DOI:
10.1039/C3GC40588E
(Critical Review)
Green Chem., 2013,
15, 1764-1772
Unusual reactions mediated by FMN-dependent ene- and nitro-reductases
Received 27th March 2013, Accepted 24th April 2013
First published on 24th April 2013
Abstract
Due to the chemical versatility of the flavin cofactor, FMN-dependent ene-reductases and nitro-reductases can catalyze or mediate a diverse spectrum of chemical reactions. Among them, two-electron transfer reactions dominate, which may proceed via sequential hydride transfer at the same or at alternate reactive sites. In addition, highly reactive intermediates are often formed, which undergo subsequent spontaneous (non-enzymatic) reactions leading to further enzymatic transformations in a cascade. Besides the well-known reductive processes involving alkenes and nitro groups at the expense of a reduced flavin cofactor, redox-neutral processes including disproportionation and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-bond isomerization reactions are catalyzed by OYE homologues. Unusual flavin-dependent biotransformations are reviewed with a special focus on the OYE family of flavoproteins (ene-reductases) and oxygen-insensitive FMN-dependent nitro-reductases.
C-bond isomerization reactions are catalyzed by OYE homologues. Unusual flavin-dependent biotransformations are reviewed with a special focus on the OYE family of flavoproteins (ene-reductases) and oxygen-insensitive FMN-dependent nitro-reductases.
1. Introduction
Enzymes often rely on the presence of cofactors as molecular shuttles to transfer redox equivalents (e.g. electrons, hydrides), unstable species (carbanions, e.g. enolates, organometallics) or toxic molecules (e.g. ammonia) to catalyze a great variety of chemical reactions, as amino acid side chains alone cannot mediate all chemical transformations to sustain the essential processes of life. Among the redox-enzymes employed in biotransformations, the majority depends on nicotinamide acting as a hydride donor for CO and CC bond reductions.1 A smaller number uses a metal, most prominent Fe2+/3+, but also Mn2+/3+, V5+ and others, for single-electron transfer.2 Flavoenzymes, in particular, owe their redox activity to the ability of the flavin cofactor—either flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD)—to mediate one- or two-electron transfer processes, which are enabled by resonance-stabilized radical and/or anionic reactive intermediates.3 In contrast to nicotinamide, which can only participate in two-electron (hydride) transfer, the diversity of flavoenzyme chemistry is reflected by an astonishingly broad spectrum of reactions, ranging from oxidations, such as mono-, di-, and peroxidations, as well as halogenation and C–C formation, to an array of (one- or two-)electron reductions involving alkenes, arenes, nitro-groups and nitro-esters.3,4 The origin of this chemical versatility lies in the tricyclic isoalloxazine ring system of flavins. Although the redox potential for two-electron reduction of flavin lies at −200 mV, it can be modulated by the enzyme environment—often via (bi)covalent attachment of the flavin onto the protein5—to span a range from −400 mV to +60 mV, thus conferring diverse reactivity to the enzyme. Additionally, flavins react with molecular oxygen to affect CC-epoxidation, C–H-hydroxylation and R-SO3H-, R-NO2-, alcohol-, amine-, thioether- and Baeyer–Villiger-oxidations. Interestingly, flavoenzymes are also known to be involved in redox-neutral processes, such as CC-isomerization, magnetoreception and light-responsiveness.6Most of the flavin-dependent enzyme-catalyzed transformations have chemical counterparts. However, these often require conditions which do not allow sustainable and/or environmentally friendly processes.7 In particular, non-enzymatic catalyzed CC-bond isomerization reactions often rely on the use of (transition)metal-based catalysts based on Ru, Rh, Pd, Ir,8 or AlCl3.9 Similarly, the catalytic asymmetric hydrogenation of alkenes traditionally involves transition-metal catalysts,10 as does the reduction of nitroaromatics,11 while the classical Nef-reaction requires the presence of a strong aqueous acid.12
2. Flexibility in hydride transfer
2.1 Reduction of activated alkenes
The most prominent application of flavoproteins is the two-electron reduction of activated CC bonds by the so-called ene-reductases, which are homologues of the Old Yellow Enzyme (OYE) family of flavoproteins. Electronic activation is provided by an electron withdrawing group such as carbonyl (ketone/aldehyde/acid/ester/lactone/cyclic imide), nitro or nitrile.13 The asymmetric reduction proceeds via transfer of [2H] in a Michael-type addition reaction operating in a trans-fashion. The hydride is provided by FMNH2, while the proton (usually delivered from a conserved tyrosine residue) ultimately derives from the solvent. The oxidized flavin is reduced again at the expense of a nicotinamide cofactor to close the catalytic cycle (Fig. 1). Alternatively, flavin may be recycled via non-conventional electrochemical or light-mediated methods14 or by a non-natural hydride donor in a nicotinamide-free system.15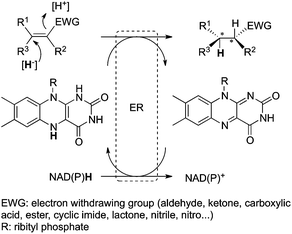 |
| | Fig. 1 Asymmetric reduction of activated alkenes by ene-reductases (ER). | |
Although only a few studies have addressed the technical aspects of ene-reductase-catalyzed biotransformations, some reactions have been investigated with a focus on productivity and upscaling. TTN values are generally low (usually <103, as estimated from conversion data reported for non-optimized reactions), a phenomenon which has been associated with enzyme deactivation by the substrate, which significantly limits substrate loading.16 More sophisticated reaction engineering techniques, however, led to significant improvements in productivity, as shown in the preparation of ethyl (S)-2-ethoxy-3-(p-methoxyphenyl)propanoate using isolated OYEs with in situ substrate-feeding product-removal (SFPR). This technique allowed elevated substrate loading and gave a productivity of 56 g L−1 d−1 on the preparative scale.17 The use of ene-reductases on a large scale was also reported for the reduction of citraconic acid dimethyl ester. The reaction was successfully scaled up to 70 g in a biphasic system, using cell paste containing the biocatalyst, in conjunction with a coupled-enzyme recycling system based on i-propanol and carbonyl reductase.18
Related to OYEs by the same FMN-cofactor, oxygen-insensitive nitro-reductases (classified as type I, see Section 2.4) catalyze the sequential two electron-reduction of the nitro group.19,20 The catalytic activities of these two enzyme classes overlap to some extent, as ene-reductases have shown reducing activities on nitro-compounds21 while nitro-reductases were partly active in the reduction of CC bonds.22
In addition to these ‘classic’ CC- and nitro-reduction activities, several unusual biotransformations have been found to be mediated by these flavin-dependent ene- and nitro-reductases, which are highlighted in this review. First, the flexibility in the regio-selectivity of hydride transfer onto C vs. N accounts for the catalytic promiscuity23 of these enzymes as demonstrated by CC vs. NO2-reduction; second, the high reactivity of intermediates obtained in the reductive step is often responsible for spontaneous (non-enzymatic) follow-up reactions leading to the formation of unusual products. Last, in addition to hydride transfer from/to nicotinamide, the FMN cofactor can also function as an acid/base catalyst in redox-neutral isomerization reactions, or it may transfer a hydride directly between two substrate molecules in a disproportionation reaction, bypassing the otherwise essential nicotinamide cofactor.
2.2 Reduction of activated alkynes
Ene-reductases from the OYE family have been extensively studied for their catalytic activity in the CC bond reduction of activated alkenes7,24 and were implemented in various asymmetric synthetic strategies for the generation of chiral molecules in enantiopure form.25 In addition, OYE homologues were shown to catalyze the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond reduction of electronically related ynones at the expense of NAD(P)H. This stereoselective reductive reaction furnished the corresponding trans-configurated enone, which was subsequently reduced to the fully saturated ketone (Fig. 2).
C bond reduction of electronically related ynones at the expense of NAD(P)H. This stereoselective reductive reaction furnished the corresponding trans-configurated enone, which was subsequently reduced to the fully saturated ketone (Fig. 2).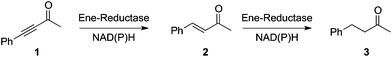 |
| | Fig. 2 Reduction of ynone by ene-reductases. | |
Recombinant E. coli strains over-expressing OYE1 (Saccharomyces pastorianus), OYE2 (Saccharomyces cerevisiae), OYE3 (Saccharomyces cerevisiae) or NCR (Zymomonas mobilis) were used in the reduction of 4-phenyl-3-butyn-2-one (1) in the presence of NAD(P)H in excess. Depending on the relative rate of both steps, the reaction yielded (E)-4-phenyl-3-buten-2-one (2) and 4-phenyl-2-butanone (3) in various ratios: while NCR predominantly furnished the fully reduced ketone without a trace of intermediate enone, OYE3 showed a strong substrate preference for the ynone over the enone.26
2.3 Reduction of activated aromatics
The ability of flavoproteins to deliver a hydride onto electron-deficient carbon atoms is not restricted to CC and CC-bonds, but may likewise take place on electron-deficient aromatics. Elucidation of biodegradation pathways of polynitro-aromatics, such as the toxic explosive 2,4,6-trinitrotoluene (TNT) (4), showed catalytic promiscuity of ene-reductases on nitro-aromatics. Apart from nitro-reduction of TNT (leading to the nitroso- and hydroxylamino-products, see Section 2.4), PETN reductase (pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2) could also direct a nucleophilic hydride attack directly onto the electron-deficient aromatic ring system. The latter process is remarkable, because it goes in hand with (energy-requiring) destruction of aromaticity. The first intermediate thus obtained is a mono-hydride-Meisenheimer complex (5), which was further reduced to an unstable di-hydride-Meisenheimer complex (6) (Fig. 3).27 Disappearance of the latter is dependent on the enzyme concentration and is accompanied by elimination of nitrite along with unidentified compounds.29 Alternatively, dimerization occurred with products derived from the concurrent nitro-reduction reaction (see Section 2.4) to yield secondary diarylamines accompanied by loss of nitrite, for which several pathways have been suggested.28,30 This activity, favored by the enhanced electrophilicity of the aromatic system by three nitro-substituents, was also found in XenB (Pseudomonas fluorescens),29 NemA (N-ethylmaleimide reductase from E. coli)29 and OPR1 (Arabidopsis thaliana).30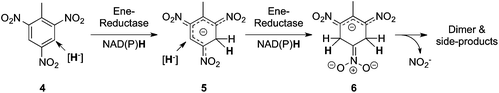 |
| | Fig. 3 Ring reduction of polynitroaromatics catalyzed by ene-reductases. | |
2.4 Nitro-group reduction: nitroaromatics
The biodegradation of nitroaromatics usually proceeds via nitro-group reduction, catalyzed by flavin-dependent nitro-reductases. Two types of these enzymes have been identified, mostly in bacterial strains.11 Type II nitroreductases catalyze nitro-group reduction via single-electron transfer yielding an unstable nitro-anion free radical. The latter undergoes spontaneous re-oxidation by O2 producing superoxide, which accounts for the oxygen-sensitivity of these enzymes.31 In contrast, oxygen-insensitive (type I) nitroreductases act via a two-electron transfer and include NfsA and NfsB from E. coli,32 SnrA and CnR from Salmonella enterica serovar Typhimurium TA1535,33 PnrA and PnrB from Pseudomonas putida,34 and YcnD from Bacillus subtilis (NfrA2).35 On nitroaromatics, they catalyze two sequential two-electron reductions at the expense of NAD(P)H, which [in the case of nitrobenzene (7)] leads to the formation of the corresponding hydroxylamine (9) via the formation of nitroso-benzene (8) as a modestly stable intermediate. The complete reduction of a nitroaromatic to the corresponding aniline (10) is a complex process because it requires the sequential delivery of three hydride equivalents. It is therefore not surprising that only a single case of such a six-electron reduction has been reported with Salmonella typhimurium nitroreductase, where nitrobenzene was reduced all the way to aniline.13 In the majority of cases, aromatic nitro-group reduction proceeds in a less controlled fashion and encompasses spontaneous (non-enzymatic) condensation of reactive intermediates, such as nitrosobenzene (8) and phenylhydroxylamine (9), to yield substantial amounts of azoxybenzene (11) as a side-product, which explains the low yields observed (Fig. 4).13 Nitroreductases are regarded as typical detoxification enzymes because they act on a remarkably broad range of nitroaromatics, such as nitrofurazone, nitrofurantoin or 5-(1-aziridinyl)-2,4-dinitrobenzamide (an anticancer prodrug known as CB195436).26,37 2,4,6-Trinitrotoluene (7) is widely accepted by most nitroreductases and OYE homologues, which sequentially reduce the nitro-group to the corresponding nitroso- and hydroxylamino-compounds.15,38 Condensation of the latter products with intermediates originating from the competing pathway via reduction of the aromatic ring (see Section 2.3) usually leads to the formation of various dimers.39 Overall, the outcome of the reductive biodegradation of nitro-aromatics is not only determined by the relative rates of enzymatic and spontaneous steps, but also by the stability of intermediates towards conditions in a given environment (such as pH, temperature, and concentrations).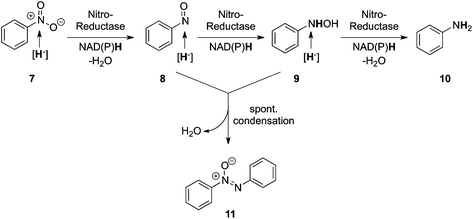 |
| | Fig. 4 Reductive biodegradation of nitrobenzene initiated by Salmonella typhimurium nitroreductase. | |
It is noteworthy that nitroreductases and ene-reductases, though structurally different, display reciprocal substrate promiscuity. While OYE homologues are strong in the nitro-group reduction of TNT, Salmonella typhimurium nitroreductase was dominant in the CC bond reduction of activated alkenes, however strongly favoring the nitro-moiety as the electron-withdrawing group, such as 1-nitrocyclohexene and trans-β-nitrostyrene.13
2.5 Nitro-group reduction: nitroaliphatics (bio-Nef-reaction)
Like aromatic nitro-groups, also aliphatic nitro analogs (12) were readily reduced by OYEs. Several ene-reductases could transfer a hydride directly onto the substrate nitrogen atom, yielding a highly unstable aliphatic sec-nitroso compound (13) which spontaneously tautomerized to form the corresponding more stable oxime (14). The latter was further attacked at the nitrogen atom by FMNH2 to form a water-labile imine (15), which rapidly hydrolyzed to yield the corresponding carbonyl compound (16) and ammonia as the final stable end-products of this cascade (Fig. 5). Overall, this resembles a biocatalytic equivalent to the Nef-reaction. The instability of the intermediates prevented isolation and/or detection of the nitroso and imine products.12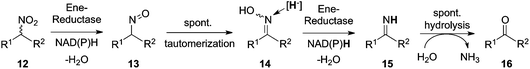 |
| | Fig. 5 Bio-Nef reaction mediated by ene-reductases on sec-nitro-aliphatics. | |
In the case of nitroalkenes, the bio-Nef pathway and the traditional CC bond reduction occurred sequentially, as exemplified with 1-nitro-2-phenylpropene (17). All ene-reductases tested delivered a mixture of 1-nitro-2-phenylpropane (obtained as the initial product from alkene reduction), and oxime and/or aldehyde (both resulting from subsequent reduction of the nitro-alkane via the Nef-pathway). Interestingly, the most active enzymes were those previously identified as xenobiotic reductases, naturally acting on nitro-compounds: XenA (Pseudomonas putida II-B 86), NerA (glycerol trinitrate reductase from Agrobacterium radiobacter) and PETN reductase. 12-Oxophytodienoic acid reductase (OPR3, Solanum lycopersicum) and morphinone reductase (Pseudomonas putida M10) also showed significant activities.
2.6 Nitro-group reduction: nitroalkenes (oxazete formation)
While the reduction of activated alkenes by ene-reductases is usually highly chemo-selective with hydride transfer onto Cβ, flexibility in the substrate binding may afford alternate binding modes favoring hydride attack on the nitrogen atom in the case of β,β-disubstituted nitroalkenes (e.g.17), where hydride delivery on Cβ is sterically impeded (compare with Section 2.5). In turn, this leads to two competing reactions, i.e. CC bond vs. nitro-group reduction, which potentially occur simultaneously on the same substrate, as opposed to the bio-Nef reaction on nitroalkenes, where CC bond and nitro-group reductions are sequential. Simultaneous competing CC-bond and nitro-group reductions were observed in the reduction of 1-nitro-2-phenylpropene (17): reduction of the nitro group to the corresponding nitroso-alkene (18) was followed by fast s-cis/s-trans-isomerization, affording a highly unstable s-cis-nitroso-alkene (19). The latter spontaneously underwent electrocyclization to rac-4-methyl-4-phenyl-1,2-oxazete (20) as the major product (Fig. 6). At elevated temperature, the modestly stable 1,2-oxazete underwent retro-[2 + 2]-cycloaddition to furnish HCN and acetophenone (21).40 From a range of ene-reductases tested, oxazete formation was predominant with XenA, while PETN reductase favored the bio-Nef pathway after reduction of the CC bond. Overall, only β,β-disubstituted nitroalkenes were found to undergo reduction of the nitro-group to form the corresponding racemic oxazete. NerA, XenA, PETN reductase and OPR3 were identified as a subset of OYE homologues showing structural similarities most likely responsible for their dual activity on nitroalkenes with regard to CC and nitro-group reductions. In particular, the presence of ‘hydrophobic hot-spots’ in the region around loop β5 and loop β6 was identified as determinant for specific nitro group reduction, allowing two concurrent binding modes required for dual catalytic activity.41 Along with the favored substrate binding mode, the enzyme kinetics eventually determine the biodegradation pathways of nitroalkenes, as the bio-Nef reaction only occurs after CC bond reduction, while oxazete formation results from preferential nitro-reduction over CC bond reduction.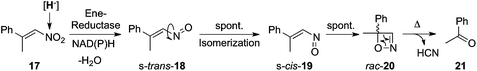 |
| | Fig. 6 Oxazete formation mediated by ene-reductases on β,β-disubstituted nitroalkene (17). | |
2.7 Reductive nitrate ester cleavage
Like nitro-aromatics, nitrate esters are primarily man-made xenobiotics which have been extensively used as explosives during the two World Wars. Glycerol trinitrate (GTN) (22), also erroneously termed nitroglycerine, and pentaerythritol tetranitrate (PETN) (23) are two common organic nitrate esters, for which biodegradation pathways involving OYE homologues have been identified. They both proceed via reductive denitration in a net two-electron transfer, liberating nitrite and the corresponding alcohols (Fig. 7). PETN reductase, NerA, OYE1 as well as XenA and XenB, for instance, catalyze the reductive liberation of 1–2 mol of nitrite from GTN at the expense of NAD(P)H, while 2 mol of nitrite are usually obtained from PETN.42 Mechanistically, the reduction occurs via hydride attack onto the N atom of nitrate ester (24), followed by spontaneous tautomerization to yield the corresponding nitrite ester (25). The latter is hydrolytically very labile and is non-enzymatically converted to the alcohol with concomitant release of nitrite. The possibility that the mechanism proceeds via a radical mechanism has been postulated.33,43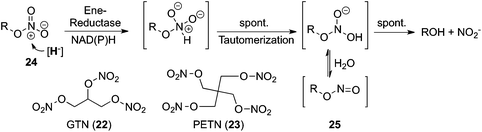 |
| | Fig. 7 Reductive nitrate ester cleavage catalyzed by ene-reductases; GTN: glycerol trinitrate, PETN: pentaerythritol tetranitrate. | |
Additionally, PETN reductase was also found to be active on structurally related N-nitramines, such as hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX, ‘Royal Demolition Explosive’) and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX), however to a much lesser extent compared to nitrate esters.44
2.8 Reductive dehydrohalogenation of activated haloalkenes
In an attempt to render insufficiently activated α,β-unsaturated carboxylic esters more reactive towards ene-reductases, the effects of additional electron-withdrawing halogen substituents on the CC bond were investigated. The latter were expected to enhance CC bond activation and thus increase ene-reductase activity on such substrates. While this was found to be extremely efficient on α-halo-α,β-unsaturated carboxylic esters of type 30, this strategy failed for β-halo-analogs 26 and 28 due to the high instability of the corresponding saturated β-halo-esters 27 and 29 obtained from the bioreduction. The enhanced acidity of the α-proton combined with the presence of a halogen on Cβ led to fast dehydrohalogenation via spontaneous elimination of hydrohalous acid. On α,β,β-trisubstituted halo-acrylic esters (26) the sequence proceeded until complete removal of halogen on Cβ took place (Fig. 8). Enzyme-based stereocontrol in the last CC bond reduction step (30) allowed the formation of both methyl 2-halopropionate enantiomers (31) in high enantiopurity (up to >99% ee).45 Overall, this pathway resembles a reductive dehalogenation of β-halogenated acrylic esters.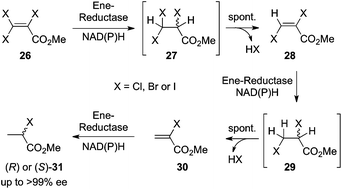 |
| | Fig. 8 Sequential reductive dehydrohalogenation of β-haloacrylic ester derivatives mediated by ene-reductases. | |
2.9 Reduction of O2 forming H2O2: Weitz–Scheffer oxidation (activated alkenes and thioethers)
Although the physiological role of ene-reductases has not been clearly identified yet, it is believed that OYE homologues are generally involved in oxidative stress response as detoxification enzymes.46 The nature of the physiological reductant has been determined, and most enzymes accept NADH and NADPH, often equally well, although some differences in kinetic parameters have been occasionally observed.29 However, oxidants capable of reacting with the enzyme-bound reduced flavin are very diverse, highlighting the catalytic promiscuity of this class of enzymes.15,47 Molecular oxygen has also been found to act as the substrate, although its reactivity is usually lower compared to activated alkenes.37 In some cases, however, this so-called ‘NAD(P)H oxidase activity’ predominates, thus producing hydrogen peroxide, which spontaneously reacts with compounds such as enones (32) and thioethers (34), thereby producing the corresponding epoxides (33) and sulfoxides (35) (Fig. 9). Since this Weitz–Scheffer reaction48 does not require the enzyme's active site, the products are formed in a non-stereoselective fashion and racemic mixtures are obtained. Though molecular oxygen is known to be accepted as oxidant by several ene-reductases,37,49 this side reaction was particularly predominant with YcnD (nitro-reductase from Bacillus subtilis), Lot6p [NAD(P)H:FMN-dependent reductase from Saccharomyces cerevisiae active on azo-compounds and quinones] and YhdA (NADPH:FMN-dependent oxidoreductase from Bacillus subtilis with azoreductase activity), all displaying the typical ping-pong bi-bi mechanism of OYE homologues. Ketoisophorone (36), citral (37), cyclohexenone (40) and menadione (38) were epoxidized to various extent (up to 77% conversion with ketoisophorone using YcnD) while thioanisole (39) was selectively oxidized to the racemic sulfoxide (35) in up to 80% conversion with YcnD, but no sulfone was detected. Interestingly, non-activated CC bonds were not reactive (e.g. in styrene, α-methylstyrene, or the isolated CC bond in citral). Performing the reaction in the presence of H2O2-degrading catalase or under an inert (argon) atmosphere suppressed product formation, thus confirming the chemical nature of this pathway.50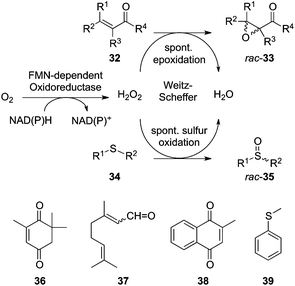 |
| | Fig. 9 Weitz–Scheffer epoxidation of enones and oxidation of thioethers mediated by NAD(P)H:FMN-dependent oxidoreductases in the presence of O2. | |
3. Redox-neutral hydrogen transfer
3.1 Disproportionation of activated alkenes
While investigating the formation of long wavelength charge-transfer bands upon mixing of OYE1 with phenolic compounds, Massey et al. discovered that OYE1 catalyzed the oxidative aromatization of several compounds, including 2-cyclohexenone (40), 3-oxodecalin-4-ene (ODE) (42) and 3-oxodecalin-4-ene-10-carboxaldehyde (ODEC), via a dehydrogenation reaction. With 2-cyclohexenone or ODE, a redox-neutral disproportion (‘dismutation’) reaction was observed (Fig. 10A/B): while one molecule of substrate was reduced to the corresponding saturated ketone—cyclohexanone (41) or 3-oxodecalin (43), respectively—as expected from the well-known ene-reductase activity, one equivalent of 40 or 42 was dehydrogenated yielding an unstable dienone, which spontaneously tautomerized to the aromatic species phenol or 3-hydroxy-6,7,8,9-tetrahydronaphthalene (HTN) (44), respectively. Overall, this reaction seems to be a shared characteristic of OYE homologues51 and does not proceed with net consumption of NAD(P)H, as FMN shuttles electrons directly between the two substrate molecules serving as hydrogen-acceptor and -donor.52 The synthetic potential of this system was investigated further by testing appropriate hydrogen-donor molecules in order to by-pass the requirement for nicotinamide cofactor and a second enzyme for its recycling in the reduction of various activated alkenes (Fig. 10C).9N-Phenyl-2-methylmaleimide (45) could be asymmetrically reduced by YqjM to (R)-N-phenyl-2-methylsuccinimide (46) in a decent conversion (51%) and >99% ee, using equimolar amounts of cyclohexane-1,4-dione (47) as a sacrificial hydrogen-donor. Dimethyl citraconate (48) was reduced to a lower extent (20% conversion) yielding dimethyl (R)-2-methylsuccinate (49) with excellent stereoselectivity (>99% ee). Overall, cyclohexane-1,4-dione (47) appeared to be a superior hydrogen donor than 3-methylcyclohexenone and offers an excellent coupled-substrate FMN-recycling approach for nicotinamide-free CC bond reduction protocols, where the driving force lies in the spontaneous aromatization of the oxidized co-product.53 In this process, the flavin cofactor plays a dual redox-role, being able to oxidize and reduce two different substrates in an ordered and sequential fashion.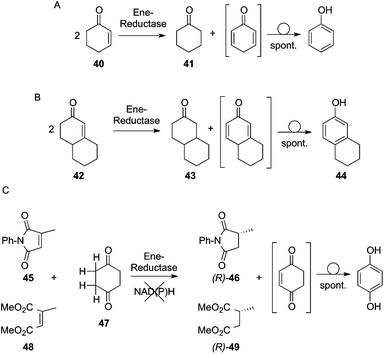 |
| | Fig. 10 A: Disproportionation of cyclohexenone; B: disproportionation of 3-oxodecalin-4-ene (ODE) catalyzed by ene-reductases; C: nicotinamide-independent reduction of enones with cyclohexane-1,4-dione as a hydrogen donor. | |
3.2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond isomerization
C bond isomerization
Examples of reduced FMN involved in non-redox rearrangement or isomerization reactions include isopentenyl-diphosphate isomerases type 2 (IDIs-2),54 which catalyze the reversible isomerization of isopentenyl diphosphate (IPP) (50) to dimethylallyl diphosphate (DMAPP) (51), both compounds being central activated intermediates in the mevalonate pathway for the biosynthesis of terpenoids (Fig. 11A). Likewise, cis–trans isomerases catalyzing (E/Z)-isomerization of alkenes are involved in the biosynthesis of β-carotenoids,55 such as lycopene-β-cyclase.56 IDIs-2 act through acid–base catalysis via a protonation/deprotonation sequence mediated by the flavin.57 In a related fashion, FMNH2 was shown to act as an acid/base catalyst hydrogen-shuttle in an unexpected CC-isomerization reaction catalyzed by ene-reductases (Fig. 11B). OYE homologues EBP1 (Candida albicans), OYE1, OYE2, and KYE (Kluyveromyces lactis) were found to mediate the isomerization of a terminal alkene (α-methylene-γ-butyrolactone) (52) to the more stable internal olefin isomer 3-methylfuran-2(5H)-one (53). The competing CC-bond reduction of both isomers leading to 2-methyl-γ-butyrolactone (54) was detected as a minor pathway (66–78% of isomerization vs. 20–27% of reduction product). With OYE2, the reduction of the thermodynamically more stable endocyclic isomer 53, which cannot be isomerized, was found to be about three times slower than the direct reduction of the exo-methylene lactone 52, and 18 times slower than CC-isomerization. Though theoretically nicotinamide-independent, this reaction requires a catalytic amount of NADH to get initiated, as the first step is the reduction of flavin, which is required for CC-isomerization activity, similar to IDI-2 catalysis, which can only be triggered by the reduced form of flavin.48 The mechanism was suggested to start by hydride attack of FMNH2 onto Cβ of the exo-methylene moiety in 52 (Fig. 11C). The enolate 55 thus formed is stabilized by hydride abstraction by FMN at the endo-Cβ′ position to produce the thermodynamically favored endo-isomer 53, while the oxidized flavin is recycled in a redox-neutral mode. Alternatively, protonation of the enolate at Cα furnishes the reduced product 54, causing a net consumption of NADH via regeneration of reduced flavin.58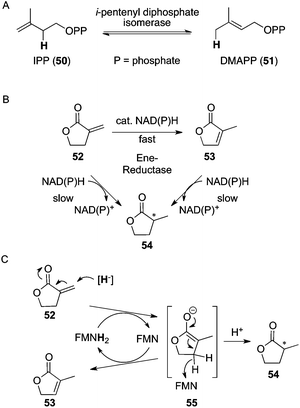 |
| | Fig. 11 A: Redox-neutral CC-bond isomerization between isopentenyl diphosphate (IPP) (50) and dimethylallyl diphosphate (DMAPP) (51); B: FMNH2-dependent enzymatic CC-isomerization and competing CC-reduction of α,β-unsaturated lactones catalyzed by ene-reductases; C: mechanistic proposal for isomerization. | |
4. Conclusion
Ene-reductases from the OYE family of flavoproteins and flavin-dependent nitroreductases are extremely versatile catalysts, as they not only display broad substrate tolerance but can also mediate diverse chemical reactions. Their catalytic promiscuity is connected to their physiological role. Ene-reductases, for instance, are suspected to act as detoxification enzymes in situations of oxidative stress and therefore have evolved to accept a broad range of activated alkenes serving as Michael-acceptors. Nitroreductases have evolved as natural remediation tools in contaminated environments with a major role in the reduction of toxic nitro-aromatics. Based on these similar reductive detoxification properties, it is not surprising that some xenobiotic reductases share catalytic characteristics of both ene- and nitro-reductases. In addition to redox catalysis, these flavin-dependent enzymes are also involved in chemo-enzymatic cascade-reactions, where the reactivity of intermediates often opens unexpected deviations to new pathways. Finally, examples of unusual reactions involving the flavin as a dual (non-)redox catalyst highlight the importance of these reductases beyond their well-studied traditional reductive activity.Acknowledgements
This project was performed within the DK ‘Molecular Enzymology’ and financial support from the Austrian Science Fund FWF (Vienna, project W9) is gratefully acknowledged.References
-
(a) F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC;
(b) S. M. A. De Wildeman, T. Sonke, H. E. Schoemaker and O. May, Acc. Chem. Res., 2007, 40, 1260–1266 CrossRef CAS;
(c) Y. Huang, N. Liu, X. R. Wu and Y. J. Chen, Curr. Org. Chem., 2010, 14, 1447–1460 CrossRef CAS;
(d) W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS;
(e) G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS;
(f) M. Kataoka, K. Kita, M. Wada, Y. Yasohara, J. Hasegawa and S. Shimizu, Appl. Microbiol. Biotechnol., 2003, 62, 437–445 CrossRef CAS;
(g) R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS;
(h) M. Hall, W. Kroutil and K. Faber, in Asymmetric Synthesis – The Essentials II, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, 2012, pp. 221–232 Search PubMed;
(i) D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS;
(j) J. H. Seo, D. Kyung, K. Joo, J. Lee and B.-G. Kim, Biotechnol. Bioeng., 2011, 108, 253–263 CrossRef CAS.
-
(a) P. A. Frey and A. D. Hegeman, in Enzymatic Reaction Mechanisms, Oxford University Press, New York, 2007, ch. 3 & 4, pp. 129–252 Search PubMed;
(b) D. E. T. Pazmino, M. Winkler, A. Glieder and M. W. Fraaije, J. Biotechnol., 2010, 146, 9–24 CrossRef;
(c) V. B. Urlacher and S. Eiben, Trends Biotechnol., 2006, 24, 324–330 CrossRef CAS;
(d) V. B. Urlacher and M. Girhard, Trends Biotechnol., 2012, 30, 26–36 CrossRef CAS;
(e) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS;
(f) J. G. Leahy, P. J. Batchelor and S. M. Morcomb, FEMS Microbiol. Rev., 2003, 27, 449–479 CrossRef CAS;
(g) V. L. Pecoraro, M. J. Baldwin and A. Gelasco, Chem. Rev., 1994, 94, 807–826 CrossRef CAS;
(h) K. Wieghardt, Angew. Chem., Int. Ed. Engl., 1989, 28, 1153–1172 CrossRef;
(i) A. Butler, Curr. Opin. Chem. Biol., 1998, 2, 279–285 CrossRef CAS;
(j) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS.
- V. Massey, FASEB J., 1995, 9, 473–475 CAS.
-
(a) P. Sobrado, Int. J. Mol. Sci., 2012, 13, 14219–14242 CrossRef CAS;
(b) M. W. Fraaije and A. Mattevi, Trends Biochem. Sci., 2000, 25, 126–132 CrossRef CAS;
(c) V. Massey, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS;
(d) K. H. van Pee and E. P. Patallo, Appl. Microbiol. Biotechnol., 2006, 70, 631–641 CrossRef;
(e) X. P. Chen and K. H. van Pee, Acta Biochim. Biophys. Sin., 2008, 40, 183–193 CrossRef CAS.
-
(a) M. W. Fraaije, W. J. H. Van Berkel, J. A. E. Benen, J. Visser and A. Mattevi, Trends Biochem. Sci., 1998, 23, 206–207 CrossRef CAS;
(b) M. W. Fraaije, R. H. H. van den Heuvel, W. J. H. van Berkel and A. Mattevi, J. Biol. Chem., 1999, 274, 35514–35520 CrossRef CAS;
(c) M. W. Fraaije, R. H. H. van den Heuvel, A. Mattevi and W. J. H. van Berkel, J. Mol. Catal. B: Enzym., 2003, 21, 43–46 CrossRef CAS.
-
(a) S. Bornemann, Nat. Prod. Rep., 2002, 19, 761–772 RSC;
(b) S. O. Mansoorabadi, C. J. Thibodeaux and H. W. Liu, J. Org. Chem., 2007, 72, 6329–6342 CrossRef CAS;
(c) C. T. Lin, D. E. Robertson, M. Ahmad, A. A. Raibekas, M. S. Jorns, P. L. Dutton and A. R. Cashmore, Science, 1995, 269, 968–970 CrossRef CAS;
(d) W. R. Briggs and J. M. Christie, Trends Plant Sci., 2002, 7, 204–210 CrossRef CAS;
(e) P. Muller and M. Ahmad, J. Biol. Chem., 2011, 286, 21033–21040 CrossRef.
-
(a) C. J. Pereira, Chem. Eng. Sci., 1999, 54, 1959–1973 CrossRef CAS;
(b) C. Bolm, O. Beckmann and O. A. G. Dabard, Angew. Chem., Int. Ed., 1999, 38, 907–909 CrossRef CAS.
-
(a) E. J. Corey and J. W. Suggs, J. Org. Chem., 1973, 38, 3224–3224 CrossRef CAS;
(b) G. J. Boons, A. Burton and S. Isles, Chem. Commun., 1996, 141–142 RSC;
(c) T. J. Donohoe, T. J. C. O'Riordan and C. P. Rosa, Angew. Chem., Int. Ed., 2009, 48, 1014–1017 CrossRef CAS.
- A. M. Linsenmeier and S. Brase, Eur. J. Org. Chem., 2012, 6455–6459 CrossRef CAS.
-
(a) P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC;
(b) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1999–2007 CrossRef;
(c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
-
(a) A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS;
(b) G. Wienhofer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS.
-
(a) M. M. Grau, J. C. van der Toorn, L. G. Otten, P. Macheroux, A. Taglieber, F. E. Zilly, I. W. C. E. Arends and F. Hollmann, Adv. Synth. Catal., 2009, 351, 3279–3286 CrossRef CAS;
(b) F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
-
(a) C. Stueckler, T. C. Reiter, N. Baudendistel and K. Faber, Tetrahedron, 2010, 66, 663–667 CrossRef CAS;
(b) C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernandez, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180–183 CrossRef CAS.
-
(a) J. F. Chaparro-Riggers, T. A. Rogers, E. Vazquez-Figueroa, K. M. Polizzi and A. S. Bommarius, Adv. Synth. Catal., 2007, 349, 1521–1531 CrossRef CAS;
(b) D. J. Bougioukou, A. Z. Walton and J. D. Stewart, Chem. Commun., 2010, 46, 8558–8560 RSC.
- M. Bechtold, E. Brenna, C. Femmer, F. G. Gatti, S. Panke, F. Parmeggiani and A. Sacchetti, Org. Process Res. Dev., 2012, 16, 269–276 CrossRef CAS.
- D. Mangan, I. Miskelly and T. S. Moody, Adv. Synth. Catal., 2012, 354, 2185–2190 CrossRef CAS.
-
(a) F. J. Peterson, R. P. Mason, J. Hovsepian and J. L. Holtzman, J. Biol. Chem., 1979, 254, 4009–4014 CAS;
(b) D. W. Bryant, D. R. Mccalla, M. Leeksma and P. Laneuville, Can. J. Microbiol., 1981, 27, 81–86 CrossRef CAS.
- M. Roldan, E. Perez-Reinado, F. Castillo and C. Moreno-Vivian, FEMS Microbiol. Rev., 2008, 32, 474–500 CrossRef CAS.
- K. Durchschein, B. F. D. Silva, S. Wallner, P. Macheroux, W. Kroutil, S. M. Glueck and K. Faber, Green Chem., 2010, 12, 616–619 RSC.
- Y. Yanto, M. Hall and A. S. Bommarius, Org. Biomol. Chem., 2010, 8, 1826–1832 CAS.
-
(a) R. J. Kazlauskas, Curr. Opin. Chem. Biol., 2005, 9, 195–201 CrossRef CAS;
(b) U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032–6040 CrossRef CAS.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 CrossRef CAS.
-
(a) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef CAS;
(b) C. K. Winkler, G. Tasnadi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381–389 CrossRef CAS.
- A. Muller, R. Sturmer, B. Hauer and B. Rosche, Angew. Chem., Int. Ed., 2007, 46, 3316–3318 CrossRef.
-
(a) C. E. French, S. Nicklin and N. C. Bruce, Appl. Environ. Microbiol., 1998, 64, 2864–2868 CAS;
(b) N. K. Hannink, S. J. Rosser, C. E. French and N. C. Bruce, Water Air Soil Pollut. Focus, 2003, 3, 251–258 CAS.
-
(a) P. van Dillewijn, R. M. Wittich, A. Caballero and J. L. Ramos, Appl. Environ. Microbiol., 2008, 74, 6703–6708 CrossRef CAS;
(b) R. M. Wittich, A. Haidour, P. Van Dillewijn and J. L. Ramos, Environ. Sci. Technol., 2008, 42, 734–739 CrossRef CAS.
- J. W. Pak, K. L. Knoke, D. R. Noguera, B. G. Fox and G. H. Chambliss, Appl. Environ. Microbiol., 2000, 66, 4742–4750 CrossRef CAS.
- E. R. Beynon, Z. C. Symons, R. G. Jackson, A. Lorenz, E. L. Rylott and N. C. Bruce, Plant Physiol., 2009, 151, 253–261 CrossRef CAS.
-
(a) G. S. Nyanhongo, M. Schroeder, W. Steiner and G. M. Gubitz, Biocatal. Biotransform., 2005, 23, 53–69 CrossRef CAS;
(b) J. Whiteway, P. Koziarz, J. Veall, N. Sandhu, P. Kumar, B. Hoecher and I. B. Lambert, J. Bacteriol., 1998, 180, 5529–5539 CAS;
(c) S. I. Liochev, A. Hausladen and I. Fridovich, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3537–3539 CrossRef CAS.
- J. Rau and A. Stolz, Appl. Environ. Microbiol., 2003, 69, 3448–3455 CrossRef CAS.
- M. R. Nokhbeh, S. Boroumandi, N. Pokorny, P. Koziarz, E. S. Paterson and I. B. Lambert, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2002, 508, 59–70 CrossRef CAS.
- A. Caballero, J. J. Lazaro, J. L. Ramos and A. Esteve-Nunez, Environ. Microbiol., 2005, 7, 1211–1219 CrossRef CAS.
- A. Morokutti, A. Lyskowski, S. Sollner, E. Pointner, T. B. Fitzpatrick, C. Kratky, K. Gruber and P. Macheroux, Biochemistry, 2005, 44, 13724–13733 CrossRef CAS.
-
(a) A. Christofferson and J. Wilkie, Biochem. Soc. Trans., 2009, 37, 413–418 CrossRef CAS;
(b) P. R. Race, A. L. Lovering, S. A. White, J. I. Grove, P. F. Searle, C. W. Wrighton and E. I. Hyde, J. Mol. Biol., 2007, 368, 481–492 CrossRef CAS.
- P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256–13264 CrossRef CAS.
-
(a) M. Hall, Y. Yanto and A. S. Bommarius, in The Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology, ed. M. Flickinger, Wiley, 2010, pp. 2234–2247 Search PubMed;
(b) R. E. Williams, D. A. Rathbone, N. S. Scrutton and N. C. Bruce, Appl. Environ. Microbiol., 2004, 70, 3566–3574 CrossRef CAS.
- B. A. Stenuit and S. N. Agathos, Appl. Microbiol. Biotechnol., 2010, 88, 1043–1064 CrossRef CAS.
- K. Durchschein, W. M. F. Fabian, P. Macheroux, K. Zangger, G. Trimmel and K. Faber, Org. Biomol. Chem., 2011, 9, 3364–3369 CAS.
- G. Oberdorfer, A. Binter, S. Wallner, K. Durchschein, M. Hall, K. Faber, P. Macheroux and K. Gruber, ChemBioChem, 2013, 14, 836–845 CrossRef CAS.
-
(a) C. E. French, S. Nicklin and N. C. Bruce, J. Bacteriol., 1996, 178, 6623–6627 CAS;
(b) D. S. Blehert, K. L. Knoke, B. G. Fox and G. H. Chambliss, J. Bacteriol., 1997, 179, 6912–6920 CAS;
(c) H. Khan, R. J. Harris, T. Barna, D. H. Craig, N. C. Bruce, A. W. Munro, P. C. E. Moody and N. S. Scrutton, J. Biol. Chem., 2002, 277, 21906–21912 CrossRef CAS;
(d) Y. Meah, B. J. Brown, S. Chakraborty and V. Massey, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8560–8565 CrossRef CAS.
- P. S. Y. Wong and J. M. Fukuto, Drug Metab. Dispos., 1999, 27, 502–509 CAS.
- H. Nivinskas, J. Sarlauskas, Z. Anusevicius, H. S. Toogood, N. S. Scrutton and N. Cenas, FEBS J., 2008, 275, 6192–6203 CrossRef CAS.
- G. Tasnadi, C. K. Winkler, D. Clay, M. Hall and K. Faber, Catal. Sci. Technol., 2012, 2, 1548–1552 CAS.
-
(a) T. B. Fitzpatrick, N. Amrhein and P. Macheroux, J. Biol. Chem., 2003, 278, 19891–19897 CrossRef CAS;
(b) R. M. Kohli and V. Massey, J. Biol. Chem., 1998, 273, 32763–32770 CrossRef CAS.
- R. E. Williams and N. C. Bruce, Microbiology, 2002, 148, 1607–1614 CAS.
- E. Weitz and A. Scheffer, Ber. Dtsch. Chem. Ges., 1921, 54, 2344–2353 CrossRef.
-
(a) O. Warburg and W. Christian, Biochem. Z., 1933, 263, 228–229 CAS;
(b) J. Strassner, A. Furholz, P. Macheroux, N. Amrhein and A. Schaller, J. Biol. Chem., 1999, 274, 35067–35073 CrossRef CAS.
- N. J. Mueller, C. Stueckler, M. Hall, P. Macheroux and K. Faber, Org. Biomol. Chem., 2009, 7, 1115–1119 CAS.
- J. Buckman and S. M. Miller, Biochemistry, 1998, 37, 14326–14336 CrossRef CAS.
- A. D. N. Vaz, S. Chakraborty and V. Massey, Biochemistry, 1995, 34, 4246–4256 CrossRef CAS.
- C. Stueckler, T. C. Reiter, N. Baudendistel and K. Faber, Tetrahedron, 2010, 66, 663–667 CrossRef CAS.
- K. Kaneda, T. Kuzuyama, M. Takagi, Y. Hayakawa and H. Seto, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 932–937 CrossRef CAS.
- Q. J. Yu, S. Ghisla, J. Hirschberg, V. Mann and P. Beyer, J. Biol. Chem., 2011, 286, 8666–8676 CrossRef CAS.
- Q. J. Yu, P. Schaub, S. Ghisla, S. Al-Babili, A. Krieger-Liszkay and P. Beyer, J. Biol. Chem., 2010, 285, 12109–12120 CrossRef CAS.
-
(a) H. Unno, S. Yamashita, Y. Ikeda, S. Sekiguchi, N. Yoshida, T. Yoshimura, M. Kusunoki, T. Nakayama, T. Nishino and H. Hemmi, J. Biol. Chem., 2009, 284, 9160–9167 CrossRef CAS;
(b) C. J. Thibodeaux, W. C. Chang and H. W. Liu, J. Am. Chem. Soc., 2010, 132, 9994–9996 CrossRef CAS;
(c) J. Calveras, C. J. Thibodeaux, S. O. Mansoorabadi and H. W. Liu, ChemBioChem, 2012, 13, 42–46 CrossRef CAS.
- K. Durchschein, S. Wallner, P. Macheroux, K. Zangger, W. M. F. Fabian and K. Faber, ChemBioChem, 2012, 13, 2346–2351 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence