed by a loss of entropy due to the restriction of translational and rotational degrees of freedom of the water molecules. In the case of ion solvation, dative bonds between charged molecules and water molecules were formed, while polar and water molecules build up hydrogen bonds. In both cases enthalpy profit compensates entropic losses, which indicates the formation of a solvation shell. In contrast, the aim of hydrophobic and water molecules is minimal contact between each other. To realize this, a cluster of water molecules is formed around the non-polar components resulting in higher (local) concentration and higher pressure in water.73,74
On water metathesis without additives
Accordingly, metathesis reactions were carried out in pure water. Polshettiwar and Varma demonstrate high activity in several RCM reactions mediated by common Grubbs II 2b in water. N-Substituted diallylamine substrates 37 and 63 offer conversion values up to 95% at higher temperature of 45 °C and a short reaction time of 2 h (Fig. 14),75 an example of an efficient and simple metathesis reaction in water using commercially available catalysts, without the need of addition of a co-solvent or other additives.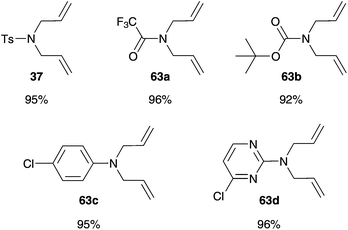 | ||
| Fig. 14 Conversion values of RCM of several N-substituted diallylamines 37 and 63 mediated by 2b at 45 °C after 2 h in water.75 | ||
Besides the simple mechanical mixing of the reaction mixture, the use of microwave and ultrasonic irradiation are further suitable methods for performing metathesis reaction in water. Microwave irradiation has become a beneficial method in several organic reactions for shortening reaction times and to increase the product yield or even influence the product contribution compared to common heating methods.76–78 While through conventional methods the reaction components are heated from the outside, microwave irradiation is a heating method from the inside. Further non-thermal effects of microwave irradiation are still under discussion.78 There are also several published microwave assisted RCM and CM reactions in organic solvents.79 Therefore, it would be interesting to study microwave assisted metathesis reactions in water.
Botta and co-workers used microwave irradiation in an aqueous enyne CM of alkyne derivatives 64 and enol-ether 65 with subsequent hydrolysis, mediated by CuSO4, to form croton aldehydes 66.80 The overall reaction is divided into three steps: first CM end up in an E:Z ratio of 2:1, followed by hydrolysis reaction, and after that an isomerisation reaction mediated by I2 in DCM to form E-isomer 66. While CM is a highly challenging metathesis reaction, because several proposed side-reactions can occur and in this example further equilibrium reactions besides metathesis are involved, in most cases α,β-unsaturated carbonyl compounds 66 were acquired in promising yields of the E-isomer up to 68% after irradiation of 3 × 10 min (Scheme 12).80
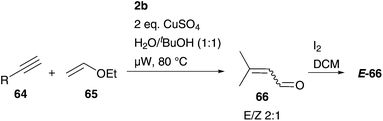 | ||
| Scheme 12 CM of alkyne derivatives 64 and enol-ether 65, followed by hydrolysis and isomerization reaction to form crotonaldehydes 66.80 | ||
An alternative activation method which is efficiently used in numerous organic reactions is ultrasonic irradiation.76,81 The activating effect of this method is induced by acoustic cavitation. While an acoustic pressure wave is formed and propagates through the reaction media, it induces the formation, growth and collapse of micrometre-sized bubbles. During this collapse, extreme conditions inside the cavity and at the interfaces occur, which include high temperature and high pressure, and can influence a small portion of molecules in the reaction mixture. This energy transfer caused by acoustic waves can enhance mechanical effects in heterogeneous processes and can induce new reactivities.77,81
The acoustic emulsification effect was also used in the metathesis reaction in neat water. Grela and co-workers performed RCM and CM of water-insoluble substrates with hydrophobic catalysts in water using ultrasonic irradiation to support the reaction (Scheme 13).82 RCM substrates were converted up to quantitative yields, including the formation of five- and six-membered rings, while the formation of larger rings failed. Moreover, also challenging the CM reaction of electron-deficient substrates results in high yields under smooth reaction conditions. It is expected that these high values are caused by a protection effect. This was assumed because catalytic species are separated from the water molecules through cavitation in organic emulsion droplets.82 Experiments in the absence of any solvent and sonification decrease the conversion value of the desired product and even oligomerization as a side-reaction occurs compared to results in water and ultrasonic irradiation.82 This demonstrates that ultrasonic experiments of metathesis reactions in neat water are very promising and non-sophisticated attempts for several RCM and even CM.
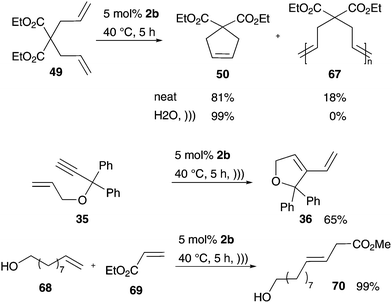 | ||
| Scheme 13 RCM and CM reaction mediated by Grubbs II 2b in neat water under ultrasonic irradiation.82 | ||
On water metathesis with additives
Besides methods for heterogeneous metathesis in water in the absence of additives, the reaction can also be supported by the addition of different beneficial additives. This also includes the use of common Grubbs type catalysts 1b, 2b and 3 with tagged or non-tagged additives for heterogeneous catalysis in water.Aqueous micelles
A smooth and well-known method for promoting heterogeneous reactions in water is based on building spherical aggregates in water, i.e. aqueous micelles.83 The formation of micelles of amphiphiles is dependent on several conditions, for instance the hydrophobic tail chain has to reach a certain length (>10 C-atoms), and the critical micelle concentration (CMC), which means the lowest concentration of the amphiphile to form micelles and temperature. There are different ways of amphiphile aggregation, which leads to monolayers at the water–air interface, or cavities introduced in water with the hydrophobic tail in the inside and a hydrophilic head at the interface, such as spheres, rods, worms, and vesicles (Fig. 15).84 Compared to the concentration in the surrounding water phase, micelles can act as hosts for hydrophobic components in water and therefore enhance concentration of reactants. This can lead to an acceleration of the reaction and furthermore to selectivity effects.83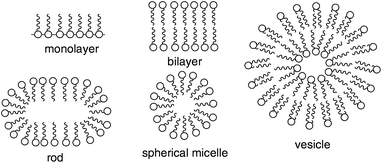 | ||
| Fig. 15 Aqueous micelles and vesicles.84 | ||
Metathesis in aqueous emulsions is the most common and applied method for performing heterogeneous metathesis in water, with a research period of nearly 20 years. In the beginning of using Grubbs type catalysts in the presence of surfactants, the research focuses mainly on polymerisation reactions.
ROMP in aqueous micelles induced by surfactants
Grubbs investigated pre-catalyst 1b in aqueous ROMP of norbornene derivatives 70–72 in the presence of emulsifier dodecyltrimethylammonium bromide (DTAB) (Fig. 16).85 All ROMP reactions were carried out in solution (DCM or DCE), suspension (water–DCM or DCE 5:1), and emulsion (water–DCM or DCE 5:1, DTBA) media. While the PDI values gained in ROMP of 71 were comparably low in all media, only experiments in solution and in suspension showed living polymerization. Results show that aqueous ROMP with addition of DTAB yielded nearly monodisperse latexes of all monomers 70–72. Nevertheless, all reactions were carried out under inert conditions.85
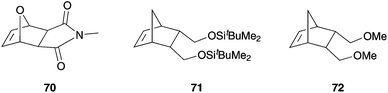 | ||
| Fig. 16 Norbornen monomers 70–72 polymerised in aqueous emulsion by Grubbs I 1b and addition of DTAB.85 | ||
Following this concept, Kiessling and co-workers polymerised carbohydrate substituted norbornene monomers in aqueous emulsion to yield neoglycopolymers, which are critical components of diverse biological processes (Fig. 17).86 Experiments on ROMP in a MeOH–CH2Cl2 solution without emulsifier DTAB end up in low conversion and growing polymer chains precipitate in this solvent-mixture. In contrast to this, aqueous emulsion conditions (water–DCE 2:1) with DTAB show even living polymerisation of monomers 73 and 74 and produce polymers with higher molecular weight.86
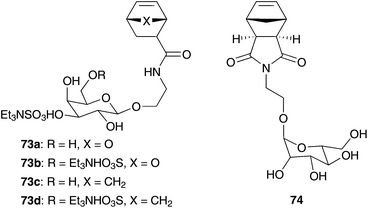 | ||
| Fig. 17 Monomers 73 and 74 polymerised in aqueous emulsion by Grubbs I 1b and addition of DTAB.86 | ||
Besides cationic DTBA amphiphile, also anionic structures such as sodium dodecyl sulphate (SDS) were used as surfactants in aqueous ROMP reactions. Claverie and co-workers polymerised cyclooctadiene (COD) and cyclooctene (COE) with Grubbs I 1bvia a miniemulsion-technique.87 The catalyst is dissolved in a minimal amount of toluene and then added dropwise to a water–monomer mixture displayed with SDS, to obtain encapsulated 1b in toluene droplets. Polymerization takes place if monomers diffuse through the water media to the catalyst droplets. The yield increases with increasing amount of toluene used for the droplet phase.87
This mini-emulsion technique was also used by Gnanou and co-workers in ROMP of norbornene (NB) mediated by Grubbs I 1b and using ionic SDS and sterically poly(styrene-b-ethylene oxide) (PS-b-PEO) as the stabilizer.88 They investigated the effect of using a mini-emulsion of norborene and a mini-emulsion of catalyst 1b. In ROMP of the NB-mini-emulsion, the catalysts were added as solution in toluene, aqueous emulsion or aqueous mini-emulsion. While in all cases monomer was completely converted, the ROMP product coagulates. In contrast, this coagulation was avoided by using a mini-emulsion of catalyst 1b in toluene to produce stable polynorbornene lattices.88
A further approach of ROMP mediated by 1b and SDS as surfactants was investigated by Mecking. They used a mini-emulsion of 1b and added a mini-emulsion of monomer. With this approach, ROMP of norbornene, COD and COE were performed with high molecular weight of the polymers.89
In accordance with this double-mini-emulsion technique of Mercking, Héroguez and Gnanou transformed this method into a tandem reaction of ROMP of norbornene (NB) and atom-transfer radical polymerisation (ATRP) of methyl methacrylate (MMA) to yield polymeric composite nanoparticles.90 They used an efficient Grubbs I precatalyst 1b for both reactions, to initiate ROMP and mediate ATRP. The first micro-emulsion consisted of the two monomers NB and MMA and ethyl-2-chloropropionate as the initiator for ATRP, while the second micro-emulsion consisted of the hydrophobic catalyst 1b dissolved in toluene. The reaction started with mixing the two emulsions and heating the reaction. This is a nice example of an efficient one-pot, one-catalyst approach to prepare graft-copolymers under smooth reaction conditions.
Clapham and Janda performed aqueous ROMP of norborbene derivatives 75 and 76 mediated by Grubbs I 1b and II 2b pre-catalysts and acacia gum was used as a surfactant (Fig. 18).91 The suspension consisted of water in the presence of a surfactant and NaCl to avoid aggregation and DCE with dissolved monomers 75 and various cross-linkers 76. By adding the dissolved catalyst in MeOH to the suspension, monomers were polymerized to resins in good yields. In following reactions, the polymers were functionalized to obtain polymeric supports for the solid-phase support (SPOS), which can be used in several organic reactions.91
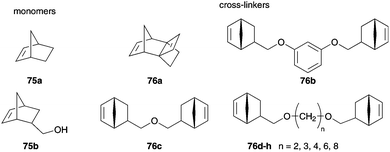 | ||
| Fig. 18 Monomers 75 polymerized in aqueous suspension mediated by Grubbs pre-catalysts 1b and 2b with acacia gum as a surfactant.91 | ||
Mingotaud and co-workers published aqueous micellar ROMP of norbornene derivative mediated by hydrophobic Hoveyda–Grubbs pre-catalyst 3b. Micelles were generated by dodecyl trimethyl ammonium (DTAC) or cetyl trimethyl ammonium (CTAC) chloride with hydrophobic catalyst 3b inside, which was additionally confirmed via UV-vis experiments. Under optimized reaction conditions fast ROMP of norbornene derivative was possible with high conversion.92
RCM and CM in aqueous micelles induced by surfactants
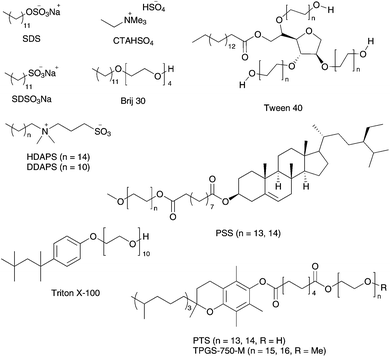
Considering several promising ROMP experiments in aqueous micellar media, further metathesis types such as RCM and CM were investigated in aqueous media in the presence of surfactants.Davis and Sinou used Grubbs I 1b in RCM of diallyl malonate 49 in aqueous micellar media induced by several tested surfactants.93 While catalytic activity of 1b reached 51% conversion of the substrate even in the absence of any surfactant in water under inert conditions, SDS intensely improved the conversion value up to 97%. In contrast, neutral (Brij 35 and Tween 40), zwitterionic (HDAPS and DDAPS), cationic (CTAHSO4) and SDS derivative (SDSO3Na) amphiphiles show no influence on conversions, compared to metathesis reaction without a surfactant (Table 9). Results of further tri- and tetra-substituted substrates for RCM under the same reaction conditions even demonstrate that a surfactant may not be essential for successful catalysis reaction. Therefore, these results are not only examples of effective aqueous micellar metathesis but also of heterogeneous metathesis in neat water without additive.93
| Surfactant | t [h] | Conv. [%] |
|---|---|---|
|
||
| — | 1 | 51 |
| SDS | 1 | 97 |
| SDSO3Na | 0.5 | 46 |
| CTAHSO4 | 1 | 66 |
| Brij 35 | 0.5 | 43 |
| Tween 40 | 0.5 | 59 |
| HDAPS | 1 | 66 |
| DDAPS | 0.5 | 44 |
The first CM reactions influenced by aqueous micelles were done by Lipshutz and co-workers. Several neutral surfactants and SDS were used in CM of allylbenzene and tert-butylacrylate in water and even under air using Grubbs precatalysts 1b and 2b.94 While pre-catalyst 1b was inactive in CM, pre-catalyst 2b demonstrates conversion around 60% even without additive, as well as with SDS and neutral surfactants TPGS, PSS, Triton X-100, Brij 30 and PEG-600. α-Tocopherol-based diester of sebacic acid, PTS improved CM up to 97% yield. With this promising combination of 2b and PTS in water, several further CM reactions were successfully carried out, giving high yields and high E-selectivity (Scheme 14).94
 | ||
| Scheme 14 Selected CM and RCM mediated by 2b in water in the presence of a surfactant PTS.95 | ||
Following this efficient metathesis reaction “duo” of Grubbs II 2b and PTS as surfactants, the same conditions were used in the performed RCM reactions of several lipophilic substrates.95 In this way, 5-, 6- and 7-membered rings as well as tri-substituted diallylamines were yielded in high values up to 99%, after a short reaction time of 3 h (Scheme 14). These results even improved RCM using Grubbs I 1b and SDS93 by decreased amounts of the catalyst of 2% and the surfactant of only 1.5–2.5%.95
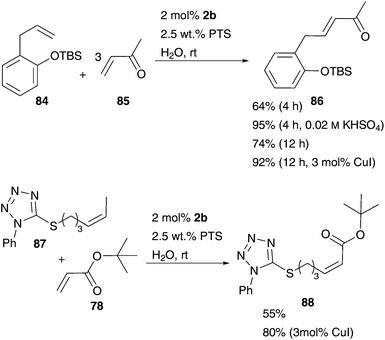
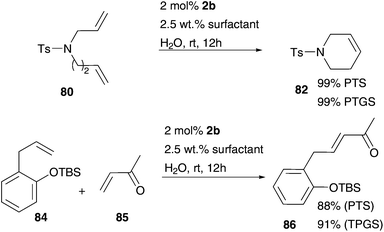
Owing to improvements in acidified aqueous ROMP of former studies,62,65Lipshutz investigated the influence of pH influencing salts in aqueous CM of Grubbs II precatalyst 2b and PTS as the surfactant.96 Varying the solvent and the pH-value, the best results in CM of allylbenzene derivative 84 and excess of methyl vinyl ketone 85 were obtained using water as a neat solvent and KHSO4 as an additive with a pH of 2.0 (Scheme 15). Also CuI-salts were used to improve the conversion value in CM reactions. In this way, even challenging CM of substrates 87 and 78 were performed under smooth reaction conditions, in water under aerobic conditions with high yields (Scheme 15). Furthermore, PTS not only improves the catalytic activity by spontaneous micelle building, but can also be recycled easily. After complete conversion of the substrates, dienes and the catalyst were extracted with diethyl ether, while PTS remains in aqueous phase and can be used in the next reaction cycle. However, even after 8 cycles conversion remained unchanged high, in each cycle not only the substrate is added, but also the catalyst 2b (Scheme 15).96
 | ||
| Scheme 15 Selected CM mediated by Grubbs II 2b and improved by PTS as a surfactant and KHSO4 or CuI.96 | ||
Owing to these successful applications of PTS as a surfactant in aqueous heterogeneous metathesis reactions, Lipshutz and co-workers investigated a new amphiphile on the basic structure of PTS with easier synthetic access and at least the same catalytic impact.97 TPGS-750-M is based on the same α-tocopherol unit exploited in PTS, but bears a longer methylated PEG chain (Fig. 19). This change leads to larger nanoparticles in water of 53–65 nm with a higher percentage of rod-like particles, compared to 24 nm with PTS, which best accommodate reactants in metathesis reactions. Both surfactants enhanced the catalytic activity of Grubbs II 2b in RCM and CM reactions carried out in water under smooth reaction conditions, while TPGS-750-M in all experiments demonstrate the same or even slightly higher conversion values (Scheme 16).97
 | ||
| Fig. 19 Several surfactants used in RCM and CM in water. | ||
 | ||
| Scheme 16 Comparison of the influence of surfactants PTS and TPGS-750 M in aqueous RCM and CM mediated by 2b.97 | ||
Although the formed micelles can be characterized in shape, size and functionality and the hydrophilic–lipophilic balance of amphiphiles (relative amount of its hydrophilic to lipophilic component) can be determined, it is difficult to predict an ideal surfactant.84,97 To get a better insight into the mechanism of metathesis reaction carried out in aqueous micelles, Charnay, Colacino and co-workers accomplished a reaction monitoring of RCM via1H NMR measurements.98 In heterogeneous RCM of hydrophilic substrates 37 and 49 with Grubbs I pre-catalyst 1b, gemini cationic surfactants 89 were chosen, because of their enhanced surface activity compared to the corresponding monocationic species and high influence to yield up to quantitative conversion (Fig. 20).
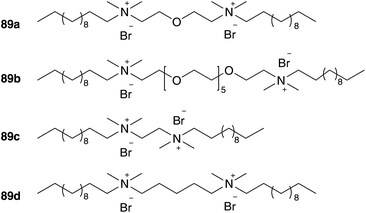 | ||
| Fig. 20 Investigated dimeric surfactants 89 for reaction monitoring.98 | ||
During reaction monitoring, water and substrate showed a biphasic system in the absence of the surfactants, while the addition of 89 leads to solubilization of the substrate in micelles formed by 89. By the addition of the catalyst, surfactant molecules arranged with the positive charged heads on the catalyst surface, which indicates more an adsorbed surfactant layer than a micellar building. In reactive media, the substrate and the catalyst get into contact and product 38 or 50 is generated. Cyclic products were not detected in micellar medium, indicating that the product leaves the micelle towards the bulk solvent (Fig. 21).98
Aqueous micelles induced by polymer tagged catalysts
All the above discussed examples are based on aqueous micelles, which are induced by added surfactants. Alternatively, micelle induced components can be tagged at the catalyst. For this, hydrophobic polymers can be used. An advantage compared to non-tagged micelle induced surfactants is that the catalytic species remains in the inside of the micelle core and recycling of the catalyst is more easily practicable.99Nuyken and Buchmeiser designed a Hoveyda–Grubbs type catalyst tagged with an amphiphilic poly(2-oxazoline)-derived block co-polymer 90, which is fixed via halogen exchange.100 This asarone type catalyst101 showed high metathesis activity in cyclopolymerization of diethyl dipropargyl malonate 91 under aqueous micellar conditions and even diminishes the PDI value of generated latex particles 92 compared to results of non-immobilized catalysts turned out in organic solvents (Scheme 17).100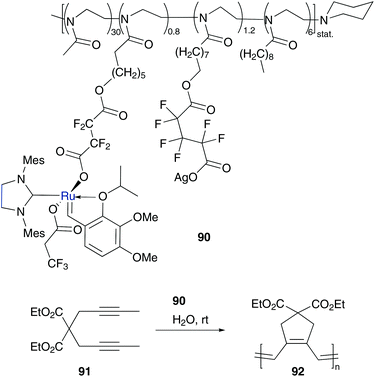 | ||
| Scheme 17 Cyclopolymerization of monomer 91 mediated by polymer tagged catalyst 90 under aqueous micellar conditions.100 | ||
A further block co-polymer supported catalyst was synthesized by Elias and Vigalok; it consisted of PEG bearing polymer fixed through phosphine ligands of ruthenium catalyst 93 (Fig. 22).102 Amphiphilic character arises from the water soluble PEG chain fixed on a hydrophobic polypeptide block. In aqueous ROMP, reaction of norbornene 75a mediated by catalyst 93 resulted in 74% of trans-alkene polymer with a lower PDI value and a higher conversion rate compared to the polypeptide free polymer-catalyst or polymer free Hoveyda–Grubbs catalyst 3a due to micellar conditions.
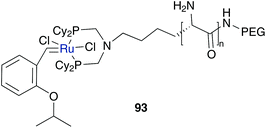 | ||
| Fig. 22 Amphiphilic block co-polymer tagged at ruthenium catalyst 93.102 | ||
Aqueous micelles induced by catsurfs
In contrast to amphiphilic polymers also smaller molecules can be bound on ruthenium catalyst to introduce micelle character. Catalysts bearing a surfactant are called catsurfs (for the catalyst and the surfactant) or inisurfs (for the initiator and the surfactant). The first inisurf molecule applied in aqueous metathesis reaction was developed by Mingotaud, Sykes and co-workers.103 They synthesized catalyst 94 bearing long hydrophobic chains at the phosphine ligands. In this way the catalytic ruthenium centre acts as a “hydrophilic” head. Instead of monolayer micelles, bilayer liposomes are used and the catalyst is incorporated into the outer phase of the liposome membrane with the catalytic centre directed to the outer aqueous phase. This system was used in ROMP of norbornene monomers 95 dissolved in an aqueous buffer solution (Fig. 23). Polymerization occurs at the surface of liposomes to generate polymer nodules with a controlled shape of diameter up to 10 μm. The shape of polymer nodules is dependent on the hydrophilicity of the monomer; while nodules of monomer 95a end up in a more spherical shape, a high hydrophilic monomer 95b gave predominantly elongated shape nodules.103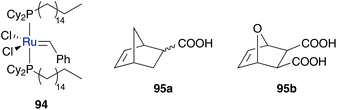 | ||
| Fig. 23 Aqueous ROMP of monomer 95 mediated by inisurf 94 incorporated into the liposome membrane.103 | ||
This concept of a long hydrophobic alkyl chain tagged on a less hydrophobic catalytic head to introduce amphiphilic character was resumed by the synthesis of further catsurfs 96–98 in following years by the group of Mingotaud and Grela (Fig. 24).104,105 To enhance air-stability, NHC bearing ruthenium catalysts were applied and micellar conditions were used instead of bilayered vesicles (Fig. 25).
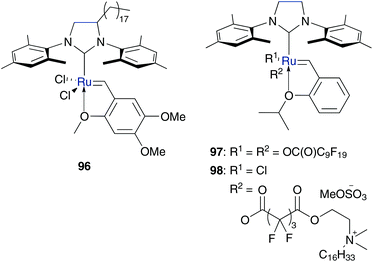 | ||
| Fig. 24 Catsurfs designed by Mingotaud (96, 97) and Grela (98).104,105 | ||
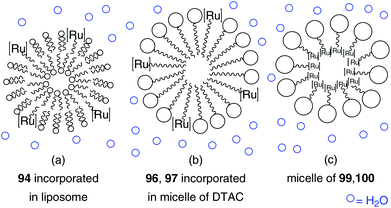 | ||
| Fig. 25 Schematic illustration of catsurf incorporation into liposome (94) and micelles with (96, 97) and without (99, 100) the addition of external surfactant.103,104,106,107 | ||
Surface activities of catalysts 96 and 97 were demonstrated by the formation of Langmuir films at the air–water interface. While asarone complex 96 is tagged on the NHC backbone with a long alkyl chain, 97 is directly tagged on the ruthenium centre with perfluorodecanoic acid derivative. Both catsurfs were tested in benchmark RCM of substrate 49 and ROMP of hydrophilic monomer 95b under aqueous micellar conditions with the addition of the auxiliary surfactant dodecyl trimethylammonium chloride (DTAC). While both systems show high catalytic activity in polymerization of monomer 95b, only catalyst 97 showed improved catalytic activity in RCM under micellar conditions compared to homogeneous conditions in CH2Cl2. This is due to localization of the reagents. Hydrophilic monomer 95b is located in the aqueous phase and therefore in close contact with the catalytic centre. In contrast, hydrophobic substrate 49 is located in the inside of the micelle shield of the catalyst at the micelle surface. However, in the presence of DTAC, a ligand exchange of pseudohalide perfluorocarboxylate moieties of 97 with chloride anions of DTAC occurs to generate Hoveyda–Grubbs catalyst 3bin situ. In this way, the system changed to micellar conditions with catalyst and added surfactant, and elucidates enhanced activity of 97 compared to 96.104
In the case of catsurf 98 the mentioned ligand lability also exists in the presence of chloride anions. However, using 98 in aqueous metathesis reaction, there is no need of addition of an external surfactant, because catsurf 98 and hydrophobic substrates already formed stable emulsions by themselves. This system demonstrate efficient RCM and CM reactions of hydrophobic substrates under smooth reaction conditions, in air and at a low temperature of 30 °C.105
Lipshutz and Ghorai designed catsurfs 99 and 100 with reverse polarity, with water-soluble side chains, which is tagged at a hydrophobic compound with the catalytic ruthenium centre at the end to generate aqueous micelles with the catalytic species in the inside (Fig. 26).106,107 The surfactant PQS consists of hydrophilic PEG chains to ensure water solubility, bound through sebacic acid on hydrophilic ubiquinol to confirm lipophilic media to solubilize organic substrates and act as linkage to the Hoveyda–Grubbs catalyst 3. Catalysts of first (3a) as well as of second (3b) generation were covalently bound to PQS, while in the case of 3b the side chain of the ubiquinol moiety was reduced, due to synthetic reasons. Both surfactants 99 and 100 build up nano-micelles of 44 nm in neat water. Phosphine bearing catalyst 99 demonstrates efficient RCM reactions of several lipophilic substrates to form five-, six- and even seven-membered rings in pure water as well as in seawater of the Pacific Ocean without any essential difference in conversion values.106 NHC variation 100 was particularly used in RCM of substituted substrates and shows a high catalytic activity with conversion values up to 99% at room temperature and even 70% of tetra-substituted diallyl amine at higher temperature. Furthermore, compound 100 catalyses CM reactions of several substrates in high yields and with high selectivity.107 Besides high catalytic activity, both catalysts can easily be recycled by a simple extraction of the organic substrates and products with diethyl ether, while the catalyst species remain in aqueous solution.106,107
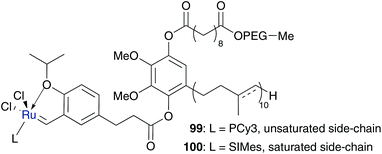 | ||
| Fig. 26 Catsurf 99 and 100 with PQS as a covalently bound surfactant.106,107 | ||
Dendrimers as unimolecular aqueous micelles
Micelles can be generated as illustrated in Fig. 15 by several surfactants to form a cavity. In contrast amphiphilic dendrimers can be used as unimolecular micelles.108 While there are some examples of metathesis reactions supported by dendrimers in organic solvents, this technique is rarely applied in aqueous solution.109Astruc and co-workers synthesized dendrimer 101 containing a hydrophobic core and hydrophilic triethylene glycol chains at the termini for application in aqueous metathesis reactions (Fig. 27).110 Aqueous CM and enyne metathesis reactions mediated by the Hoveyda–Grubbs catalyst 3b can be substantially improved by the addition of only 0.083 mol% of dendrimer 101. In this way, RCM reactions can be performed even without the need of additive.75,93 However, the catalyst amount can be reduced to less than 0.1 mol% by consistently good conversion values via addition of only 0.083 mol% of dendrimer 101. Further improvement is high and easy recyclability of the aqueous solution containing the water-soluble dendrimer 101. Via filtration of the catalyst and extraction of the products with diethyl ether the dendrimeric solution can be used ten times without a significant decrease in conversion values. In this way, several substrates can be efficiently converted under air, at room temperature, and in pure water.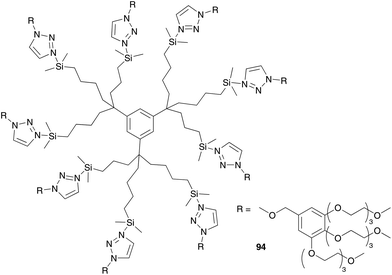 | ||
| Fig. 27 Amphiphilic dendrimer 101 for aqueous metathesis reaction.110 | ||
Supramolecular additives
The concept of aqueous micelles is a highly efficient method to improve heterogeneous metathesis reaction; however, supramolecular additives without micellar character are also promising candidates. Schatz and co-workers investigated the impact of various supramolecular additives in RCM of N-tosyl diallyl amine 37 in pure water catalysed by Grubbs II 2b (Fig. 28).111 With this, sulfocalix[n]arenes 102 show the beneficial influence of catalytic activity from 75% without additive to 99% conversion. One visible effect of additives 102 is (micro)solubilisation of organic compounds in the reaction mixture. This might be a reason for the enhanced catalytic activity. Furthermore, it is expected that sulfocalix[n]arenes 102 act as a phosphine scavenger, because of their high affinity.112 The first step in the catalytic cycle is dissociation of the phosphine ligand to create a catalytically active species. Dissociated phosphine is now protonated in water and can then be caught by additive to interfere with re-dissociation to the catalyst and therefore increase catalytic species in the reaction mixture.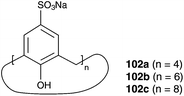
Immobilized catalysts
Recycling of the catalyst and removal of ruthenium traces in the product are very important for industrial applications. Therefore, several strategies have been taken into account dealing mainly with supported catalysts.113 One opportunity is immobilization of the catalyst on solid supports. The challenge with this method is to retain catalytic selectivity and activity. This might be a reason for only limited examples of such catalytic systems, especially for metathesis reactions in water.Bowden and co-workers generated heterogeneous catalysts based on commercially available Grubbs catalysts 1b and 2b, which are occluded in polydimethylsiloxan (PDMS) slabs.114 For metathesis reactions, substrates were dissolved in a methanol–water mixture and diffused into a PDMS slab, where the catalytic reaction occurs. Neither PDMS nor the catalyst is soluble in the aqueous solution and therefore they remain in the PDMS slab. In this way heterogeneous and homogeneous catalysis are combined without the need of synthesis of a new catalyst. Furthermore, the PDMS slab can act as an “active membrane”, because of its hydrophobic character polar reaction compounds were excluded that otherwise would impact the reactivity of the occluded catalyst. Several RCM and homocoupling reactions were efficiently accomplished with this system, without catalyst traces remaining in the product.114
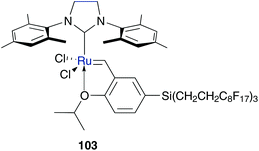
Bannwarth and co-workers investigated a further system that consisted of the non-covalent incorporation of ruthenium catalyst into an amphiphilic polymer co-network (APCN).115 This network consists of two immiscible covalently bound phases, the fluorophilic phase of perfluoropolyether (PFPE) and the hydrophilic phase of poly-(2-hydroxyethylacrylate) (PHEA) to form a bicontinuous nanophase separated polymer network. In this network perfluoro tagged Hoveyda type catalyst 103 was incorporated (Fig. 29). In the dry state of APCN, both phases approximately consist of the same volume. By adding hydrophilic solvent to the APCN, the hydrophilic phase swells and the fluorophilic phase collapses, while by adding fluorophilic solvent the process is reversed. With this property, fluorophilic catalyst 103 can easily be incorporated into the APCN by treatment with diethyl ether; the fluorophilic phase PFPE swells and allows the dissolved catalyst 103 to enter the network. After drying, the PFPE shrinks to its original volume and simultaneously encapsulates the catalyst. By the addition of water-soluble substrates in hydrophilic solvent, the hydrophilic phase PHEA swells and the substrate can contact the catalytic centre. With this system several RCM were performed in different aqueous solvent mixtures, while results in pure water showed best results with a low substrate loading of 0.002 M. However, recycling of this amphiphilic system decreases the catalytic activity. This demonstrates an interesting method of only a few examples in aqueous metathesis mediated by incorporated catalyst.115
Conclusions
Olefin metathesis in aqueous media offers a new, broad research area targeted towards smooth and biocompatible reaction conditions. Metathesis of homogeneous catalysis using a water-soluble catalyst as well as heterogeneous catalysis demonstrates efficient results in neat water or in water-mixtures. In this way, RCM and CM are even possible with multiple substituted substrates and aqueous ROMP of highly functional monomers can be polymerized, especially under micellar conditions. An important goal is recycling of the catalyst from the economic point of view as well as for the “green-ness” factor. Especially in pharmaceutical or bioactive products it is essential to remove metal impurities (<10 ppm) due to toxicity and potential side-effects. In several examples, catalyst-recycling in aqueous metathesis is intended using different strategies, such as tagging or incorporation of the catalyst, but often causes losses in catalytic activity. Catalyst-recycling remains difficult and will be aspired in further research. Although industrial applications of metathesis in organic solvents are plenty,116 there are already several promising and interesting applications in protein modification,117 drug discovery,118 and polymer chemistry31,119 and recent examples are dealing with dynamic combinatorial chemistry, all performed with aqueous metathesis.120Notes and references
- R. H. Grubbs, Handbook of Metathesis, Wiley-VCH, Weinheim, 1st edn, 2003, vol. 1 Search PubMed.
- N. Calderon, Acc. Chem. Res., 1972, 5, 127–132 CrossRef CAS.
- A. W. Anderson, N. G. Merckling and P. H. Settlage, USP, 2721189, 1995 Search PubMed.
- (a) R. L. Banks and G. C. Bailey, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170–173 CrossRef CAS; (b) F. W. Michelotti and W. P. Keaveney, J. Polym. Sci., 1965, A3, 895–905 Search PubMedA. Fürstner and O. R. Thiel, J. Org. Chem., 2000, 65, 1738–1742 CrossRef CAS; R. L. Pederson, I. M. Fellows, T. A. Ung, H. Ishihara and S. P. Hajela, Adv. Synth. Catal., 2002, 344, 728–735 CrossRef; J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef.
- M. Schuster and S. Blechert, Angew. Chem., Int. Ed. Engl., 1997, 36, 2036–2056 CrossRef.
- F. D. Mango and J. H. Schachtschneider, J. Am. Chem. Soc., 1967, 89, 2484–2486 CrossRef CAS; G. S. Lewandos and R. Pettit, Tetrahedron Lett., 1971, 11, 789–793 CrossRef; R. H. Grubbs and T. K. Brunck, J. Am. Chem. Soc., 1972, 94, 2538–2540 CrossRef; C. G. Biefeld, H. A. Eick and R. H. Grubbs, Inorg. Chem., 1973, 12, 2166–2170 CrossRef.
- J.-L. Hérisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161–176 CrossRef.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef.
- K. J. Ivin, Olefin Metathesis, Academic Press, London, New York, 1st edn, 1983 Search PubMed.
- R. H. Grubbs, J. Macromol. Sci., Part A: Pure Appl. Chem., 1994, A31, 1829–1833 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875–3886 CrossRef CAS.
- (a) S. T. Nuygen, L. K. Johnson and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 3974–3975 CrossRef; (b) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., 1995, 107, 2179–2178 ( Angew. Chem., Int. Ed. , 1995 , 34 , 2039–2041 ) CrossRef; (c) J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791–799 CrossRef CAS.
- A. Fürstner, Alkene Metathesis in Organic Synthesis, Springer, Berlin, Heidelberg, New York, 1st edn, 1998 Search PubMed.
- (a) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS; (b) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS; (c) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS; (d) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef CAS; G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef.
- S. B. Garber, J. S. Kingsbury and B. L. Gray, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS; S. Gessler, S. Randl and S. Blechert, Tetrahedron Lett., 2000, 41, 9973–9976 CrossRef.
- C.-J. Li and T.-H. Chan, Comprehensive Organic Reactions in Aqueous Media, Wiley-Interscience, New Jersey, 2nd edn, 2007 Search PubMed.
- D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., Int. Ed., 2007, 46, 3798–3800 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- P. A. Grieco, Organic Synthesis in Water, Blackie Academic & Professional, London, 1998 Search PubMed.
- V. Grignard, Compt. Rend., 1900, 130, 1322–1324 CAS.
- A. Reformatsky, Ber., 1887, 20, 1210–1211 CrossRef.
- P. Barbier, Compt. Rend., 1899, 128, 110–111 CAS.
- C.-J. Li and T.-H. Chan, Organic Reactions in Aqueous Media, Wiley-Interscience, New York, 1st edn, 1997 Search PubMed.
- B. Cornils and W. A. Herrmann, Aqueous-Phase Organometallic Catalysis, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- E. M. Leitao, S. R. Dubberley, W. E. Piers, Q. Wu and R. McDonald, Chem.–Eur. J., 2008, 14, 11565–11572 CrossRef CAS; K. Vehlow, S. Gessler and S. Blechert, Angew. Chem., Int. Ed., 2007, 46, 8082–8085 CrossRef; S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961–7968 CrossRef.
- (a) M. B. Dinger and J. C. Mol, Organometallics, 2003, 22, 1089–1095 CrossRef CAS; (b) M. B. Dinger and J. C. Mol, Eur. J. Inorg. Chem., 2003, 2827–2833 CrossRef CAS.
- For recent reviews, see: (a) J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS; (b) D. Burtscher and K. Grela, Angew. Chem., Int. Ed., 2009, 48, 442–454 CrossRef CAS; (c) B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2008, 41, 59–72 CAS; (d) S. Zaman, O. J. Curnow and A. D. Abell, Aust. J. Chem., 2009, 62, 91–100 CrossRef CAS; (e) K. Grela, Ł. Gułajski and K. Skowerski, Alkene Metathesis in Water, in Metal-catalyzed reactions in Water, ed. P. H. Dixneuf and V. Cadierno, Wiley-VCH, Weinheim, 2013, pp. 291–336 Search PubMed; (f) S. Kobayashi, Science of Synthesis: Water in Organic Synthesis, Georg-Thieme Verlag KG, 2012, Section 3.6, vol. 5, pp. 225–256 Search PubMed.
- F. W. Michelotti and W. P. Keaveney, J. Polym. Sci., 1965, A3, 895–905 Search PubMed; R. E. Rinehart and H. P. Smith, Polym. Lett., 1965, 3, 1049–1052 CrossRef CAS.
- B. M. Novak and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 960–961 CrossRef CAS.
- B. M. Novak and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 7542–7543 CrossRef CAS.
- (a) S. Y. Lu, P. Quayle, F. Heatley, C. Booth, S. G. Yeates and J. C. Padget, Macromolecules, 1992, 25, 2692–2697 CrossRef CAS; (b) K. H. Mortell, M. Gringas and L. L. Kiessling, J. Am. Chem. Soc., 1994, 116, 12053–12054 CrossRef CAS; (c) K. H. Mortell, R. V. Weatherman and L. L. Kiessling, J. Am. Chem. Soc., 1996, 118, 2297–2298 CrossRef CAS.
- M. A. Hillmeyer, C. Lepetit, D. V. McGrath, B. M. Novak and R. H. Grubbs, Macromolecules, 1992, 25, 3345–3350 CrossRef.
- N. C. Batista, J. L. Silva Sá, B. R. McGarvey, D. W. Franco and B. S. Lima-Neto, React. Kinet. Catal. Lett., 2011, 102, 49–65 CrossRef CAS.
- B. Mohr, D. M. Lynn and R. H. Grubbs, Organometallics, 1996, 15, 4317–4325 CrossRef CAS.
- R. H. Grubbs, in Aqueous Organometallic Chemistry and Catalysis, ed. I. T. Horváth and F. Joó, Kluwer, Amsterdam, 1995, pp. 15–22 Search PubMed.
- B. M. Novak, Ph. D. Thesis, Aqueous Ring – Opening Metathesis Polymerization, California Institute of Technology, 1989 Search PubMed.
- D. M. Lynn, B. Mohr and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 1627–1628 CrossRef CAS.
- T. A. Kirkland, D. M. Lynn and R. H. Grubbs, J. Org. Chem., 1998, 63, 9904–9909 CrossRef CAS.
- D. M. Lynn, B. Mohr, R. H. Grubbs, L. M. Henling and M. W. Day, J. Am. Chem. Soc., 2000, 122, 6601–6609 CrossRef CAS.
- S. Zaman, O. J. Curnow and A. D. Abell, Aust. J. Chem., 2009, 62, 91–100 CrossRef CAS.
- M. Saoud, A. Romerosa and M. Peruzzini, Organometallics, 2000, 19, 4005–4007 CrossRef CAS.
- A. N. Roberts, A. C. Cochran, D. A. Rankin, A. B. Lowe and H.-J. Schanz, Organometallics, 2007, 26, 6515–6518 CrossRef CAS.
- M. A. Dunbar, S. L. Balof, A. N. Roberts, E. J. Valente and H.-J. Schanz, Organometallics, 2011, 30, 199–203 CrossRef CAS.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- Ł. Gułajski, A. Michrowska, R. Bujok and K. Grela, J. Mol. Catal. A: Chem., 2006, 254, 118–123 CrossRef.
- K. Grela, S. Harutyunyan and A. Michrowska, Angew. Chem., 2002, 114, 4210–4212 ( Angew. Chem., Int. Ed. , 2002 , 41 , 4038–4040 ) CrossRef CAS; A. Michrowska, R. Bujok, S. Harutyunyan, V. Sashuk, G. Dolgonos and K. Grela, J. Am. Chem. Soc., 2004, 126, 9318–9325 CrossRef.
- (a) A. Michrowska, Ł. Gułajski, Z. Karczmarska, K. Mennecke, A. Kirschning and K. Grela, Green Chem., 2006, 8, 685–688 RSC; (b) Ł. Gułajski, A. Michrowska, J. Naroznik, Z. Karczmarska, L. Rupnicki and K. Grela, ChemSusChem, 2008, 1, 103–109 CrossRef.
- (a) B. de Clercq and F. Verpoort, Tetrahedron Lett., 2002, 43, 9101–9104 CrossRef CAS; (b) B. de Clercq and F. Verpoort, J. Organomet. Chem., 2003, 672, 11–16 CrossRef CAS; (c) B. Allaert, N. Dieltiens, N. Ledoux, C. Vercaemst, P. Van Der Voort, C. V. Stevens, A. Linden and F. Verpoort, J. Mol. Catal. A: Chem., 2006, 260, 221–226 CrossRef CAS.
- J. B. Binder, I. A. Guzei and R. T. Raines, Adv. Synth. Catal., 2007, 349, 395–404 CrossRef CAS.
- (a) D. Rix, H. Clavier, Y. Coutard, Ł. Gułajski, K. Grela and M. Mauduit, J. Organomet. Chem., 2006, 691, 5397–5405 CrossRef CAS; (b) D. Rix, F. Caïjo, I. Laurent, Ł. Gułajski, K. Grela and M. Mauduit, Chem. Commun., 2007, 3771–3773 RSC.
- K. Skowerski, G. Szczepaniak, C. Wierzbicka, Ł. Gułajski, M. Bieniek and K. Grela, Catal. Sci. Technol., 2012, 2, 2424–2427 CAS.
- J. P. Jordan and R. H. Grubbs, Angew. Chem., Int. Ed., 2007, 46, 5152–5155 CrossRef CAS.
- T. Rölle and R. H. Grubbs, Chem. Commun., 2002, 1070–1071 RSC.
- S. J. Connon and S. Blechert, Bioorg. Med. Chem. Lett., 2002, 12, 1873–1876 CrossRef CAS.
- M. T. Zarka, O. Nuyken and R. Weberskirch, Macromol. Rapid Commun., 2004, 25, 858–862 CrossRef CAS.
- Q. Yao, Angew. Chem., Int. Ed., 2000, 39, 3896–3898 CrossRef CAS.
- S. Zaman and A. D. Abell, Tetrahedron Lett., 2009, 50, 5340–5343 CrossRef CAS.
- S. Zaman, H. Chen and A. D. Abell, Tetrahedron Lett., 2011, 52, 878–880 CrossRef CAS.
- J. P. Gallivan, J. P. Jordan and R. H. Grubbs, Tetrahedron Lett., 2005, 46, 2577–2580 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS.
- S. H. Hong and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 3508–3509 CrossRef CAS.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS.
- K. Breitenkamp and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5715–5721 CrossRef CAS.
- D. Samanta, K. Kratz, X. Zhang and T. Emrick, Macromolecules, 2008, 41, 530–532 CrossRef CAS.
- C. Mayer, D. G. Gillingham, T. R. Ward and D. Hilvert, Chem. Commun., 2011, 47, 12068–12070 RSC.
- C. Lo, M. R. Ringenberg, D. Gnandt, Y. Wilson and T. R. Ward, Chem. Commun., 2011, 47, 12065–12067 RSC.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS; (b) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., 2005, 117, 3339–3342 ( Angew. Chem., Int. Ed. , 2005 , 44 , 3275–3279 ) CrossRef.
- S. J. Connon, M. Rivard, M. Zaja and S. Blechert, Adv. Synth. Catal., 2003, 345, 572–575 CrossRef CAS.
- M. Zaja, S. J. Connon, A. M. Dunne, M. Rivard, N. Buschmann, J. Jiricek and S. Blechert, Tetrahedron, 2003, 59, 6545–6558 CrossRef CAS.
- J. B. Binder, J. J. Blank and R. T. Raines, Org. Lett., 2007, 9, 4885–4888 CrossRef CAS.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS.
- M. C. Pirrung, Chem.–Eur. J., 2006, 12, 1312–1317 CrossRef CAS.
- P. T. Anastas, T. C. Williamson and R. Breslow, Green Chemistry, Oxford University Press, 1998 Search PubMed.
- V. Polshettiwar and R. S. Varma, J. Org. Chem., 2008, 73, 7417–7419 CrossRef CAS.
- R. S. Varma, Green Chem., 2008, 10, 1129–1130 RSC.
- A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131–1141 RSC.
- (a) C. O. Kappe, Angew. Chem., 2004, 116, 6408–6443 ( Angew. Chem., Int. Ed. , 2004 , 43 , 6250–6284 ) CrossRef; (b) M. A. Herrero, J. M. Kremsner and C. O. Kappe, J. Org. Chem., 2008, 73, 36–47 CrossRef CAS; (c) L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199–9223 CrossRef CAS.
- (a) D. Dallinger, M. Irfan, A. Suljanovic and C. O. Kappe, J. Org. Chem., 2010, 75, 5278–5288 CrossRef CAS; (b) S. Garbacia, B. Desai, O. Lavastre and C. O. Kappe, J. Org. Chem., 2003, 68, 9136–9139 CrossRef CAS; (c) K. G. Mayo, E. H. Nearhoof and J. J. Kiddle, Org. Lett., 2002, 4, 1567–1570 CrossRef CAS; (d) B. Nosse, A. Schall, W. B. Jeong and O. Reiser, Adv. Synth. Catal., 2005, 347, 1869–1874 CrossRef CAS.
- D. Castagnolo, L. Botta and M. Botta, J. Org. Chem., 2009, 74, 3172–3174 CrossRef CAS.
- (a) G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180–196 RSC; (b) G. Cravotto and P. Cintas, Chem.–Eur. J., 2007, 13, 1902–1909 CrossRef CAS; (c) C. Einhorn and J.-L. Luche, Synthesis, 1989, 787–813 CrossRef CAS; (d) J. L. Luche, C. Einhorn, J. Einhorn and J. V. Sinisterra-Gago, Tetrahedron Lett., 1990, 31, 4125–4128 CrossRef CAS.
- Ł. Gułajski, P. Śledz, A. Lupa and K. Grela, Green Chem., 2008, 10, 271–274 RSC.
- T. Dwars, E. Paetzhold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS.
- M. N. Khan, Micellar Catalysis, CRC Press, Boca Raton, FL, 1st edn, 2006 Search PubMed.
- D. M. Lynn, S. Kanaoka and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 784–790 CrossRef CAS.
- (a) D. D. Manning, X. Hu, P. Beck and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 3161–3162 CrossRef CAS; (b) M. Kanai, K. H. Mortell and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 9931–9932 CrossRef CAS.
- J. P. Claverie, S. Viala, V. Maurel and C. Novat, Macromolecules, 2001, 34, 382–388 CrossRef CAS.
- D. Quémener, V. Héroguez and Y. Gnanou, Macromolecules, 2005, 38, 7977–7982 CrossRef.
- V. Monteil, P. Wehrmann and S. Mecking, J. Am. Chem. Soc., 2005, 127, 14568–14569 CrossRef CAS.
- (a) C. Airaud, V. Héroguez and Y. Gnanou, Macromolecules, 2008, 41, 3015–3022 CrossRef CAS; (b) C. Airaud, E. Ibarboure, C. Gaillard and V. Héroguez, Macromol. Symp., 2009, 281, 31–38 CrossRef CAS; (c) C. Airaud, E. Ibarboure, C. Gaillard and V. Héroguez, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4014–4027 CrossRef CAS.
- B. S. Lee, S. Mahajan, B. Clapham and K. D. Janda, J. Org. Chem., 2004, 69, 3319–3329 CrossRef CAS.
- A.-F. Mingotaud, C. Mingotaud and W. Moussa, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2833–2844 CrossRef CAS.
- K. J. Davis and D. Sinou, J. Mol. Catal. A: Chem., 2002, 177, 173–178 CrossRef CAS.
- B. H. Lipshutz, G. T. Aguinaldo, S. Ghorai and K. Voigtritter, Org. Lett., 2008, 10, 1325–1328 CrossRef CAS.
- B. H. Lipschutz, S. Ghorai and G. T. Aguinaldo, Adv. Synth. Catal., 2008, 350, 953–956 CrossRef.
- B. H. Lipshutz, S. Ghorai, W. W. Y. Leong, B. R. Taft and D. V. Krogstad, J. Org. Chem., 2011, 76, 5061–5073 CrossRef CAS.
- B. H. Lipschutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef.
- L. Laville, C. Charnay, F. Lamaty, J. Martinez and E. Colacino, Chem.–Eur. J., 2012, 18, 760–764 CrossRef CAS.
- M. R. Buchmeiser, Chem. Rev., 2009, 109, 303–321 CrossRef CAS.
- J. O. Krause, M. T. Zarka, U. Anders, R. Weberskirch, O. Nuyken and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965–5969 CrossRef CAS.
- K. Grela and M. Kim, Eur. J. Org. Chem., 2003, 963–966 CrossRef CAS.
- S. Elias and A. Vigalok, Adv. Synth. Catal., 2009, 351, 1499–1504 CrossRef CAS.
- N. Jarroux, P. Keller, A.-F. Mingotaud, C. Mingotaud and C. Sykes, J. Am. Chem. Soc., 2004, 126, 15958–15959 CrossRef CAS.
- A.-F. Mingotaud, M. Krämer and C. Mingotaud, J. Mol. Catal. A: Chem., 2007, 263, 39–47 CrossRef CAS.
- R. Gawin, P. Czarnecka and K. Grela, Tetrahedron, 2010, 66, 1051–1056 CrossRef CAS.
- B. H. Lipshutz and S. Ghorai, Org. Lett., 2009, 11, 705–708 CrossRef CAS.
- B. H. Lipshutz and S. Ghorai, Tetrahedron, 2010, 66, 1057–1063 CrossRef CAS.
- (a) G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS; (b) G. R. Newkome, C. N. Moorefield, G. R. Baker, A. L. Johnson and R. K. Behera, Angew. Chem., 1991, 103, 1205–1207 ( Angew. Chem., Int. Ed. Engl. , 1991 , 30 , 1176–1179 ) CrossRef CAS.
- D. Astruc, A. K. Diallo, S. Gatard, L. Liang, C. Ornelas, V. Martinez, D. Méry and J. Ruiz, J. Org. Chem., 2011, 7, 94–103 CAS.
- A. K. Diallo, E. Boisselier, L. Liang, J. Ruiz and D. Astruc, Chem.–Eur. J., 2010, 16, 11832–11835 CrossRef CAS.
- T. Brendgen, T. Fahlbusch, M. Frank, D. T. Schühle, M. Seßler and J. Schatz, Adv. Synth. Catal., 2009, 351, 303–307 CrossRef CAS.
- M. B. Seßler, Ph. D. Thesis, Metathese in Wasser, Friedrich-Alexander Universität, Erlangen, 2011 Search PubMed.
- H. Clavier, K. Grela, A. Kirschning, M. Mauduit and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 6786–6801 CrossRef CAS.
- M. T. Mwangi, M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2006, 128, 14434–14435 CrossRef CAS.
- E. M. Hensle, J. Tobis, J. C. Tiller and W. Bannwarth, J. Fluorine Chem., 2008, 129, 968–973 CrossRef CAS.
- P. Naresh, M. Sirisha, K. Vijay and N. Srinath, Int. J. Pharm. Technol., 2011, 3, 1748–1767 Search PubMed; A. Fürstner, Chem. Commun., 2011, 47, 6505–6511 RSC; B. B. Marvey, C. K. Segakweng and M. H. C. Vosloo, Int. J. Mol. Sci., 2008, 9, 615–625 CrossRef CAS.
- Y. A. Lin, J. M. Chalker and B. G. Davis, ChemBioChem, 2009, 10, 959–969 CrossRef CAS; Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef; J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef.
- K. C. Nicolaou, R. Hughes, S. Y. Cho, N. Winssinger, H. Labischinski and R. Endermann, Chem.–Eur. J., 2001, 7, 3824–3843 CrossRef CAS.
- K. Müller, R. Dyllick-Brenzinger, M. Limbach and B. Sturm, WO, 2011051374, 2011 Search PubMed.
- L. Hunter, G. C. Condie and M. M. Harding, Tetrahedron Lett., 2010, 51, 5064–5067 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |