 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of flow in green chemistry and engineering
Stephen G.
Newman
and
Klavs F.
Jensen
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu; Fax: (+1) 617-258-8992; Tel: (+1) 617-253-4589
First published on 26th April 2013
Abstract
Flow chemistry and continuous processing can offer many ways to make synthesis a more sustainable practice. These technologies help bridge the large gap between academic and industrial settings by often providing a more reproducible, scalable, safe and efficient option for performing chemical reactions. In this review, we use selected examples to demonstrate how continuous methods of synthesis can be greener than batch synthesis on a small and a large scale.
 Stephen G. Newman | Stephen G. Newman was born in Newfoundland, Canada in 1985. He obtained a Bachelor of Science with a major in Chemistry from Dalhousie University in 2008. In 2012, he graduated with a PhD from the University of Toronto under the supervision of Professor Mark Lautens. His thesis research was on the development of new palladium-catalyzed carbon–carbon and carbon–heteroatom bond forming reactions. He is currently an NSERC Postdoctoral Fellow in the laboratory of Professor Klavs F. Jensen, using flow chemistry to safely investigate chemical reactions under industrially relevant conditions that are difficult to obtain in batch. |
 Klavs F. Jensen | Klavs F. Jensen is Warren K. Lewis Professor and Head of the Chemical Department at the Massachusetts Institute of Technology. He received his chemical engineering education from the Technical University of Denmark (MSc) and University of Wisconsin-Madison (PhD). His research interests revolve around microfabrication, testing, and integration of microsystems for chemical and biological discovery, synthesis and processing. Catalysis, chemical kinetics and transport phenomena are also topics of interest along with development of simulation approaches for reactive chemical and biological systems. He is a member of the US National Academy of Engineering and the American Academy of Arts and Science. |
Introduction
The flask has been the most important piece of equipment in chemistry labs for hundreds of years. It is versatile, durable, chemical and thermal resistant, and inexpensive, making it very convenient for running reactions on the milligram to gram scale. On a large scale, however, the flask becomes inefficient, and the petrochemical, polymer, and bulk chemical industries often turn to tubes and pipes as their vessel, which allows continuous operation with better control over heating and mixing. In contrast, the pharmaceutical and fine chemical industries frequently use stirred tank reactors to perform reactions. This contrast is perhaps due to the nature of the different industries – pharmaceutical development occurs gradually over a number of years, starting at the milligram or gram scale and increasing stepwise by several orders of magnitude until ton scale production is required. As such, scale up is generally done by increasing the size of the reactor rather than performing a time-consuming and costly ground-up redesign of the process to ensure that it is the most green and efficient at the required scale.On a large scale, the goals of green chemistry and industry align. High safety, low waste generation, and energy efficiency are not suggestions but requirements of a good process. A fundamental goal of green chemistry and engineering is to address these issues earlier in a project's lifetime, ideally all the way down to academic reaction development and medicinal chemistry. Flow chemistry fits into this regime. Flow reactors often offer significant improvements in mixing and heat management, scalability, energy efficiency, waste generation, safety, access to a wider range of reaction conditions, and unique opportunities in heterogeneous catalysis, multi-step synthesis, and more.1–3 Indeed, further development of methods in continuous processing was identified as the most important area of research in green chemistry and engineering for the pharmaceutical industry.4
Since running reactions in flow requires detailed consideration of the chemical reaction as well as reactor design, transport phenomena (mass and heat transfer), and scalability, it is uniquely positioned at the interface of chemistry and engineering. As such, researchers in the field should be aware of both green chemistry5 and green engineering4,6 issues. In this review, we will discuss how flow chemistry and continuous processing can contribute to the development of more efficient, environmentally friendly processes. The first section will focus on recent methods for performing continuous chemistry on a small scale. The second section will focus on how innovations in small scale research can be readily scaled to kg- or even tonne-scale processes. Most of the discussion will be on reactors composed of tubes or micro structured systems with etched channels, as this is where the majority of recent innovations in flow chemistry have been made. Discussions on what kind of chemistries can be run continuously and what equipment is available to do so are thoroughly covered elsewhere.7–10 We will instead focus on when and why one should utilize flow for sustainable synthesis.
How flow chemistry can make processes greener
Increasing reaction efficiency through access to a wider range of reaction conditions
Efficient utilization of energy and time is fundamental to green chemistry and engineering. These factors are directly related to the rate of a chemical reaction, as a fast reaction will require less operating time. Economical use of space is also important, and fast reactions may allow for a smaller reactor to be utilized, particularly in continuous processes. The most straightforward way to increase reaction rate is with an increase in temperature; however, in a batch reactor, this is generally limited to the atmospheric boiling point of the solvent or reagents. In a flow reactor, pressure and temperature can be safely manipulated far beyond atmospheric conditions. Analogous to microwaves synthesis,11 reactions done in flow are often faster than in the corresponding batch reactions, which gives improved energy, time, and space efficiency.12 In contrast to typical microwave reactors, a closed system is not required, greatly facilitating scale-up.Methylation reactions of nitrogen and oxygen nucleophiles such as indoles and phenols are important transformations often carried out on a large scale using hazardous reagents such as methyl iodide or dimethyl sulfate (Scheme 1). Dimethylcarbonate (DMC) has been recognized as a green, albeit less reactive alternative. Due to the relatively low boiling point (90 °C) and reduced reactivity of this reagent, methylation reactions with DMC are generally slow.13 The use of an autoclave or microwave may allow higher temperatures to be used, accessing faster rates; however, this makes scale-up challenging. To access fast reaction rates and improve scalability, Tilstam used a flow reactor to do phenol and N-heterocycle methylations with DMC.14 A simple set-up was used consisting of a high pressure syringe pump, stainless steel tubing, a GC oven, and a back pressure regulator to ensure that the DMC stayed in solution. At 220 °C, yields up to 97% could be obtained with reaction times as short as ten minutes. A report by the Kappe and Holbrey group found similar results using an ionic liquid catalyst.15 While more energy may be required to reach these elevated temperatures, the use of insulation to prevent heat loss, and recycling of the energy given off from exothermic reactions all contribute greatly to energy efficiency on a commercial scale.16 Perhaps most importantly, the reduction in size and operating time of the vessel offers great improvements in sustainability. Indeed, the output of a reactor with respect to its size and operating time was identified as the most impactful component of a good process by researchers at Boehringer Ingelheim.17
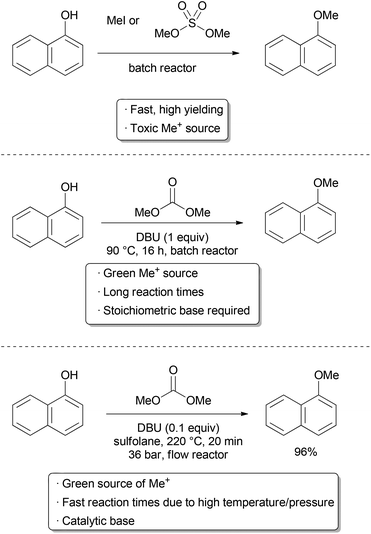 | ||
| Scheme 1 Methylation with DMC. | ||
Rapid, exothermic reactions are challenging to do in batch reactors. Reagents such as organometallics, strong bases, and highly active electrophiles are often added slowly to a reaction mixture under energy-intensive cryogenic conditions to prevent an uncontrollable exotherm. Quenching of these high-energy reagents may again require low temperature. This issue is scale dependent,18 and without proper precautions, both the likelihood and hazard of a runaway reaction increase with the size of a reactor. The high surface area to volume ratio found in flow reactors makes heat transfer more efficient than in batch, allowing rapid removal of thermal energy given off. These features serve to give the chemist or engineer more control over reaction temperature and reduces the risk of thermal runaway. Many instances have been reported of reactions being performed safely at 0 °C or room temperature in flow that would require cryogenic conditions in batch.19–21 This has a further benefit on the overall processing time, as the reaction will occur faster at the elevated temperature and inefficient cooling and warming steps are avoided. A remarkable example demonstrating these principles is the room temperature Swern oxidation reaction by Yoshida and co-workers (Scheme 2).22 The Swern reaction is a reliable procedure for converting alcohols to ketones and aldehydes using DMSO activated by an electrophile (typically COCl2 or TFAA) as the oxidant. In batch, the reaction takes place over three exothermic steps, each of which requires dropwise addition of reagents at cryogenic temperatures.23,24 When converting the process to flow, the Yoshida group found that the Swern oxidation could be done at room temperature with good yields and purity. Moreover, instead of having reaction times on the order of minutes or hours, the whole process was completed in seconds. They attributed the success of their process to the precise temperature control that can be obtained in flow systems, as well as the ability to quickly transfer unstable intermediates to subsequent steps. Using only a series of syringe pumps, stainless steel tubing, and commercial micromixers, they could prepare over 10 grams of material per hour. Being able to perform reactions on species with very short lifetimes is another general advantage of performing reactions in flow.25
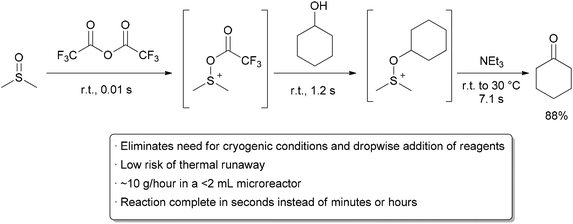 | ||
| Scheme 2 Room temperature Swern oxidation. | ||
The ability to rapidly remove heat generated in an exothermic reaction allows the chemist to run reactions not only at higher temperatures, but also at higher concentrations. This is of great benefit not only to the reduction of waste through solvent usage, but also increased reaction rates and simplified purification. In some instances, reactions may even be run neat, as was demonstrated by Löwe and co-workers in the Michael addition of secondary amines to acrylonitrile and ethyl acrylate (Scheme 3).26 When performed in batch, this highly exothermic reaction was found to require dropwise addition of the amine nucleophile to the Michael acceptor in ethanol over several hours to ensure safe removal of the heat generated. In a flow reactor, the two reactants could be mixed neatly in a micromixer, decreasing the reaction time from several hours to several minutes while maintaining high yields. The combined effects of decreased reaction time and increased concentration mean that, for identical reactor volumes, the flow reactor could produce 235 times more material per hour.
 | ||
| Scheme 3 Solvent-free Michael addition. | ||
It is important to understand both the benefits and limitations of continuous processing. The above cases demonstrate how a wider range of temperatures, pressures, and concentrations (i.e. novel process window)27 are available in flow reactors, greatly enhancing the efficiency of a reaction. However, there are cases where performing reactions quickly may not be ideal. For example, many enantioselective reactions require low temperatures and long reaction times to ensure high asymmetric induction. While there are many continuous examples of asymmetric catalysis,28 it can be argued that flow chemistry is not advantageous for a wide range of such time-consuming homogeneous transformations. Another limitation of flow is the challenge in handling solids. In some cases, the use of suboptimal solvents, high dilution, or special precautions may be required for preventing clogging of the reactor. Batch reactors have comparatively few issues with handling slurries and sparingly soluble reactants and products. As always, the relative merits of using flow or batch conditions should be evaluated on a case-by-case basis.16
Safe, small scale access to supercritical fluids
The ability to safely access high temperatures and pressures in flow reactors has implications not only on the rate of chemical reactions, but also on the types of solvents one can use. Many green solvents such as methanol and acetone have boiling points too low for certain batch applications, whereas performing reactions at high pressure in a flow reactor may allow for their safe use at elevated temperatures. Supercritical fluids are particularly interesting, since these solvents are entirely inaccessible without high pressure conditions. The use of supercritical fluids in a flow system offers numerous advantages over batch reactors. Reactions may be performed on a small scale, improving safety and reducing the amount of material required. Depending on the type of reactor, it may be possible to visualize the reaction to evaluate the phase behaviour. Moreover, the reaction can be analyzed and the temperature and pressure subsequently changed without stopping the reaction and cleaning the vessel, as is necessary in a simple autoclave. Continuous methods for utilizing supercritical fluids for extraction,29 chromatography,30 and as a reaction medium31 have all been commercialized, particularly for supercritical carbon dioxide (scCO2).32 Academic examples using scMeOH, scH2O, and scCO2 for continuous reactions such as hydrogenations, esterifications, oxidations, and Friedel–Crafts reactions have been reported.33A recent example that illustrates many of the green advantages of performing supercritical fluid chemistry in flow is in the ring opening of phthalic anhydride with methanol by Verboom and co-workers (Scheme 4).34 They designed a microreactor with a volume of just 0.32 μL that can withstand very high pressures. The exceptionally small channel causes a large build-up of pressure, and supercritical conditions with pressures of up to 110 bar and temperatures up to 100 °C can occur inside the reactor, giving an ‘on-chip’ phase transition. The channel size increases near the outlet, allowing the fluid to expand to atmospheric conditions. Thus, the total volume of scCO2 under high pressure is exceptionally small, alleviating the major hazards of operating under supercritical conditions. The reaction was thoroughly studied on this small scale, allowing the authors to determine rate constants at several different temperatures and pressures.
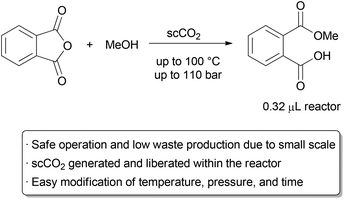 | ||
| Scheme 4 Small scale continuous use of supercritical fluids. | ||
Near- and supercritical water (scH2O) can be an interesting green solvent only obtainable at very high temperature (Tc = 374 °C) and pressure (Pc = 221 bar). It is commonly used for complete oxidation of organic waste materials to CO2; however, it has also been shown to be an effective solvent for selective oxidations.35 Given the harshness of the reaction conditions, it is not surprising that side product formation is common and highly dependent on the reaction time. For fast reactions in a batch reactor, precise control of reaction time is challenging, as the vessel takes time to heat and cool. In contrast, rapid heating, cooling, and quenching can be accomplished in a continuous process, allowing for well defined reaction times. Fine tuning of the temperature, pressure, and time is also easier in a continuous process, as these variables can be changed without stopping and starting the reaction between samples. Thus, more data points can be obtained with less material and fewer heating and cooling cycles. The Poliakoff group used these advantageous to perform a detailed study on the oxidation of p-xylene to terephthalic acid in scH2O, a reaction carried out on industrial scale in acetic acid (Scheme 5).36 By using a flow reactor, reaction times as low as 9 seconds could be used. The equivalents of oxygen could also be finely varied on a small scale through the controlled thermal decomposition of H2O2. Studying this aerobic oxidation with such precision in a batch process would prove highly challenging. Under optimal conditions, excellent selectivity for the desired product could be obtained. Further research by the same group identified improved conditions for this transformation.37
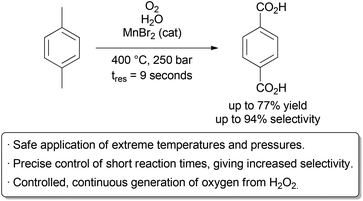
Heterogeneous catalysis and catalyst recycling
Heterogeneous catalysis is widely used in the synthesis of bulk and fine chemicals. In a general, small scale batch reaction, the catalyst, reactants, and solvent are stirred together until completion of the reaction, after which the bulk liquid is separated by filtration. The catalyst can then be collected for either recycling or disposal. In a continuous process, the catalyst can be fixed in space and the reaction mixture allowed to flow over it. The reaction and separation are thus combined in a single step, and the catalyst remains in the reactor for easy recycling. Beyond facilitating separation, the catalyst may have improved lifetime due to decreased exposure to the environment, and reaction rates and turnover numbers can be enhanced through the use of high concentrations of a catalyst with continuous recycling. The benefits of flow are seemingly obvious, yet it has only recently become a widely adopted method for bench-scale synthesis.38The most common application of continuous heterogeneous catalysis is in hydrogenation reactions,39 where the handling and separation of solid precious metal catalysts is not only tedious but hazardous under batch conditions. Moreover, the mixing between the three phases in a hydrogenation is generally quite poor. The use of a flow reactor gives a higher interfacial area between phases and thus more efficient reactions. For example, Ley and co-workers found that the hydrogenation of alkene 1 to 2 was challenging in batch, requiring multiple days at 80 bar of H2 (Scheme 6).40 Using a commercially available H-Cube® reactor, the reaction time was shortened to 4 hours, the pressure reduced to 60 bar, and manual separation and recycling of the catalyst from the reaction was unnecessary. The increased efficiency is due to a combination of improved mixing of the three phases, as well as the continuous recycling and high local concentration of the catalyst. The H-Cube offers a further safety advantage because it generates hydrogen gas on demand from water, obviating the need for a high pressure H2 tank.
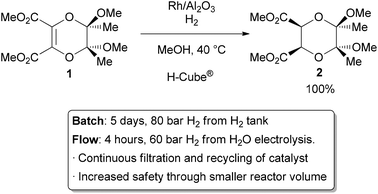 | ||
| Scheme 6 Hydrogenation with an immobilized heterogeneous catalyst. | ||
Homogeneous catalysis has many advantages over heterogeneous catalysis, such as increased activity and selectivity, and mechanisms of action that are more easily understood. Unfortunately, the difficulty associated with separating homogeneous catalysts from the product is a significant hindrance to their large scale application. In an attempt to combine the high activity of homogeneous catalysis with the practical advantageous of heterogeneous catalysis, there has been much research into immobilizing homogeneous catalysts on solid supports.41 This is generally achieved by linking the catalyst to the surface of an insoluble solid such as silica or polymer beads. As was the case in batch hydrogenation reactions, the process of separating and purifying the catalyst is inefficient, potentially dangerous, and may lead to degradation and loss of material. Performing these reactions in a flow system can help overcome these problems.42 A highly efficient example has been demonstrated by van Leeuwen and co-workers, who sought to immobilize a catalyst used in transfer hydrogenation reactions (Scheme 7).43 Their test reaction was the asymmetric reduction of acetophenone; homogeneous reduction with ruthenium and ligand 3 provided 88% conversion and 95% enantioselectivity. The ligand was then covalently linked to silica gel through the benzyl group to form 4. Using this heterogenized system under batch conditions, conversion dropped to 38% on the same time scale, and a slight decrease in enantioselectivity occurred. A reduction in activity of a catalyst upon immobilization is common, so highly efficient recycling is required. Unfortunately, when attempting to re-use the catalyst after filtration, significant degradation and leaching occurred. The catalyst was then packed in a glass column for application in flow chemistry. After a short optimization of flow rate, 95% conversion and 90% ee were obtained. Importantly, the reaction could be run continuously for up to one week without significant degradation in conversion or enantioselectivity. The physical isolation of catalyst species on the solid support is suggested to contribute to the long catalyst lifetime. Interestingly, the basic potassium tert-butoxide additive was only required initially to activate the catalyst, and the reaction could subsequently be run without additional base, allowing the product to be isolated completely free of additives. It is important to note, on top of the decreased activity due to modification, that leaching from cleavage off the solid support and the increased cost of the catalyst due to derivatization are all potential downsides of immobilization of catalysts. In some instances, a seemingly heterogeneous catalyst has been shown to leach active homogeneous species into solution.44 However, as can be seen above, robust systems can be developed which do combine the best features of both homogeneous and heterogeneous catalysis.
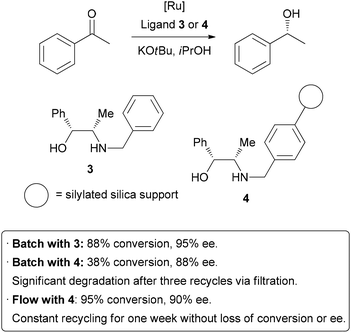 | ||
| Scheme 7 Immobilization of a homogeneous catalyst on a solid support. | ||
Another important method for recycling expensive catalysts is through the use of liquid–liquid biphasic conditions where the catalyst and reactants can be separated by extraction upon completion of the reaction. Such processes have already been utilized on the medium and large scale in a continuous or semi-continuous fashion.45,46 Recycling on a small scale is typically done through batch liquid–liquid extractions, but examples using continuous methods are increasing.47–51 A recent automated small scale recycling of a biphasic catalyst system was demonstrated by the George group in the continuous oxidation of citronellol (Scheme 8).52 A highly fluorinated porphyrin was used as the photocatalyst, and a combination of hydrofluoroether (HFE) and scCO2 was used as the solvent. Under high pressure flow conditions, a single phase was observed. Depressurization occurred after the reactor, resulting in two phases – the organic product in one, and the catalyst and HFE in the other. The denser, catalyst-containing fluorous phase was continuously pumped back through the reactor. With this method, the catalyst was recycled 10 times while maintaining 75% of its catalytic activity, giving an increase in TON of approximately 27-fold compared to previous batch conditions.53 Some leaching of the fluorinated catalyst into the organic product was observed, accounting for the decreased activity over time.
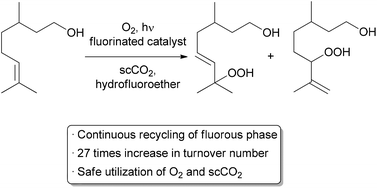 | ||
| Scheme 8 Automated recycling of a biphasic catalyst system. | ||
Telescoping multistep reactions
The synthesis of fine chemicals sometimes requires multiple reactions and tedious work-up between each step is often necessary. Purification may involve the addition of a quenching reagent, multiple aqueous and organic extractions, the addition of a drying agent, filtration, evaporation, and further purification by chromatography, distillation, or recrystallization. These operations all require significant input of energy and materials that ultimately end up as large amounts of waste. Methods and technologies that eliminate or simplify one or many of these steps can make a significant influence on the environmental impact of a multistep chemical synthesis. Continuous processing is particularly suitable for ‘telescoping’ reaction sequences, and many methods have been developed to facilitate this.54One strategy utilizes solid supported reagents packed into columns which allow starting materials to flow in and product to be collected at the outlet without requiring separation of the spent reagent. Different columns may be linked in series, allowing multistep processes to take place. Extra operations may also be necessary, such as solvent changes or the removal of unwanted side products. Methods for automating these processes have also been developed. An example from the Ley group illustrates many of these technologies in the design of a single apparatus to continuously prepare Imatinib (Gleevec) from simple starting materials (Scheme 9).55 Acid chloride 5 and aniline 6 in DCM were flowed through a cartridge containing immobilized DMAP as a nucleophilic catalyst, followed by a basic cartridge to scavenge any remaining 5. The formation of the amide 7 was monitored by an in-line UV spectrometer and subsequently added to a vial containing piperazine 8 in DMF at 50 °C, which facilitated evaporation of the DCM. Once a particular amount of 7 was obtained, as indicated by the UV spectrometer, a connected autosampler would collect this solution and pump it through an immobilized base to induce a substitution reaction, followed by an immobilized isonitrile to scavenge any remaining 8. An immobilized acid was used to ‘catch’ amine 9 through protonation, allowing unreacted 7 to go to waste. ‘Release’ of 9 through deprotonation followed by the addition of aniline 10 and a palladium catalyst facilitated a cross-coupling reaction, furnishing the crude Imatinib, which was then evaporated onto a silica gel column for automated chromatography. Pure product was isolated in 32% overall yield and >95% purity. While not explicitly demonstrated, the possibility of using this apparatus to form analogs by using modified starting materials is proposed. The ability to perform multi-step synthesis of pharmaceuticals without handling of the intermediates is particularly interesting, as exposure to these species can be hazardous.
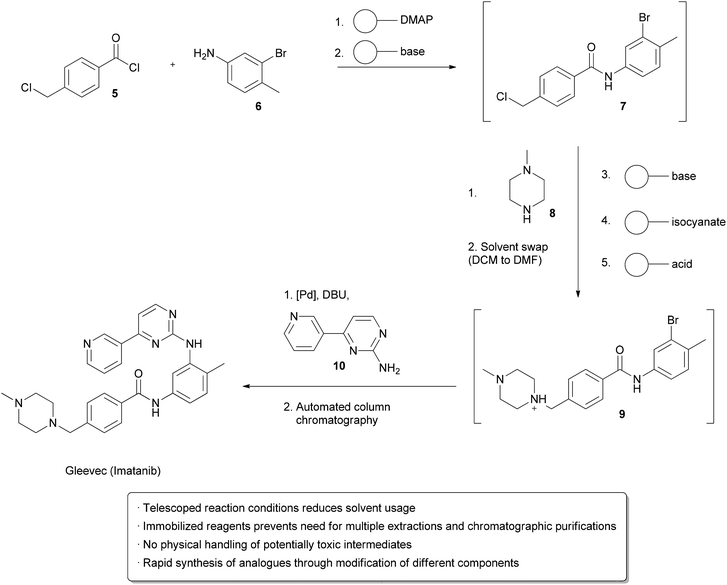
The above example utilizes packed cartridges of scavengers to effect purification. An alternative method is to more closely emulate typical batch purification operations such as distillation and extraction, but on a small, continuous scale. Several different ‘chip’ purification devices have been developed for this purpose.56–65 Some of these technologies were used together in a combined triflation/Heck reaction of phenols (Scheme 10).66 After the initial triflation step in dichloromethane, the product is combined with a stream of aqueous HCl and passed on to a chip containing a membrane that allows the organic phase to pass through while the aqueous stream is passed to waste. The purified triflate then combines with a stream of DMF and the material enters a distillation device heated to 70 °C which allows the volatile dichloromethane to be carried out of the reactor with a stream of nitrogen gas. The product then enters a final reactor where it combines with a stream of alkene and catalyst to form the Heck product. The whole reactor was operated continuously for 5.5 hours, generating approximately 32 mg of product per hour.
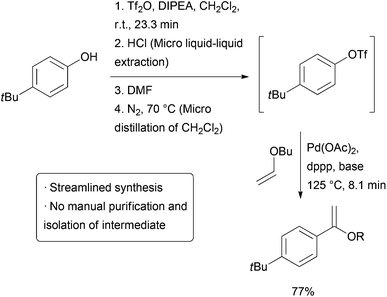 | ||
| Scheme 10 Triflation/Heck coupling facilitated by automated extraction and distillation.64 | ||
Integration of multiple reaction steps, separations, and purifications into one continuous process has great potential for avoiding energy intensive and wasteful intermediate purification. While great progress has been made, the development of a truly general set of reagents, methods, and devices still requires more research. Immobilized reagents can be wasteful to scale up, and there are significant limitations to current microreactor extraction and distillation technologies. Crystallization is another very important technique in pharmaceutical synthesis, and while there are an increasing number of methods for continuous crystallization,67,68 it is yet to be used as an intermediate purification step in an automated multi-step synthesis. Lastly, large scale applications of such complex, streamlined processes are required before a thorough assessment of their environmental impact in comparison with traditional batch routes can be made.
Readily accessible and scalable photochemistry
Light has long been recognized as a valuable tool for performing organic reactions. It acts as a traceless reagent, adding energy to a chemical system without generating waste in the process. Historically, UV light has found most application in synthesis; however recent studies have shown that visible light can affect a wide range of useful transformations as well.69,70 While photochemistry has been used on the industrial scale, it is still uncommon in the pharmaceutical industry. This is likely due to the challenges associated with performing photochemistry on a preparative scale, where the volume of the reactor is too large to allow light to penetrate through. Flow chemistry provides a more efficient method to access photochemical transformations in a range of scales with inexpensive laboratory equipment.71–73 In general, the reaction is pumped through transparent polymer tubing or a transparent chip microreactor which is irradiated with a light source. The small diameter of the channels allows good light penetration, and all molecules are exposed to similar amounts of heat and light. Since no component of the reaction is shielded from the light source, photochemical reactions performed in flow are often found to be orders of magnitude faster than the corresponding batch reactions. In a recent example, Stephenson and co-workers investigated intramolecular radical reactions with alkyl halides which traditionally require toxic reagents such a trialkyltin hydrides (Scheme 11).74 In batch using a ruthenium catalyst, a stoichiometric base, and a 15 W fluorescent lamp, a range of tin-free radical cyclization reactions could be completed on ∼0.1 mmol scale with a 12 hour reaction time. Scaling of the batch process was inefficient, and attempts at processing grams of material failed after 2 days reaction time.75 To overcome this issue, a simple flow reactor was designed with an assembly of blue LEDs (5.88 W) irradiating a coil of PFA tubing which carries the reaction mixture. The small diameter of the tubing allows for optimal absorbance of the light, and reaction times of just 1 minute were obtained, corresponding to an output of 2.88 mmol h−1. In comparison, the unoptimized batch reactor produced approximately 0.012 mmol h−1.72 The ability to obtain high outputs from small reactors makes application of green photochemistry much more efficient and scalable.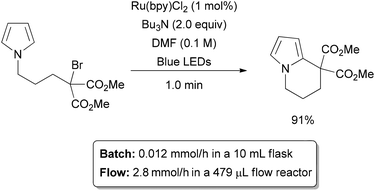 | ||
| Scheme 11 Photochemical alkylation. | ||
More data using less material and time
Reactions for the purpose of discovery, optimization, and kinetic analysis seek only to gain information about a given set of conditions, not to form large amounts of products. To minimize waste generation, it is desirable to perform these transformations on as small a scale as is practical. The μL volume sizes and the ability to easily manipulate reaction conditions such as temperature, pressure, and time make microreactor technology very effective in gathering large amounts of data with small amounts of material. Moreover, investigating the reaction on a small scale allows one to evaluate the intrinsic behaviour without worrying about issues with mixing and heat transfer. These advantages of small scale reaction analysis are further enhanced by technologies available for active monitoring of products. The output of the microreactor has been integrated with a range of analytical techniques,76 including UV,77 IR,78 Raman,79 fluorescence,80 NMR,81 HPLC,82 and MS83 devices.The continuous nature of flow chemistry and the ability to actively monitor the results of a reaction also provide a unique opportunity for automation.84 Several groups have developed methods for ‘self-optimizing’ chemical reactions,85–87 as well as techniques for obtaining useful kinetic data.88,89 In a general approach, a reactor is connected to an instrument capable of in-line monitoring of a desired property (e.g. yield). This feeds information to a computer, which is in turn connected to the syringe pumps and/or temperature controller of the reactor. Stoichiometry, temperature, and time are thus linked via a computer. An algorithm is used to suggest new conditions, a yield is obtained after completion of the reaction, and the process repeats itself until a maximum yield is identified. A recent example was carried out by Moore and Jensen, who explored a number of different algorithms for the optimization of a Paal Knorr reaction (Scheme 12).90 A 232 μL chip reactor was used along with a commercially available IR flow cell which was calibrated to determine the concentration, and thus yield, of the desired product at the outlet of the reactor. Temperatures ranging from 30 °C to 130 °C and reaction times from 2 to 30 minutes were evaluated. Stoichiometry was not investigated in this particular example to avoid trivializing the optimization through using one reagent in large excess. With the most effective algorithm, only 38 experiments were required to find the optimal conditions. The highest conversions were identified at the extreme of the parameter ranges; however, optimal production conditions were at 130 °C and 12.35 minutes, giving a conversion of 81%. Running a large number of reactions in batch is also possible with, for example, a 96-well plate. However, the sequential nature of the automated flow system with inline IR allows the maximum amount of data to be gathered with limited material, as the result of one reaction can be used to guide the next experiment. On the other hand, efficient methods for analyzing discrete variables such as solvent and catalyst have not yet been integrated into automated flow optimizations, leaving significant opportunities for further development.
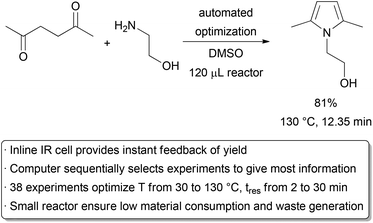 | ||
| Scheme 12 Automated optimization of temperature and time. | ||
Safe, practical use of gases in gas–liquid phase reactions
For the industrial production of bulk chemicals, gaseous reagents at elevated temperatures and pressures are frequently used, as they are often the most widely available, renewable, and atom economical sources of certain elements. Indeed, if one traces back the synthetic origin of many common organic chemicals, they are often derived from simple gases such as oxygen, carbon monoxide, carbon dioxide, methane, hydrogen, ammonia, and ethylene.91 In laboratory-scale synthesis, the use of gaseous reagents is much less common, in favor of higher molecular weight liquid and solid alternatives. This is likely due to the fact that gases are difficult to handle practically and safely, even though they may be the most environmentally friendly reagents for many chemical transformations. Thus, the development of methods and technologies for performing gas–liquid biphasic reactions on a small scale is important for sustainable synthesis. The use of microreactor technology can contribute to this goal. The reduced size of the reactor means increased safety when working under high pressures and temperatures. The flow rate of the gas can be varied, allowing controlled stoichiometry, increased product purity and reduced gas waste. A higher surface area to volume ratio of the reactor decreases the likelihood of hazardous thermal gradients. Finally, the high interfacial area between the gaseous and liquid phases improves mixing and thus the reaction rate. These factors give flow chemistry the ability to emulate industrially relevant conditions for gas–liquid phase synthesis on a scale that is practical for academic research.The palladium-catalyzed aminocarbonylation reaction is a straightforward method of preparing amides from aryl halides. Such reactions are frequently carried out in batches at relatively low pressure, sometimes with only 1 atmosphere of carbon monoxide gas provided by a balloon.92–94 In the interest of exploring conditions that cannot easily be obtained and varied in batches, the Jensen and Buchwald groups investigated aminocarbonylation reactions of aryl halides with temperatures up to 160 °C and pressures up to 14.8 bar (Scheme 13).95 A silicon microreactor was used, with the liquid and gaseous components mixing at the reactor inlet to give two-phase flow. When exploring a range of temperatures and pressures, the expected amide 11 as well as double carbonylated α-ketoamide 12 could be formed with varying selectivity. At a relatively low pressure of CO and high temperature, complete selectivity for the amide product was observed. Decreasing the temperature and increasing the pressure shifted selectivity towards the ketoamide, presumably due to larger amounts of dissolved CO. Over 100 experiments were carried out in the 78 μL microreactor, demonstrating how easy it can be to vary parameters such as temperature and pressure. The use of such a small reactor volume overcomes safety issues from working under pressure encountered in larger batch reactors. Moreover, the temperature, pressure, and time are much easier to manipulate in the flow reactor than an autoclave.
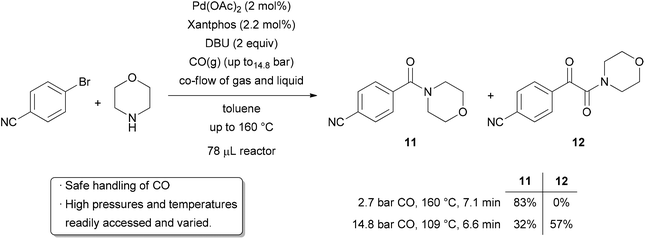 | ||
| Scheme 13 Aminocarbonylation with fine control over pressure, temperature, and time. | ||
In the above example, the liquid and gas phases flowed simultaneously through a single channel in a ‘slug flow’ regime. An alternative method of performing gas–liquid reactions is through the separation of the two phases with a gas-permeable membrane.96 The Ley group has utilized this concept in a ‘tube-in-tube’ design.97 In his reactor, a smaller inner tube is made of a gas-permeable fluoropolymer, Teflon AF. This is housed in a larger outer tube which holds pressure. Either the inner or outer tube can hold the gas, with the other holding the liquid. Thus, the gas can permeate across the inner tube into the liquid phase, but not vice versa. In a recent application of this technology, ammonia gas was used as a nitrogen source for Paal–Knorr synthesis of unprotected pyrroles (Scheme 14).98 Ammonia is perhaps the ideal source of N1, with millions of tons being produced per year. For small scale synthetic applications, however, less atom economical ammonium salts or pre-made commercially available stock solutions are generally used for practical reasons. These reagents are problematic when elevated temperatures are required, as the volatile ammonia will evaporate out of the reaction vessel. To perform the pyrrole synthesis, the reactor was divided into two sections. The first section contained the tube-in-tube setup with starting material in methanol on the outside and 3.5 bar ammonia on the inside, all cooled to 0 °C. This allowed fast diffusion of the ammonia into the organic phase. The solution then entered a heated section of the reactor where the reaction took place under high pressure to prevent outgassing of ammonia. Excellent yields could be obtained in 120 minutes from the time when reagents are mixed to the outlet. Importantly, all components of the reactor set-up are commercially available and are relatively straightforward to set up, making it ideal for small scale applications. The high cost and thermal sensitivity of Teflon AF tubing is a limitation on larger scale.
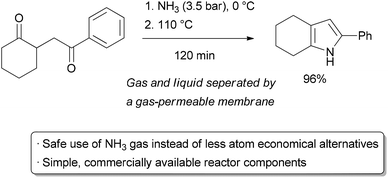 | ||
| Scheme 14 Ammonia gas in a ‘tube-in-tube’ reactor. | ||
Safe use of potentially hazardous reagents
The small size of microreactors means that only μL volumes of reactants and reagents are exposed to one another at a given time. This has significant safety implications, as the hazards of reactor failure are low, even in the worst case scenario. As such, there has been much interest in the advantages of performing useful but hazardous chemical reactions in small flow reactors. For example, reactions with both fluorine gas99 and liquid or solid fluorinating reagents,100,101 high energy organometallics,102,103 azides,104,105 and diazo compounds106,107 have all been studied in flow. A particularly useful way of increasing safety when using hazardous reagents is through their immediate synthesis, use, and quench, all within a microreactor. This avoids the need to transport and stockpile large quantities of such chemicals, instead allowing for the production of exactly the required amount of material on-site, on-demand.Diazomethane is a powerful reagent, capable of inducing useful transformations such as methylation and cyclopropanation reactions with nitrogen gas being the only side product. Unfortunately, it is also highly explosive, volatile, and toxic, and is often avoided in favour of less efficient reagents. Methods for the continuous production and utilization of diazomethane can significantly decrease the safety hazards associated with its use.108 The Stark group has developed a useful, small scale method for the rapid generation and use of diazomethane from Diazald (Scheme 15).109 For their test system, benzoic acid was methylated to methyl benzoate in a microreactor. A mixed solvent system of isopropanol and diethylene glycol ethyl ether was required to avoid the formation of solid precipitate. An excess of acid was used to ensure complete consumption of the diazomethane. Good yields were obtained with very fast reaction times, suggesting that moles of material could be produced in a day from a single microreactor. Most importantly, the total amount of diazomethane present at any given time was negligible.
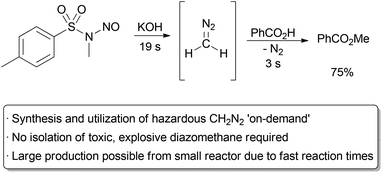 | ||
| Scheme 15 Preparation and immediate use of diazomethane. | ||
Flow chemistry on large scale
Continuous operation
Scaling up of a chemical reaction often requires wasteful reoptimization of reaction conditions due to the change in mixing and heating properties. As such, reactions which work well on the bench scale might need increased reaction times, cryogenic conditions, or may be simply impossible to perform on a process scale. A good reaction should be easily translated between different scales without requiring reoptimization. This is particularly important for green chemistry, since a green process needs to be scalable to have the greatest impact. In a continuous reaction, the most straightforward way to change the total amount of product prepared is by changing the length of time that the reactor is run. For example, increasing the operating time from 10 minutes to 1 week gives a 1000-fold increase in output. In contrast, changing the total output of a batch reaction requires either running the reaction multiple times in a small reactor or increasing the reactor size, which may require changing the conditions. Thus, a continuous reaction allows one to readily choose the amount of material produced without modifying the process or running multiple batches, meeting the green engineering principle to meet needs and minimize excess. This is particularly important in larger scale chemistry, where the cost and complexity of reaction vessels make changing the total output more challenging.Scaling out
To further increase the scale of a flow reactor while still taking advantage of the good mixing and heat transfer properties of microreactors, multiple reactors can be run simultaneously, referred to as numbering up, parallelization, or scaling out. Pumping and heating dozens of chip or tubular microreactors is difficult, so a more practical method may be to design a reactor with multiple channels which only requires a single pump and heating apparatus. The Styring group implemented this idea in a scaled up Kumada coupling (Scheme 16). In the initial single reactor screening, an immobilized nickel species was identified which could catalyze the reaction at room temperature, producing 35.4 mg product per hour.110 A simple, stainless steel column packed with the catalyst was used. To scale the process up, a vessel was designed featuring 120 capillaries of identical shape and volume to the single channel reactor.111 The entire apparatus was small enough to fit on top of a hotplate-stirrer. A positive pressure of nitrogen was used to pump the reactants through the bed of nickel catalyst, and similar reaction times and yields were obtained as was found in the single channel reactor. The reaction was run continuously for 31 hours without a noticeable change in yield and no detectable leaching of the catalyst, giving approximately 5 grams of product per hour.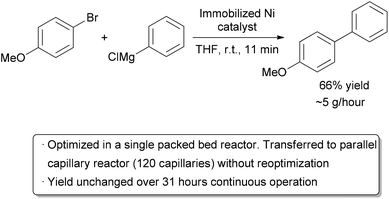 | ||
| Scheme 16 120-fold scale-out by increasing the number of reactor channels. | ||
Scaling up through increasing channel length and diameter
A microreactor has, by definition, volumes in the microliter range. While numbering up is a possibility, there are limitations to how many flow reactors can be utilized simultaneously. The cost of individual reactors, as well as the challenge in pumping liquid evenly throughout the reactors, can quickly become impractical. To scale up to the mL (mesoreactor) range, however, risks losing some of the main advantages of performing reactions in microreactors. Mixing by diffusion may no longer be sufficient, highly exothermic reactions may be challenging to control, and flow may become turbulent. These factors may prevent all molecules from experiencing the same conditions throughout the reaction. Careful engineering may be used to understand and overcome these problems. For example, heat sinks and static or dynamic micromixers can be incorporated into the system, and an understanding of the reaction kinetics and flow patterns within the reactor can also be used to modify and optimize reaction conditions. To demonstrate scale-up in reactor volume of an industrially relevant reaction, the Jensen and Jamison groups studied the opening of styrene oxide by a primary amine to give a β-amino alcohol analogous to that found in pharmaceuticals such as Salbutamol and Indacaterol (Scheme 17).112 Side products resulting from poor regioselectivity or bisalkylation must be avoided, which make the reaction complex. The system must be well understood to ensure that selectivity is not altered due to changes in mass and heat transfer on scale-up. In a 125 μL microreactor, over 100 experiments were performed in an 8 hour period to obtain kinetic data for the formation of both the mono- and bis-alkylation products. The results showed that the best yield and selectivity were obtained at high temperature and high amine to epoxide ratio. Knowledge of the kinetics and key parameters of the reactor allowed 100 fold scale-up into a 12.5 mL tubular stainless steel mesoreactor. In a single test run, 9.2 grams of product (78% yield) were obtained over 30 minutes at a 110 second residence time, demonstrating that scale-up in flow can be safe, predictable, and can avoid wasteful reoptimization. It is worth noting that the reaction was performed efficiently using ethanol as the solvent far above its boiling point, providing another example of how the availability of a greater range of reaction conditions can allow for higher reactivity and the use of green solvents.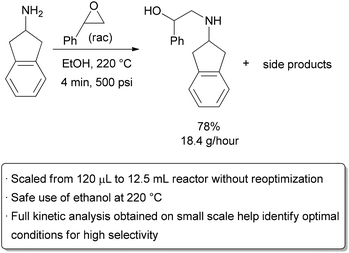 | ||
| Scheme 17 Scale-up by increasing the reactor size. | ||
We have already discussed how reactions that require dropwise addition of reagents at cryogenic temperatures in batch may be run at higher temperatures in microreactors due to the high surface area to volume ratio and good heat transfer. Researchers at Lonza and Corning designed a reactor for the purpose of retaining the benefits of μL volume reactors while allowing high production.113 Their solution was to implement multiple injection ports, emulating the common batch technique of dosing or dropwise addition of one reagent. To test their design, an exceptionally exothermic Grignard addition to an acyl chloride was chosen, with an enthalpy of 260 kJ mol−1.114 When running at a total flow rate of 80 g min−1, a respectable yield of 50% could be obtained for this challenging transformation (Scheme 18). The reactor was operated without interruption for several weeks. The local generation of hotspots was attributed as the primary reason for side-product formation, and further temperature reductions were challenging due to the poor solubility of the Grignard reagent.
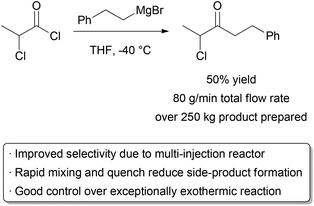 | ||
| Scheme 18 A highly exothermic Grignard reaction. | ||
Scaling of potentially hazardous gas–liquid transformations
As discussed earlier, performing reactions with hazardous gases in a continuous process offer advantages in improved rates due to increased mixing of the gas and liquid phases, as well as higher safety due to the small volumes of pressurized equipment needed. These benefits may lessen upon scaling from a microreactor to a mesoreactor, but with proper precautions, performing gas–liquid reactions in flow can still offer great improvements over batch processes. For example, oxidation reactions using O2 as the stoichiometric oxidant are highly desirable due to the high availability and low environmental impact of molecular oxygen. For these reasons, its use in academic research, particularly with transition metal catalysts, has been increasing.115–120 Oxygen is also frequently used in the bulk chemical industry, such as for the production of ethylene glycol from ethylene.121 Application to fine chemicals and pharmaceuticals, however, is much less common, in part due to safety concerns when mixing large volumes of O2 with flammable solvents. Demonstration of the safe and selective use of O2 on the mesoscale is a challenging but important goal. The Stahl group, in collaboration with researchers at Eli Lilly, sought to scale up a palladium-catalyzed aerobic oxidation of alcohols to ketones, a transformation frequently carried out using wasteful stoichiometric reagents (Scheme 19).122 Under batch conditions, the oxidation of 1-phenylethanol proceeds with a 90% yield using 1 bar of pure oxygen over 18 h on a 1 mmol scale at room temperature. Upon converting to a 5 mL flow reactor, pressure and temperature were increased, and a comparable yield could be obtained with just a 45 minute residence time. Interestingly, catalyst decomposition was problematic when using elevated temperatures in batch, suggesting that the improved gas–liquid mixing in flow is critical. The oxidation was then scaled to a 400 mL reactor at 100 °C with dilution of the O2 in N2, and further scaled to a 7 L reactor with a decrease in the loading of palladium. Dilution of the gas served as a safety feature, but also allowed increased flow rates to be used, enhancing mixing. The use of a long coil of stainless steel tubing with a relatively small inner diameter gave a further increase in linear velocities, and thus mixing, of the reactants. Nonetheless, an increase in residence time was required on each jump in scale. In the 7 L reactor, an output of approximately 52 g h−1 could be obtained. A 99.5% yield was obtained when processing 1 kg of material, demonstrating that O2 can be used safely and selectively on the mesoscale.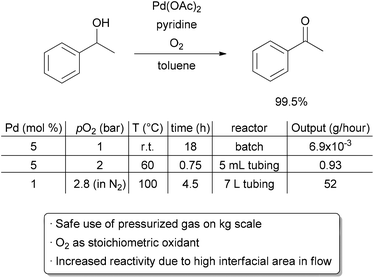 | ||
| Scheme 19 Safe scale-up of an aerobic oxidation. | ||
Multi-kilo scale-up under GMP conditions
Examples of flow processes being used to produce exceptionally large amounts of material are becoming increasingly common as industrial researchers become more knowledgeable about the benefits of continuous reactions. The above examples from academic groups serve to illustrate that reactions optimized in small reactors processing tens to hundreds of mg hour−1 of material can be scaled up to several grams per hour. Projects in process chemistry are often time-sensitive, however, and production of multiple kg of material may be needed in a short amount of time. An example of how the efficient scaling of a flow reaction can save time and reduce waste is provided by a group of researchers at Eli Lilly in their kg synthesis of a key drug intermediate under GMP conditions (Scheme 20). In batch, ketoamide 13 was condensed with NH4OAc and cyclized to form imidazole 14 at 100 °C in butanol on a 1 gram scale. However, side product formation became a significant problem on multiple runs at a 250 g scale. It was proposed that this was due to slow heat up times of the reactor with increasing scale, as lower temperatures seemed to favour increased degradation over productive cyclization. Upon switching to a 4.51 mL flow reactor, another optimization was carried out which identified methanol as a superior solvent that had been neglected in batch screening due to its low boiling point at atmospheric pressure. Scale-up to a 7.14 L reactor proceeded smoothly without the need for reoptimization, and running on this scale with a residence time of 90 minutes for a six-day continuous run provided 29.2 kg of product after recrystallization (approximately 207 g hour−1). The adoption of a flow protocol by a group of industrial researchers in a scale-up with time constraints demonstrates both the effectiveness and maturity of flow chemistry. While the given reaction was used to produce kilograms of material for a deadline, continuous operation without further optimization could produce over 1 metric tonne of product per year in a reactor that fits into a GC oven.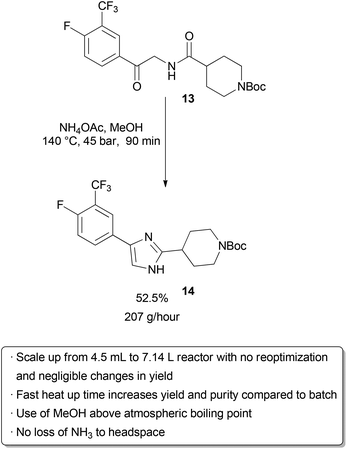 | ||
| Scheme 20 Kilogram-scale synthesis of an imidazole API precursor. | ||
Scaling up and out
For commercial-scale production, a combination of scaling up and scaling out may be the best option. Researchers at DSM and Corning found this to be the ideal solution when faced with the need to selectively mononitrate an undisclosed diol (Scheme 21).123 Nitration reactions are fast, exothermic, and relatively dangerous in large batch reactors, making them well-suited for continuous processing where potential exotherms can be controlled. Examples of aromatic nitration are particularly common.124–129 A two-phase organic/aqueous system was required, which meant that highly efficient mixing was necessary. Flow reactors made out of small tubing are known to provide good mass transfer in two-phase flow, but this becomes less efficient as the tubing size increases. For the desired large scale nitration, a commercially available glass mesoreactor by Corning was used, which contains elements specifically designed to give good mixing of immiscible fluids. The nitration reaction and quench were all performed in the 150 mL reactor, allowing processing of 13 kg of material per hour. After the reaction was optimized and deemed safe, a production scale unit was developed by numbering up, preventing any further reoptimization. Eight reactors were operated simultaneously, allowing approximately 100 kg of total flow per hour. Over 0.5 tonne of the desired nitration product were prepared under GMP conditions, demonstrating the large scale that can be accessed while still benefiting from enhanced mixing, heat transfer, and safety of flow reactor technology.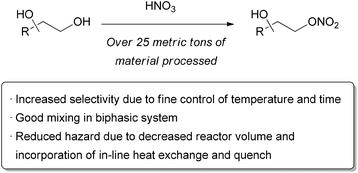 | ||
| Scheme 21 Large scale nitration using glass mesoreactors. | ||
Assessing the environmental impact of batch vs. flow
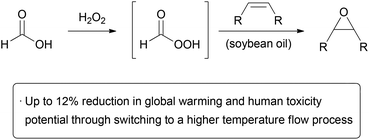
The above processes all demonstrate chemistries where continuous processing has a particular green advantage, such as increased safety, scalability, and efficiency. In many cases, reactions performed in flow simply cannot be emulated in a batch process, making the choice of reactor type obvious. It is also important to consider if there are advantages in changing a working large scale batch process into a continuous one. Such a question is not straightforward to answer, as there are many pros and cons associated with each technology. The continuous nature of flow chemistry means that time and energy can be saved from avoiding frequent loading, unloading, and cleaning of the reactor. A smaller reactor may be used, which requires less energy input for heating and cooling. Integrated mixing elements may be incorporated into the apparatus, obviating the need for large mechanical stirrers. The amount of material produced can be modified by changing the operating time rather than changing the reactor. On the other hand, microreactors are generally more costly to manufacture and have a reduced lifetime relative to stirred tanks. Moreover, while active mixing may not be necessary, a large amount of electricity is required to operate pumps, particularly if a high pressure is required. As such, running a process continuously does not automatically make it more sustainable than in batch, and a thorough analysis is required. The most common metric used to fully evaluate the environmental impact of a process is a life cycle assessment (LCA). Here, two or more processes are compared from a holistic point of view, and the comparative impact on factors such as global warming or resource depletion is evaluated. For example, the Kolbe–Schmitt synthesis of β-resorcylic acid from resorcinol and potassium bicarbonate can give significantly higher yields through the use of high pressure and temperature in a microreactor.130 A simplified life cycle assessment revealed that the total energy demand of the process was decreased despite the harsher conditions used.14 A more drastic impact of microreactor technology was observed in LCA of a large scale lithium–halogen exchange reaction. Energy demand was significantly lower than in batch, primarily due to the ability to operate at room temperature rather than under cryogenic conditions.131In a more recent example, Kralisch and co-workers looked at the environmental impact of performing the epoxidation of soybean oil in batch and flow (Scheme 22).132 Epoxidized soybean oil (ESBO) is produced commercially at a rate of approximately 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes per year, usually using hydrogen peroxide as the oxidant in the presence of a carboxylic acid (e.g. formic acid) and a mineral acid catalyst (e.g. sulfuric acid). This biphasic transformation takes place in two steps: conversion of the carboxylic acid to a peracid in the aqueous phase, followed by epoxidation in the organic phase. Ring opening of the epoxide and decomposition of hydrogen peroxide are undesired side reactions. In the industrial batch route, the oxidant is gradually added to the oil to control the exotherm. Performing such a process in flow may offer advantages in mass transfer between the two phases and improved temperature control; however, the batch process has already been demonstrated to be effective and scalable. To determine if performing the epoxidation in flow was of any environmental benefit, a systematic evaluation of the input and output of material and energy was carried out. The energy demand per mole of product was found to be lowest when performed at high temperatures in a flow reactor (T > 100 °C) due to decreased reaction times; however, this effect levelled off at T > 180 °C due to increased energy demand. In a best-case scenario, the authors note that switching the existing process to a high temperature flow reaction can give approximately 11–12% reduction in global warming and human toxicity potential. Considering that the largest factors in environmental impact of the process are from the starting material supply which cannot be reduced beyond stoichiometric quantities, this is a significant improvement. Economic feasibility was also found to be favorable due to decreased personnel costs when operating on a large scale in the continuous process. As the calculations performed on the flow system are thus far theoretical, the development of a pilot plant is required before one can be sure that the expectations can be fulfilled.
000 tonnes per year, usually using hydrogen peroxide as the oxidant in the presence of a carboxylic acid (e.g. formic acid) and a mineral acid catalyst (e.g. sulfuric acid). This biphasic transformation takes place in two steps: conversion of the carboxylic acid to a peracid in the aqueous phase, followed by epoxidation in the organic phase. Ring opening of the epoxide and decomposition of hydrogen peroxide are undesired side reactions. In the industrial batch route, the oxidant is gradually added to the oil to control the exotherm. Performing such a process in flow may offer advantages in mass transfer between the two phases and improved temperature control; however, the batch process has already been demonstrated to be effective and scalable. To determine if performing the epoxidation in flow was of any environmental benefit, a systematic evaluation of the input and output of material and energy was carried out. The energy demand per mole of product was found to be lowest when performed at high temperatures in a flow reactor (T > 100 °C) due to decreased reaction times; however, this effect levelled off at T > 180 °C due to increased energy demand. In a best-case scenario, the authors note that switching the existing process to a high temperature flow reaction can give approximately 11–12% reduction in global warming and human toxicity potential. Considering that the largest factors in environmental impact of the process are from the starting material supply which cannot be reduced beyond stoichiometric quantities, this is a significant improvement. Economic feasibility was also found to be favorable due to decreased personnel costs when operating on a large scale in the continuous process. As the calculations performed on the flow system are thus far theoretical, the development of a pilot plant is required before one can be sure that the expectations can be fulfilled.
 | ||
| Scheme 22 Life cycle analysis of batch and flow syntheses of ESBO. | ||
Advantages in large scale production from small reactors
In general, the improved mass and heat transfer available in microreactors, as well as the ability to continuously feed reagents and safely use high pressures, temperatures, and concentrations, allows increased rates of chemical production to be possible in small reactors. This has general advantages in safety and energy efficiency, but also provides the opportunity to develop more portable, small scale manufacturing plants. For example, the conversion of syngas to liquid fuels—the Fischer Tropsch process—must be operated at a very high capacity to keep the cost per barrel low. As such, it is desirable to build plants near the source of syngas to avoid long distance transportation. Researchers at Velocys have developed a multi-channel microreactor that can obtain high efficiency at a much lower production capacity than is capable with the traditional routes, reducing capital investment and operating costs (Scheme 23).133,134 Furthermore, the production capacity per unit mass of the reactor is significantly higher. This opens up the potential to transport the reactor to the site of biomass, rather than the converse. This is particularly advantageous for sources of biomass that may not be large enough to justify the development of a fixed facility. Application of this technology to the preparation of liquid fuels from natural gas reserves, particularly those offshore, has also been proposed. | ||
| Scheme 23 Microreactor technology helps overcome scale limitations in converting biomass to liquid fuels. | ||
Conclusions
Flow chemistry and continuous manufacturing have become increasingly recognized as a viable and, in many cases, superior alternative to batch processing. Continuous methods generally offer increased safety, energy efficiency, scalability, and reproducibility. In certain cases, such as heterogeneous catalysis and photochemistry, performing reactions in flow offers further advantages that cannot be emulated in a round-bottom flask or stirred tank. The application of continuous methods on a process scale has recently seen a rapid increase, demonstrating that these advantages are real and impactful. Further developments in academic labs and large scale applications from industry will expand the types of transformations that can be performed more sustainably in flow.Acknowledgements
S. G. N. thanks NSERC for a postdoctoral fellowship.References
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 Search PubMed.
- J.-i. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331–340 CrossRef CAS.
- C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 Search PubMed.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef.
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 Search PubMed.
- C. Wiles and P. Watts, Chem. Commun., 2011, 47, 6512–6535 RSC.
- J.-i. Yoshida, Flash Chemistry. Fast Organic Synthesis in Microsystems, Wiley-Blackwell, 2008 Search PubMed.
- C. Wiles and P. Watts, Micro Reaction Technology in Organic Synthesis, CRC Press, 2011 Search PubMed.
- Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH, Weinheim, 2006 Search PubMed.
- T. Razzaq and C. O. Kappe, Chem.–Asian. J., 2010, 5, 1274–1289 CAS.
- W.-C. Shieh, S. Dell and O. Repič, Org. Lett., 2001, 3, 4279–4281 CrossRef CAS.
- U. Tilstam, Org. Process Res. Dev., 2012, 16, 1974–1978 Search PubMed.
- T. N. Glasnov, J. D. Holbrey, C. O. Kappe, K. R. Seddon and T. Yan, Green Chem., 2012, 14, 3071–3076 RSC.
- S. Heubschmann, D. Kralisch, V. Hessel, U. Krtschil and C. Kompter, Chem. Eng. Technol., 2009, 32, 1757–1765 CrossRef CAS.
- R. Dach, J. J. Song, F. Roschanger, W. Samstag and C. H. Senanayake, Org. Process Res. Dev., 2012, 16, 1697–1706 Search PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS.
- V. Hessel, C. Hofmann, H. Löwe, A. Meudt, S. Scherer, F. Schönfeld and B. Werner, Org. Process Res. Dev., 2004, 8, 511–523 Search PubMed.
- A. Nagaki, Y. Tomida, H. Usutani, H. Kim, N. Takabayashi, T. Nokami, H. Okamoto and J.-i. Yoshida, Chem.–Asian J., 2007, 2, 1513–1523 CrossRef CAS.
- T. Gustafsson, H. Sörensen and F. Pontén, Org. Process Res. Dev., 2012, 16, 925–929 Search PubMed.
- T. Kawaguchi, H. Miyata, K. Ataka, K. Mae and J.-I. Yoshida, Angew. Chem., Int. Ed., 2005, 44, 2413–2416 CrossRef CAS.
- A. K. Sharma and D. Swern, Tetrahedron Lett., 1974, 15, 1503–1506 Search PubMed.
- A. K. Sharma, T. Ku, A. D. Dawson and D. Swern, J. Org. Chem., 1975, 40, 2758–2764 CrossRef.
- J.-i. Yoshida, Chem. Rec., 2010, 10, 332–341 CrossRef CAS.
- H. Löwe, V. Hessel, P. Löbe and S. Hubbard, Org. Process Res. Dev., 2006, 10, 1144–1152 Search PubMed.
- V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS.
- X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.19.
- F. Sahena, I. S. M. Zaidul, S. Jinap, A. A. Karim, K. A. Abbas, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2009, 95, 240–253 Search PubMed.
- D. J. Dixon and K. P. Jhonston, in Encyclopedia of Separation Technology, ed. D. M. Ruthven, John Wiley, 1997, 1544–1569 Search PubMed.
- P. Licence, J. Ke, M. Sokolova, S. K. Ross and M. Poliakoff, Green Chem., 2003, 5, 99–104 RSC.
- X. Han and M. Poliakoff, Chem. Soc. Rev., 2012, 41, 1428–1436 RSC.
- S. Marre, Y. Roig and C. Aymonier, J. Supercrit. Fluids, 2012, 66, 251–264 Search PubMed.
- F. Benito-Lopez, R. M. Tiggelaar, K. Salbut, J. Huskens, R. J. M. Egberink, D. N. Reinhoudt, H. J. G. E. Gardeniers and W. Verboom, Lab Chip, 2007, 7, 1345–1351 RSC.
- R. Holliday, B. Y. M. Jong and J. W. Kolis, J. Supercrit. Fluids, 1998, 12, 255–260 CrossRef CAS.
- P. A. Hamley, T. Ilkenhans, J. M. Webster, E. García-Verdugo, E. Vernardou, M. J. Clarke, R. Auerbach, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2002, 4, 235–238 RSC.
- E. Pérez, J. Fraga-Dubreuil, E. García-Verdugo, P. A. Hamley, M. L. Thomas, C. Yan, W. B. Thomas, D. Housley, W. Partenheimer and M. Poliakoff, Green Chem., 2011, 13, 2397–2407 RSC.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS.
- C. F. Carter, I. R. Baxendale, M. O'Brien, J. P. V. Pavey and S. V. Ley, Org. Biomol. Chem., 2009, 7, 4594–4597 RSC.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC.
- S. Ceylan and A. Kirschning, in Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley & Sons Ltd, 2009, pp. 379–410 Search PubMed.
- A. J. Sandee, D. G. I. Petra, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Chem.–Eur. J., 2001, 7, 1202–1208 CrossRef CAS.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Belénd and P. D. Cerà, ChemCatChem, 2012, 4, 432–445 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- W. A. Herrmann, C. W. Kohlpaintner, H. Bahrmann and W. Konkol, J. Mol. Catal., 1992, 73, 191 CrossRef CAS.
- A. B. Theberge, G. Whyte, M. Frenzel, L. M. Fidalgo, R. C. R. Wootton and W. T. S. Huck, Chem. Commun., 2009, 6225–6227 RSC.
- A. Yoshida, X. Hao and J. Nishikido, Green Chem., 2003, 5, 554–557 RSC.
- E. Perperi, Y. Huang, P. Angeli, G. Manos, C. R. Mathison, D. J. Cole-Hamilton, D. J. Adams and E. G. Hope, Dalton Trans., 2004, 2062–2064 RSC.
- S. Liu, T. Fukuyama, M. Sato and I. Ryu, Org. Process Res. Dev., 2004, 8, 477–481 Search PubMed.
- T. Fukuyama, M. T. Rahman, M. Sato and I. Ryu, Synlett, 2008, 151–163 CAS.
- J. F. B. Hall, X. Han, M. Poliakoff, R. A. Bourne and M. W. George, Chem. Commun., 2012, 48, 3073–3075 RSC.
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322 CrossRef CAS.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452 RSC.
- J. G. Kralj, H. R. Sahoo and K. F. Jensen, Lab Chip, 2007, 7, 256–263 RSC.
- R. L. Hartman, H. R. Sahoo, B. C. Yen and K. F. Jensen, Lab Chip, 2009, 9, 1843–1849 RSC.
- M. O'Brien, P. Koss, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2012, 10, 7031–7036 RSC.
- K. K. R. Tetala, J. W. Swarts, B. Chen, A. E. M. Janssen and T. A. van Beek, Lab Chip, 2009, 9, 2085–2092 RSC.
- D. M. Fries, T. Voitl and P. R. von Rohr, Chem. Eng. Technol., 2008, 31, 1182–1187 CrossRef CAS.
- S. Aljbour, H. Yamada and T. Tagawa, Top. Catal., 2010, 53, 694–699 CrossRef CAS.
- A. Smirnova, K. Shimura, A. Hibara, M. A. Proskurnin and T. Kitamori, Anal. Sci., 2007, 23, 103–107 CrossRef.
- R. C. R. Wootton and A. J. deMello, Chem. Commun., 2004, 266–267 RSC.
- A. Hibara, K. Toshin, T. Tsukahara, K. Mawatari and T. Kitamora, Chem. Lett., 2008, 1064–1065 CrossRef CAS.
- Y. Zhang, S. Kato and T. Anazawa, Lab Chip, 2010, 10, 899–908 RSC.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CAS.
- S. Lawton, G. Steele, P. Shering, L. Zhao, I. Laird and X.-W. Ni, Org. Process Res. Dev., 2009, 13, 1357–1363 Search PubMed.
- H. Zhao, J.-X. Wang, Q.-A. Wang, J.-F. Chen and J. Yun, Ind. Eng. Chem. Res., 2007, 46, 8229–8235 CrossRef CAS.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 Search PubMed.
- M. Oelgemöller and O. Shvydkiv, Molecules, 2011, 16, 7522–7550 Search PubMed.
- M. Oelgemoeller, Chem. Eng. Technol., 2012, 35, 1144–1152 Search PubMed.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef CAS.
- J. Yue, J. C. Schouten and T. A. Nijhuis, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 Search PubMed.
- F. Benito-Lopez, W. Verboom, M. Kakuta, J. G. E. Gardeniers, R. J. M. Egberink, E. R. Oosterbroek, A. van den Berg and D. N. Reinhoudt, Chem. Commun., 2005, 2857–2859 RSC.
- H. Lange, C. F. Carter, M. D. Hopkin, A. Burke, J. G. Goode, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 765–769 RSC.
- S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef CAS.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS.
- L. Ciobanu, D. A. Jayawickrama, X. Zhang, A. G. Webb and J. V. Sweedler, Angew. Chem., Int. Ed., 2003, 42, 4669–4672 CrossRef CAS.
- C. J. Welch, X. Gong, J. Cuff, S. Dolman, J. Nyrop, F. Lin and H. Rogers, Org. Process Res. Dev., 2009, 13, 1022–1025 Search PubMed.
- D. L. Browne, S. Wright, B. J. Deadman, S. Dunnage, I. R. Baxendale, R. M. Turner and S. V. Ley, Rapid Commun. Mass Spectrom., 2012, 26, 1999–2010 Search PubMed.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 Search PubMed.
- J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 7076–7080 CrossRef CAS.
- A. J. Parrott, R. A. Bourne, G. R. Akien, D. J. Irvine and M. Poliakoff, Angew. Chem., Int. Ed., 2011, 50, 3788–3792 CrossRef CAS.
- S. Krishnadasan, R. J. C. Brown, A. J. deMello and J. C. deMello, Lab Chip, 2007, 7, 1434–1441 RSC.
- B. J. Reizman and K. F. Jensen, Org. Process Res. Dev., 2012, 16, 1770–1782 Search PubMed.
- J. P. McMullen and K. F. Jensen, Org. Process Res. Dev., 2011, 15, 398–407 Search PubMed.
- J. J. S. Moore and K. F. Jensen, Org. Process Res. Dev., 2012, 16, 1409–1415 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC . Supporting information.
- J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843–4846 CrossRef CAS.
- T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS.
- A. Shoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS.
- E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS.
- T. Noël and V. Hessel, ChemSusChem, 2013, 6(3), 405–407 Search PubMed.
- A. Polyzos, M. O'Brien, T. P. Petersen, I. R. Baxendale and S. V. Ley, Angew. Chem., Int. Ed., 2011, 50, 1190–1193 CrossRef CAS.
- P. B. Cranwell, M. O'Brien, D. L. Browne, P. Koos, A. Polyzos, M. Peña-López and S. V. Ley, Org. Biomol. Chem., 2012, 10, 5774–5779 RSC.
- C. P. McPake and G. Sandford, Org. Process Res. Dev., 2012, 16, 844–851 Search PubMed.
- M. Baumann, I. R. Baxendale and S. V. Ley, Synlett, 2008, 2111–2114 CAS.
- M. Baumann, I. R. Baxendale, L. J. Martin and S. V. Ley, Tetrahedron, 2009, 65, 6611–6625 CrossRef CAS.
- D. Webb and T. F. Jamison, Org. Lett., 2012, 14, 568–571 Search PubMed.
- A. Nagaki, C. Matsuo, S. Kim, K. Saito, A. Miyazaki and J.-i. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 3245–3248 Search PubMed.
- B. Gutmann, J.-P. Roduit, D. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2010, 49, 7101–7105 CrossRef CAS.
- P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525–3528 CrossRef CAS.
- N. Zaborenko, E. R. Murphy, J. G. Kralj and K. F. Jensen, Ind. Eng. Chem. Res., 2010, 49, 4132–4139 CrossRef CAS.
- R. Fortt, R. C. R. Wootten and A. J. de Mello, Org. Process Res. Dev., 2003, 7, 762–768 Search PubMed.
- L. D. Procter and A. J. Warr, Org. Process Res. Dev., 2002, 6, 884–892 Search PubMed.
- M. Struempel, B. Ondruschka, R. Daute and A. Stark, Green Chem., 2008, 10, 41–43 RSC.
- N. T. S. Phan, D. H. Brown and P. Styring, Green Chem., 2004, 6, 526–532 RSC.
- P. Styring and A. I. R. Parracho, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.29.
- N. Zaborenko, M. W. Bedore, T. F. Jamison and K. F. Jensen, Org. Process Res. Dev., 2011, 15, 131–139 Search PubMed.
- P. Barthe, C. Guermeur, O. Lobet, M. Moreno, P. Woehl, D. M. Roberge, N. Bieler and B. Zimmerman, Chem. Eng. Technol., 2008, 31, 1146–1154 Search PubMed.
- D. M. Roberge, N. Bieler, M. Mathier, M. Eyholzer, B. Zimmermann, P. Barthe, C. Guermeur, O. Lobet, M. Moreno and P. Woehl, Chem. Eng. Technol., 2008, 31, 1155–1161 Search PubMed.
- A. John and K. M. Nicholas, J. Org. Chem., 2011, 76, 4158–4162 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS.
- C. Zhang and N. Jiao, J. Am. Chem. Soc., 2010, 132, 28–29 CrossRef.
- A. Gheorghe, T. Chinnusamy, E. Cuevas-Yañez, P. Hilgers and O. Reiser, Org. Lett., 2008, 10, 4171–4174 CrossRef CAS.
- K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854–3867 RSC.
- S. Rebsdat and D. Mayer, “Ethylene Glycol” in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000 Search PubMed.
- X. Ye, M. D. Johnson, T. Diao and M. H. Yates, Green Chem., 2010, 12, 1180–1186 RSC.
- S. Braune, P. Pochlauer, R. Reintjens, S. Steinhofer, M. Winter, O. Lobet, R. Guidat, P. Woehl and C. Guermeur, Chim. Oggi, 2009, 27, 26 Search PubMed.
- T. Schwalbe, V. Autze and G. Wille, Chimia, 2002, 56, 636–646 CrossRef CAS.
- A. A. Kulkarni, V. S. Kalyani, R. A. Joshi and R. R. Joshi, Org. Process Res. Dev., 2009, 13, 999–1002 Search PubMed.
- J. Shen, Y. Zhao, G. Chen and Q. Yuan, Chin. J. Chem. Eng., 2009, 17, 412–418 Search PubMed.
- J. R. Cage, X. Guo, J. Tao and C. Zheng, Org. Process Res. Dev., 2012, 16, 930–933 Search PubMed.
- G. Panke, T. Schwalbe, W. Stirner, S. Taghavi-Moghadam and G. Wille, Synthesis, 2003, 2827–2830 CAS.
- J. Pelleter and F. Renaud, Org. Process Res. Dev., 2009, 13, 698–705 Search PubMed.
- V. Hessel, C. Hofmann, P. Löb, J. Löhndorf, H. Löwe and A. Ziogas, Org. Process Res. Dev., 2005, 9, 479–489 Search PubMed.
- D. Kralisch and G. Kreisel, Chem. Eng. Sci., 2007, 62, 1094–1100 CrossRef.
- D. Kralisch, I. Streckmann, D. Ott, U. Krtschil, E. Santacesaria, M. Di Serio, V. Russo, L. De Carlo, W. Linhart, E. Christian, B. Cortese, M. H. J. M. de Croon and V. Hessel, ChemSusChem, 2012, 5, 300–311 CrossRef CAS.
- S. R. Deshmukh, A. L. Y. Tonkovich, J. S. McDaniel, L. D. Schrader, C. D. Burton, K. T. Jarosch, A. M. Simpson and D. R. Kilanowski, Biofuels, 2011, 2, 315–324 Search PubMed.
- S. R. Deshmukh, A. L. Y. Tonkovich, K. T. Jarosch, L. Schrader, S. P. Fitzgerald, D. R. Kilanowski, J. J. Lerou and T. J. Mazanec, Ind. Eng. Chem. Res., 2010, 49, 10833–10888 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |