 Open Access Article
Open Access ArticleA prototype device for evaporation in batch and flow chemical processes†
Benjamin J.
Deadman
,
Claudio
Battilocchio
,
Eric
Sliwinski
and
Steven V.
Ley
*
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, United Kingdom. E-mail: svl1000@cam.ac.uk; Fax: +44 (0)1223 336442; Tel: +44 (0)1223 336398
First published on 24th June 2013
Abstract
We report a convenient and efficient prototype device for evaporating, concentrating and switching solvents in continuous flow and batch processing and batch mode fashion. One of the main features of this system is the level of recyclability, whereby all solvents removed can be easily collected and reused, with reduced environmental impact.
Flow chemistry is now recognised as a valuable component of modern synthesis programmes. However, better integration of these new enabling technologies with existing batch methods will provide the necessary stimulus for a step change in applications for the future.1 The appeal of flow chemistry is due to its ability to achieve improved heat and mass transfer, reduction of waste, safe containment of hazardous compounds, automation over a 24/7 working regime and less solvent usage through reaction telescoping and continuous processing.2
However, both batch and flow methods are increasingly under scrutiny owing to the need to greatly improve downstream processing. Indeed, one of the difficulties commonly encountered when developing multi-step flow protocols is the problem of solvent compatibility between subsequent reaction steps. While it is possible to utilise immobilised scavengers to remove many reagents and by-products,3 the problem of switching between solvents still requires innovative solutions in order to realise the full potential of multi-step flow processing. In particular solvent choice, usage and recycle opportunities are important decision points in any synthesis programme whether in batch or flow.
Continuous solvent removal and switching is well established on a large industrial scale with falling film evaporators, cone evaporators and similar large scale devices serving the needs of the food, beverage, petroleum and other industries. On the other end of the spectrum, a number of forays have also been made into the design of microfluidic evaporator units but these have primarily been used as concentrators for specialised analytical devices.4–6 Despite the creativity of the devices so far developed, there are only a few examples of their application in multi-step flow synthesis.5,6 Limiting factors in many of the microfluidic evaporators previously reported include poor chemical compatibility, low flow rates and they are currently limited to easy solvent transitions from low boiling solvents such as dichloromethane (DCM) and MeOH, to extremely high boiling solvents like toluene and dimethylformamide (DMF). We believe there is a need to develop a general, multi-purpose evaporator for meso-fluidic processing in research laboratories which is compatible with existing flow chemistry equipment and with the typical product output from the reactors. Drawing inspiration from electrospray ionisation techniques, we envisaged a device which would expose a high surface spray of solution to a desolvation gas to remove solvent quickly and efficiently (Fig. 1).
 | ||
| Fig. 1 Graphical representation of the device used for the solvent evaporation/switch (see also ESI†). | ||
A prototype in-line evaporator was constructed from an Omnifit®7 glass column and Swagelok® fittings. The central channel, consisting of a 0.125 mm i.d. (1/16 inch o.d.) stainless steel tube, carries the solution to be evaporated while the surrounding larger 1/8 inch o.d. stainless steel tube transports in nitrogen gas for desolvation. Surrounding both of these tubes is a third PTFE tube (1/4 inch o.d.) which serves as an exhaust for the desolvation gas and solvent vapours. The fine spray generated from this concentric design is directed into a 15 mm borosilicate glass Omnifit® column which serves as the evaporation chamber. This column can be heated by any standard column heater commonly employed in flow synthesis, allowing the evaporator to be readily incorporated into typical flow chemistry platforms.
We found that it was beneficial to include a peristaltic pump on the concentrated liquid outlet of the device at the bottom of the evaporation chamber. This was to prevent gas from exiting with the liquid concentrate and also allowed excellent control over the flow rate of the concentrate leaving the evaporator.
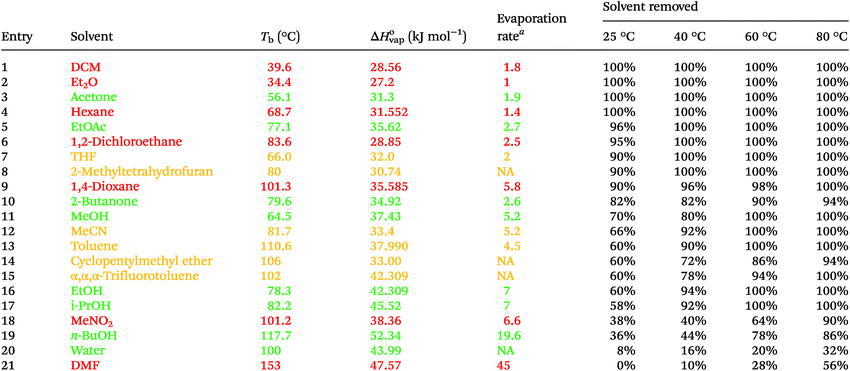
To evaluate the effectiveness of this device we examined 1 mL min−1 flow streams of a range of solvents (Table 1). The flow rate of gas was kept at 10 L min−1 by means of an adjustable flow meter. The evaporation chamber was heated at various temperatures of 25, 40, 60 and 80 °C. Pleasingly, the system was very effective at evaporating low to medium boiling point solvents (Tb < 100 °C). It was also interesting to observe that the rate of removal of a solvent was not dependent on its standard boiling point. While low boiling solvents such as diethyl ether (Et2O), hexane, DCM and acetone were completely removed at room temperature, higher boiling aprotic solvents such as EtOAc, 2-methyltetrahydrofuran and 1,4-dioxane could all be substantially removed at room temperature. By contrast, protic solvents such as MeOH, EtOH and i-PrOH were harder to evaporate and required heating above 40 °C to remove more than 80% of the solvent flow. The high boiling protic solvents n-BuOH, water, along with the aprotic DMF, were unsurprisingly more difficult to remove using the new device. Heating to 80 °C however increased the solvent removal to 86%, 32% and 56% for n-BuOH, water and DMF respectively.
 = preferred,
= preferred,  = usable,
= usable,  = undesirable)8
= undesirable)8
| a DIN 53170 standard (Et2O = 1).9 |
|---|
|
The temperature required to effect total removal of the solvent appeared to be better related to the standard enthalpy of vaporisation (ΔH°vap). In general, solvents with ΔH°vap of 32 kJ mol−1 or less could be 90% removed (or better) at room temperature, solvents with ΔH°vap between 32 and 46 kJ mol−1 could be 90% removed (or better) at 60 °C. Solvents with ΔH°vap above 46 kJ mol−1 were difficult to remove completely using this particular configuration of the in-line evaporator. There were exceptions to these observations and they should be regarded as guidelines only, since EtOAc and 1,4-dioxane were easier to remove than their ΔH°vap would suggest while water was more difficult.
For the most reliable predictor for selecting the evaporation temperature we used evaporation rate charts. These experimental measurements of solvent evaporation rates, relative to that of a diethyl ether standard, at room temperature showed a good correlation with the evaporation rates observed for the in-line evaporator.9
Removal of high boiling solvents such as water, DMF, DMSO, pyridine and N-methyl-2-pyrrolidone (NMP) is extremely slow and consumes large amounts of energy using standard rotary evaporators. In a typical reaction work up it is more time and energy efficient to use extraction methods to transfer the reaction products into a lower boiling organic solvent which is then subsequently removed by rotary evaporation. Not only does this add additional labour intensive steps to the reaction work up but it is also a major contributor to the high solvent consumption of batch mode organic synthesis. The contamination of large quantities of water with harmful solvents (e.g. DMF) is an additional problem of current work up practices in synthesis.
Finally, the extraction of water soluble or water sensitive products can be problematic. An in-line evaporator which could remove these troublesome solvents, even if only partially, would offer a much more acceptable way of removing high boiling solvents at the end of a synthesis step.
Our initial efforts to remove water and DMF had shown that our prototype device at 80 °C, could only remove 32% and 52% respectively (Table 1). We hypothesised therefore that running the device with a lower flow rate of the solvent would allow removal of more high boiling solvent. Running a 0.5 mL min−1 flow of water through the device at 80 °C increased the removal from 32% to 64%. This was still not sufficient for our needs. Therefore we investigated the dilution of the water flow with more volatile solvents to facilitate its removal in the evaporator. EtOH forms a positive azeotrope (95.63% EtOH, 4.37% water) with water. Consequently we first tested a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and ethanol at 1 mL min−1 at 80 °C. This resulted in only 64% removal of the overall solvent flow. Better results were obtained when acetone was used to dilute the water in a 1:1 ratio. The 1:1 mixture was pumped at 1 mL min−1 through the prototype to obtain a complete removal of the water–acetone mixture at 80 °C and 68% removal at 60 °C. Since acetone does not form any azeotrope with water, this enhanced evaporation can be attributed to the rapid evaporation of acetone in the solvent sprayed generating smaller droplets of residual water. The higher surface area of these water droplets then facilitates their volatilisation in the in-line evaporator.10
:1 mixture of water and ethanol at 1 mL min−1 at 80 °C. This resulted in only 64% removal of the overall solvent flow. Better results were obtained when acetone was used to dilute the water in a 1:1 ratio. The 1:1 mixture was pumped at 1 mL min−1 through the prototype to obtain a complete removal of the water–acetone mixture at 80 °C and 68% removal at 60 °C. Since acetone does not form any azeotrope with water, this enhanced evaporation can be attributed to the rapid evaporation of acetone in the solvent sprayed generating smaller droplets of residual water. The higher surface area of these water droplets then facilitates their volatilisation in the in-line evaporator.10
Another source of inefficiency in multi-step organic synthesis is the need to change solvents between subsequent reaction steps. We envisaged that the in-line evaporator should be capable of performing continuous solvent switches between subsequent flow processes. Simple switches from low boiling to high boiling solvents have already been demonstrated for systems where the difference in boiling points is greater than 40 °C (e.g. MeOH to toluene or DCM to DMF).5 Our early results had shown that the rate of evaporation in the new prototype device was not directly related to the boiling point of the solvent. As a result, the evaporator allows continuous solvent switches which are not possible with microfluidic evaporators.
In our flow synthesis of the probe for the Neurotensin receptor-1, Meclinertant (SR48692), we reported the solvent switch from toluene to methanol in a multi-step flow sequence.11 For us this represented an unfortunate problem since both the previous acylation/rearrangement steps and the subsequent aromatisation/methylation were telescoped, the only break in the sequence being the necessity to remove toluene and subsequently re-dissolve 1 in methanol (Scheme 1a).
 | ||
| Scheme 1 (a) Semi-telescoped flow scheme for the synthesis of acetophenone 2 and (b) graphical scheme for the solvent switch from toluene to MeOH involving intermediate 1, using our bespoke prototype. | ||
Using our in-line evaporator therefore, a 0.16 mL min−1 flow of a toluene solution of 1 was conveniently combined with 0.64 mL min−1 MeOH at a T-piece. The 1:4 mixture of toluene and MeOH was then passed through the evaporator at 20 °C, with a gas flow rate of 10 L min−1, such that toluene was removed at a faster rate than MeOH. The flowing solution of 1 was concentrated by the evaporator (0.13 mL min−1) and enriched in MeOH (10:1 MeOH–toluene). The evaporator was run continuously for 1 hour and the resulting solution of 1 was fed directly into the subsequent steps of the flow synthesis of Meclinertant. Drying a sample of the evaporator output revealed that 90% of the solid product (1) was recovered. The 10% loss of material was due to aggregation of undissolved residual material on the evaporator walls. We believe that this would be minimised by long term continuous processing (Scheme 1b). Nevertheless, complete recovery of any loss due to precipitating material is an issue which will be addressed in future prototypes.
Since solvent is not the only volatile ingredient in reactions which might require removal between reaction steps we decided to use the new in-line device in other applications. One such example is nitromethane, a potentially explosive but useful reagent in synthesis. The condensation of nitromethane to aldehydes (Henry reaction) to form nitro alkenes is a well known procedure and can be efficiently catalysed by many immobilised amines.12,13 Typically a large excess of nitromethane is used (often as the solvent) to drive the reaction to completion. However, the use of large quantities of nitromethane in organic synthesis can raise safety concerns since it is known to be sensitised towards detonation by additives, including amines.14
Despite these safety concerns, the nitro alkenes generated by this process are important electrophiles for Michael addition processes. For safety and to avoid competing reactions it is necessary to completely remove excess nitromethane following formation of the nitro alkene. The generation and subsequent Michael addition of nitro alkenes has been performed in flow but the lack of an in-line evaporator required the authors to resort to a batch distillation to remove the excess nitromethane.13 We saw an opportunity to streamline the generation and use of nitro alkenes in flow, with improved safety, by removing nitromethane in a continuous fashion using the new evaporator.
Flowing a 0.1 M solution of 3 in toluene–nitromethane through a column of silica supported methyl thiourea (Si-MTU) (4) heated to 80 °C provided nitro alkene 5 with 92% conversion. The resulting solution was then processed through the in-line evaporator to remove the 9 equivalents of nitromethane in excess. Diluting the 0.1 mL min−1 reaction stream with 0.9 mL min−1 hexane, which forms a positive azeotrope with nitromethane, was required to help remove the relatively non-volatile nitromethane. Exposing the combined stream to 2.7 L min−1 nitrogen gas and heating the column at 20 °C effectively reduced the nitromethane levels to 1.1 equivalents relative to 5. The 0.45 mL min−1 of solution obtained was further diluted with hexane (0.55 mL min−1) and returned for a second in-line evaporation under the same conditions. The second pass through the evaporator removed the remaining nitromethane to levels lower than 0.1 equivalents compared to 5. The toluene–hexane solution of 5 could then be directed into a flow reactor for the Michael addition of dibenzyl malonate (6), providing 7 in good yield (60%) and purity after crystallisation (Scheme 2).

In addition to improving the safety of using nitromethane on large scale, our new in-line evaporator proved to be particularly helpful for the easy recovery and recycling of the condensed fractions of the excess reagent, therefore reducing the overall environmental impact of the process (Scheme 2).
In conclusion, we have devised a convenient and efficient system for evaporating, concentrating and switching solvents in continuous processing and batch mode. We have successfully applied our prototype to a telescoped process where toluene was exchanged for MeOH. Additionally, we were able to achieve the removal of excess of potentially dangerous reagent, nitromethane, from a reaction environment in order to telescope the material to the following Michael addition. In the case of nitromethane removal, we were able to collect the excess reagent for recycling purposes. Finally, it is worth noting the recyclability achieved with this device, whereby all solvents removed can be easily collected and reused, with reduced environmental impact.
We gratefully acknowledge funding from Pfizer Worldwide Research and Development (C.B.), the EPSRC (E.S.), the Commonwealth Scholarship Commission (B.J.D.) and the BP 1702 endowment (S.V.L.). We also wish to thank Dr J. E. Davies for crystal structure determination and the EPSRC for a financial contribution toward the purchase of the X-ray diffractometer.
Notes and references
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS.
- (a) G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708–5723 CrossRef CAS; (b) C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS; (c) H. Lange, C. F. Carter, M. D. Hopkin, A. Burke, J. G. Goode, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 765–769 RSC; (d) M. Rasheed and T. Wirth, Angew. Chem., Int. Ed., 2011, 50, 357–358 CrossRef CAS; (e) S. A. M. W. van den Broek, J. R. Leliveld, R. Becker, M. M. E. Delville, P. J. Nieuwland, K. Koch and F. P. J. T. Rutjes, Org. Process Res. Dev., 2012, 16, 934–938 CrossRef CAS; (f) X. Liu, B. Ünal and K. F. Jensen, Catal. Sci. Technol., 2012, 2, 2134–2138 RSC; (g) D. L. Browne, S. Wright, B. J. Deadman, S. Dunnage, I. R. Baxendale, R. M. Turner and S. V. Ley, Rapid Commun. Mass Spectrom., 2012, 26, 1999–2010 CrossRef CAS; (h) M. O'Brien, P. Koos, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2012, 10, 7031–7036 RSC; (i) J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS; (j) D. X. Hu, M. O'Brien and S. V. Ley, Org. Lett., 2012, 14, 4246–4249 CrossRef CAS; (k) T. P. Peterson, A. Polyzos, M. O'Brien, T. Ulven, I. R. Baxendale and S. V. Ley, ChemSusChem, 2012, 5, 274–277 CrossRef; (l) J. G. Stevens, R. A. Bourne and M. Poliakoff, Green Chem., 2009, 11, 409–416 RSC; (m) P. Koos, U. Gross, A. Polyzos, M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2011, 9, 6903–6908 RSC; (n) M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 1250–1257 RSC; (o) S. Kasinathan, S. L. Bourne, P. Tolstoy, P. Koos, M. O'Brien, R. W. Bates, I. R. Baxendale and S. V. Ley, Synlett, 2011, 2648–2651 CAS; (p) C. Battilocchio, I. R. Baxendale, M. Biava, M. O. Kitching and S. V. Ley, Org. Process Res. Dev., 2012, 16, 798–810 CrossRef CAS; (q) C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2013, 15, 2278–2281 CrossRef CAS.
- R. M. Myers, K. A. Roper, I. R. Baxendale and S. V. Ley, in Modern Tools for the Synthesis of Complex Bioactive Molecules, ed. J. Cossy and S. Arseniyadis, J. Wiley, N. Y., 2012, pp. 359–394, ISBN 978-0-470-61618-5 Search PubMed.
- (a) X. Casadevall i Solvas, V. Turek, T. Prodromakis and J. B. Edel, Lab Chip, 2012, 12, 4049–4054 RSC; (b) B. Z. Cvetković, O. Lade, L. Marra, V. Arima, R. Rinaldi and P. S. Dittrich, RSC Adv., 2012, 2, 11117–11122 RSC; (c) B. Z. Cvetković and P. S. Dittrich, Anal. Bioanal. Chem., 2013, 405, 2417–2423 CrossRef; (d) A. Hibara, K. Toshin, T. Tsukahara, K. Mawatari and T. Kitamori, Chem. Lett., 2008, 1064–1065 CrossRef CAS; (e) J. Leng, B. Lonetti, P. Tabeling, M. Joanicot and A. Ajdari, Phys. Rev. Lett., 2006, 96, 84503–84506 CrossRef; (f) G. C. Randall and P. S. Doyle, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10813–10818 CrossRef CAS; (g) Y. Zhang, S. Kato and T. Anazawa, Chem. Commun., 2009, 2750–2752 RSC.
- (a) R. L. Hartman, H. R. Sahoo, B. C. Yen and K. F. Jensen, Lab Chip, 2009, 9, 1843–1849 RSC; (b) M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452 RSC; (c) R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CrossRef CAS; (d) M. D. Hopkin, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2013, 11, 1822–1839 RSC.
- W.-Y. Tseng, J. Cho, X. Ma, K. Mahal, A. Chatziioannou and R. M. van Dam, Proceedings of the 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences, 2010, pp. 1010–1012 Search PubMed.
- http://www.omnifit.com/ .
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- DIN 53170 – Solvents for paints and varnishes – Determination of the evaporation rate. Deutsches Institut für Normung.
- (a) CRC handbook of chemistry and physics, ed. W. M. Haynes, CRC Press/Taylor and Francis, 93rd edn, 2013 Search PubMed; (b) E.-K. Hilmen, (November 2000). Separation of Azeotropic Mixtures: Tools for Analysis and Studies on Batch Distillation Operation. Norwegian University of Science and Technology, Dept. of Chemical Engineering. Retrieved 24 March 2007.
- C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, Chem.–Eur. J., 2013, 19, 7917–7930 CrossRef CAS.
- (a) N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS; (b) M. Lakshmi Kantam and P. Sreekanth, Catal. Lett., 1999, 57, 227–231 CrossRef; (c) S. L. Poe, M. Kobaslija and D. T. McQuade, J. Am. Chem. Soc., 2007, 129, 9216–9221 CrossRef CAS; (d) F. Shang, H. Liu, J. Sun, B. Liu, C. Wang, J. Guan and Q. Kan, Catal. Commun., 2011, 12, 739–743 CrossRef CAS; (e) K. Motokura, M. Tada and Y. Iwasawa, Catal. Today, 2009, 147, 203–210 CrossRef CAS; (f) K. Motokura, M. Tada and Y. Iwasawa, J. Am. Chem. Soc., 2007, 129, 9540–9541 CrossRef CAS; (g) Y. Sohtome, Y. Hashimoto and K. Nagasawa, Adv. Synth. Catal., 2005, 347, 1643–1648 CrossRef CAS; (h) R. Ballini, G. Bosica, D. Livi, A. Palmieri, R. Maggi and G. Sartori, Tetrahedron Lett., 2003, 44, 2271–2273 CrossRef CAS; (i) K. Motokura, M. Tada and Y. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 9230–9235 CrossRef CAS; (j) G. Demicheli, R. Maggi, A. Mazzacani, P. Righi, G. Sartori and F. Bigi, Tetrahedron Lett., 2001, 42, 2401–2403 CrossRef CAS; (k) D. Macquarrie, R. Maggi, A. Mazzacani, G. Sartori and R. Sartorio, Appl. Catal., A, 2003, 246, 183–188 CrossRef CAS; (l) M. I. Burguete, H. Erythropel, E. Garcia-Verdugo, S. V. Luis and V. Sans, Green Chem., 2008, 10, 401–407 RSC; (m) A. Palmieri, S. V. Ley, A. Polyzos, M. Ladlow and I. R. Baxendale, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.23.
- L. Soldi, W. Ferstl, S. Loebbecke, R. Maggi, C. Malmassari, G. Sartori and S. Yada, J. Catal., 2008, 258, 289–295 CrossRef CAS.
- Y. A. Gruzdkov and Y. M. Gupta, J. Phys. Chem. A, 1998, 102, 2322–2331 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details about the construction and operation of the prototype evaporator; a video file of the prototype device removing toluene. CCDC 940593. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3gc40967h |
| This journal is © The Royal Society of Chemistry 2013 |