 Open Access Article
Open Access ArticleEfficient three-component coupling catalysed by mesoporous copper–aluminum based nanocomposites†
Jana
Dulle
a,
K.
Thirunavukkarasu
b,
Marjo C.
Mittelmeijer-Hazeleger
c,
Daria V.
Andreeva
*a,
N. Raveendran
Shiju
*c and
Gadi
Rothenberg
*c
aPhysical Chemistry II, University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany. E-mail: daria.andreeva@uni-bayreuth.de; Fax: +49 (0)921 55 2059; Tel: +49 (0)921 55 2750
bNational Centre for Catalysis Research, Indian Institute of Technology-Madras, Chennai-600 036, India
cVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: n.r.shiju@uva.nl; g.rothenberg@uva.nl; Fax: +31 (0)20525 5604; Tel: +31 (0)20525 6515 Web: http://www.hims.uva.nl.hcsc
First published on 31st January 2013
Abstract
Traditional synthesis methods for propargylamines have several drawbacks. A recently developed alternative route is the so-called “A3 coupling” in which an alkyne, an aldehyde, and an amine are coupled together. Typically, these reactions are catalysed by homogeneous gold salts, organogold complexes or silver salts. But these homogeneous catalysts are expensive and their separation is difficult. Here we report the discovery that solid Cu/Al/oxide mesoporous “sponges” are excellent A3 coupling catalysts. These materials are robust, inexpensive, and easy to make. They give good to excellent yields (87–97%) for a wide range of substrates. Being heterogeneous, these catalysts are also easy to handle and separate from the reaction mixture, and can be recycled with no loss of activity.
One of the biggest challenges in organic chemistry is mimicking the complexity of biosynthesis in vitro. Nature excels at matching multiple simple substrates in one-pot reactions that give complex molecules with high yields.1–4 We chemists can also do this, but typically using a host of stoichiometric reagents and protecting groups, and often generating much waste in the process.
The synthesis of propargylic amines is a good example. These amines are key intermediates for making drugs and agrochemicals.5–8 They are traditionally synthesized by nucleophilic attack of lithium acetylides or Grignard reagents on imines or their derivatives.9,10 However, such reagents must be used stoichiometrically, causing large amounts of waste. They are also moisture-sensitive, and require strictly controlled reaction conditions.
An elegant alternative route is the so-called “A3 coupling” (eqn (1)). This catalytic reaction between an alkyne, an aldehyde, and an amine (hence the three A's) gives water as the only by-product.7,11 The reaction is run in a liquid phase, using homogeneous catalysts such as gold salts,12 organogold complexes,13 silver salts,14–17 and Hg2Cl2.18 Among these, the gold salts show the highest activity. The problem is that rapid reduction of cationic gold species to inactive metallic atoms is unavoidable when gold salts activate alkynes/alkenes.19–21 Moreover, using homogeneous catalysts brings inherent complications in catalyst separation and reuse. The ideal solution, therefore, would be replacing the homogeneous gold salt with an effective, yet inexpensive, heterogeneous catalyst.22–35 Many of the heterogeneous catalysts are supported/immobilized gold nanoparticles.36 Zhang et al. reported that supported gold is active for the A3 coupling reaction.21 Also, gold nanoparticles embedded in a mesoporous carbon nitride support was shown to be an efficient heterogeneous catalyst for the synthesis of propargylamines, although its catalytic efficiency has been demonstrated for very few substrates.37 Another active catalyst is gold nanoparticles supported on nanocrystalline MgO.38 The silver salt of 12-tungstophosphoric acid (Ag3PW12O40) was also used to catalyse the A3-coupling.39 Gold and copper thin films were used as catalysts in microwave-assisted continuous-flow organic synthesis (MACOS) of propargylamines40. Liu et al. reported gold functionalized IRMOF-3 catalysts for the one-pot synthesis of propargylamines.41 In another report, gold nanoparticles impregnated on alumina was used as a catalyst for propargylamine synthesis in a flow reactor.34 Considering the costs, it would be better to have an active catalyst containing no precious metals. A recent review by Peshkov et al. summarised the homogeneous and heterogeneous catalysts utilized in A3-coupling.42
We now report the discovery that copper/aluminum (Cu/Al) based mesoporous nanocomposites are excellent A3 coupling catalysts. These materials are robust, inexpensive, and easy to make. They are highly active in the coupling reaction of aldehydes, amines, and alkynes, giving propargylamines selectively with water as the only by-product.
The catalysts are essentially RANEY®-type materials, though our method for making them differs from the RANEY® process.43 We create first a Cu/Al alloy, and then use intense sonication for fragmenting and partially oxidising the particles (see the Experimental section for detailed procedures).44–47
 | (1) |
This simple and waste-free route is based on our recent finding that ultrasound can cause re-structuring of metal alloys due to cavitation induced microphase separation (Fig. 1). During sonication the metal alloy particles undergo the sonomechanical and sonochemical effects stimulated by cavitation. These effects lead to fragmentation, surface reactions, and formation of pores. The metals in the alloy have different chemical (standard electrode potentials) and physical (melting points and hardness of the oxide layer) properties. Thus, the rates of the above processes are different for the alloy components. In the case of Cu/Al, this gives a mesoporous aluminum-based framework, with homogeneously distributed oxidized copper particles. This morphology of the Cu/Al particles has several attractive features for heterogeneous catalysis: increased surface area, mesopores, and unique textural stability.
 | ||
| Fig. 1 Sonochemical formation of mesoporous Cu/Al based particles. Intensive sonication of the initial alloy particles leads to fragmentation, porosity, and surface oxidation. | ||
Benzaldehyde, piperidine, and phenylacetylene were used as model substrates to study the catalytic activity of the Cu/Al catalyst in A3 coupling. Our catalyst gave high benzaldehyde conversion (∼99%), as well as a high yield of the coupling product (see Table 1). Control experiments confirmed that no conversion was found in the absence of catalyst under otherwise identical conditions. Compared with commercial Cu2O (Cu+ is predominant in our catalyst; see below the XPS characterization) the sample was not as active and selective to A3 coupling, under the same conditions. Homocoupling of the alkyne was also observed in this case and selectivity to the A3 coupled product was only 60%. Indeed, homogeneous copper salts were also reported to be active as catalysts, but again these gave only moderate conversions and selectivities.48–51 Moreover, difficulties in recovering the catalyst from the reaction mixture limit their use.
|
|
||||
|---|---|---|---|---|
| Entry | Ra (aldehyde) | R′2NH (amine) | R′′ (alkyne) | Yield (GC/isolated)b |
| a Reaction conditions: aldehyde (1.0 mmol), amine (1.2 mmol), and alkyne (1.3 mmol), Cu/Al (0.12 mmol Cu), toluene (1.7 ml), 100 °C, 22 h; b mol% of A3 coupling product yield based on aldehyde starting material (entries 1–12, complete conversion monitored by GC); c Cu2O catalyst, 75% conv.; d Cu/Al, 90 °C, 22 h (GC conv. 92%); e Cu/Al, 70 °C, 22 h (GC conv. 82%). | ||||
| 1 | Ph | Piperidine | Ph | 99/94 |
| 2 | 4-CH3C6H4 | Piperidine | Ph | 93/87 |
| 3 | Cyclohexyl | Piperidine | Ph | 97/90 |
| 4 | 3-ClC6H4 | Piperidine | Ph | 96 |
| 5 | 3-OHC6H4 | Piperidine | Ph | 92 |
| 6 | 3-NO2C6H4 | Piperidine | Ph | 88 |
| 7 | Ph | Morpholine | Ph | 94/88 |
| 8 | Ph | Pyrrolidine | Ph | 90 |
| 9 | Ph | Piperidine | Hexyl | 92 |
| 10 | Ph | Piperidine | 4-CH3C6H4 | 95 |
| 11 | Ph | Piperidine | 4-CH3OC6H4 | 90 |
| 12 | 4-OCH3C6H4 | Piperidine | Ph | 94 |
| 13 | Ph | Piperidine | Ph | 45c |
| 14 | Ph | Piperidine | Ph | 92d |
| 15 | Ph | Piperidine | Ph | 81e |

To examine the scope of the A3 coupling reaction, we studied different combinations of aldehydes, amines, and alkynes (see Table 1). Aromatic aldehydes as well as cyclohexanecarboxaldehyde (entry 3) gave high yields. The reactions with morpholine and pyrrolidine (entries 7 and 8) also gave >90% yields of the A3 coupled product.
Kantam et al. showed that CuO nanoparticles gave a good yield for A3 coupling, while commercially available bulk Cu2O and bulk CuO gave poor yields.35 The yield was 82% using benzaldehyde, piperidine and phenylacetylene using toluene as the solvent. The yield varied from 8% to 84% when the substrates were changed. Recently, Albaladejo et al. reported that Cu2O on titania is a good catalyst for A3 coupling.52 The catalyst was prepared by supporting copper nanoparticles, made by the addition of CuCl2 to a suspension of lithium and 4,4′-di-tert-butylbiphenyl in THF, on TiO2. They obtained 32% yield (using toluene as the solvent) and 98% yield (using neat substrate) starting from benzaldehyde, piperidine and phenylacetylene. For other substrates, the yield of the A3 coupled products varied, from 52% to 98%. Notwithstanding these results, our Cu/Al catalyst gives complete conversion to the A3 product in toluene, as well as high activity for different substrates as well, with 88%–99% yield. This matches and in some cases outperforms gold catalysts published elsewhere. Thus, Wei et al. reported that AuCl, AuI, AuBr3, and AuCl3 showed good activities for A3 coupling.12 The yield of propargylamines ranged from 53% to 99%, depending on the substrates. Zhang et al. showed that Au/CeO2 catalyses A3 coupling and the yield varies from 25%–99%, again depending on the substrates.21 Another report shows that gold nanoparticles embedded in a mesoporous carbon nitride catalyses the A3 coupling, yielding 51% after 12 h and 62% after 24 h.37
We believe that the reaction mechanism (Scheme 1) involves the formation of copper acetylide, as was proposed for A3 coupling with cationic gold under homogeneous conditions.12,28 Recently, Albaladejo et al. demonstrated that A3 coupling using heterogeneous Cu/TiO2 also involves copper acetylide formation.52 The same mechanism can be invoked in our system. Thus, the C–H bond of the alkyne is activated by a Cu(I) species to give a copper acetylide intermediate (a), which reacts with the immonium ion (b) generated in situ from the aldehyde and secondary amine to give the corresponding propargylamine (c) and regenerate the catalytically active site.
 | ||
| Scheme 1 Tentative mechanism of the A3 coupling. The Cu(I) species activates the C–H bond of the alkyne to give a copper acetylide intermediate (a), which reacts with the immonium ion (b) generated in situ from the aldehyde and secondary amine to give the corresponding propargylamine (c). | ||
Understanding the structure and the morphology of the catalyst surface is the key to understanding its activity. Therefore, we carried out a series of characterisation experiments. Electron microscopy (Fig. 2a and 2c) showed that the initial particles of ∼120 μm are broken into ∼40 μm pieces after 60 min of sonication. The SEM and TEM images show a porous interfacial layer. Combining this with N2 adsorption studies (Fig. 2d) we confirm the formation of a porous outer surface and modification of the inner structure, increasing the specific surface area to 34 m2 g−1. The BET isotherm is type II.53 We see that the monolayer coverage is completed and multilayer N2 adsorption starts at a relative pressure of ca. 0.03. This shows that the material has both micropores and mesopores. The meso and micropore areas were 11.2 m2 g−1 and 22.8 m2 g−1 and the meso and micropore volumes were 0.03 cm3 g−1 and 0.01 cm3 g−1 (pore size distribution is given in ESI†).
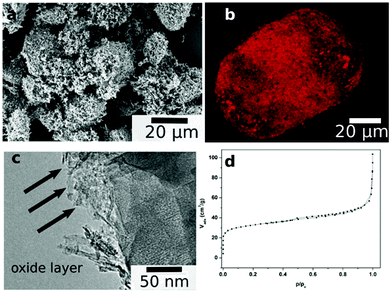 | ||
| Fig. 2 Scanning electron micrograph (a), 3D confocal microscopy reconstruction of the particle loaded with rhodamine B (picture taken in fluorescent mode) showing the porosity throughout the catalyst particle (b), transmission electron micrograph (c), and nitrogen adsorption isotherm (d) of the Cu/Al catalyst, showing the morphology and porous structure. | ||
We also examined the inner structure of the material by monitoring the spatial distribution of a fluorescent dye inside the particles. Fig. 2b shows a 3D reconstruction of confocal scanning fluorescence microscopy (CSFM) images of a catalyst particle loaded with rhodamine B. Here, too, we can see the porous inner structure.
The powder X-ray diffraction (PXRD) pattern of the initial alloy showed intense peaks for Al and CuAl2 (Fig. 3). After sonication, the PXRD reveals the peaks corresponding to highly ordered Al(OH)3 (bayerite) crystallites and CuO, thus, partial oxidation of the alloy. The different Cu/Al mixed phases also can be distinguished in the PXRD pattern of the modified particle, indicating an increased interaction between the two metals.
 | ||
| Fig. 3 PXRD patterns of the initial particles (a) and particles sonicated for 60 min (b). | ||
Using XPS, we observed mixed oxidation states for Cu on the catalyst surface (Fig. 4 and Table 2). The majority (72%) of the copper on the surface was Cu+, with a binding energy (BE) of 932.5 eV. The rest is Cu2+ (BE = 933.7 eV). Indeed, low intensity satellite features (exclusively due to Cu2+) support the minor contribution from the latter. However, because Cu0 and Cu+ states have the same BE, we also recorded the X-ray-excited Auger electron spectroscopy (XAES) for the Cu LMM region. The Cu LMM peak can be resolved into two distinct peaks (see Fig. 4, inset) at 914.2 eV (kinetic energy, KE) and 917.7 eV. These correspond to Cu+ and Cu2+, respectively. The presence of Cu2+ was further confirmed by the satellite peaks (denoted by arrows in Fig. 4). The absence of an ∼918.5 eV KE feature in the spectra shows that there is no Cu0 in the sample.54,55 Moreover, the negative shift in the BE of the Al 2p core level (73.7 eV) compared with the Al 2p of the Al2O3 support alone (∼74 eV) indicates a metal-to-support electron transfer process. The interaction between the Cu and the support and consequent modification of the electronic environment may be responsible for the high selectivity of the catalyst, compared to the bulk Cu2O. Since the XRD did not show peaks corresponding to Cu2O, it is either amorphous or highly dispersed on the Al surface.
 | ||
| Fig. 4 Cu 2p XP spectrum of Cu/Al; the inset shows the Cu LMM XAE spectrum (tabulated data are provided in Table 2). | ||
To test catalyst reusability, we ran five consecutive reaction cycles. After each cycle, the catalyst was filtered, washed with acetone and water and dried at 403 K. The results were similar to the original reaction and no significant deactivation was observed, confirming the reusability of our catalyst (Fig. 5).
 | ||
| Fig. 5 Catalyst recycling studies in the coupling of phenyl acetylene, benzaldehyde and piperidine. Reaction conditions: aldehyde (1.0 mmol), amine (1.2 mmol), and alkyne (1.3 mmol), Cu/Al (0.12 mmol Cu), toluene (1.7 ml), 100 °C, 22 h. After each cycle, the catalyst was filtered, washed with acetone and water and dried at 403 K. | ||
Conclusions
We showed here that the A3 coupling reaction can run in the presence of a solid catalyst that contains only copper, aluminum, and oxygen. This catalyst is stable. It is readily recovered after the reaction and can be recycled several times without deactivation. The process is simple and general, giving propargylamines in good to excellent yields for a variety of substrates. Finally, the fact that our catalyst contains neither noble nor heavy or toxic metals opens true opportunities for practical application.Experimental section
Materials and instrumentation
Aluminium shot (irregular, <15 mm, 99.9%; metal basis) and copper beads (2–8 mm, 99.9995%; trace metals basis) were purchased from Alfa Aesar and Sigma Aldrich, respectively, and used as received. Water was purified using a three-stage Millipore Milli-Q Plus 185 purification system (final resistivity >18.2 MΩ cm).Transmission and scanning electron microscopes (TEM, Zeiss 922 EFTEM operating at 200 kV and Zeiss 1530 FE-SEM respectively) in combination with an ultra-microtome (Ultracut E Reichert Jung, thickness 50 nm) were applied to characterise the optical response, structure, and size of the Cu/Al powder.
Powder X-ray diffraction data were collected on a Stoe STADI P X-ray diffractometer using Cu Kα1 radiation at room temperature. The N2 adsorption–desorption isotherms were measured at 77 K on a vacuum gas sorption apparatus Surfer (Thermo Scientific), after evacuation at 300 °C for 24 h. The surface area was calculated by the BET method. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a multiprobe system (Omicron Nanotechnology, Germany) equipped with a dual Mg/Al X-ray source and a hemispherical analyzer operating in constant analyzer energy (CAE) mode. The spectra were obtained with a pass energy of 50 eV for the survey scan and 20 eV for individual scans. A Mg Kα X-ray source was operated at 300 W and 15 kV. The base pressure in the analyzing chamber was maintained at 1 × 10−10 mbar. The data were processed with the Casa XPS program (Casa Software Ltd, UK), and calibrated with reference to the adventitious carbon peak (284.9 eV) in the sample. Peak areas were determined by integration employing a Shirley-type background. Peaks were considered to be a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mix of Gaussian and Lorentzian functions. The relative sensitivity factors (RSF) provided by the manufacturer were used for quantifying elements.
:30 mix of Gaussian and Lorentzian functions. The relative sensitivity factors (RSF) provided by the manufacturer were used for quantifying elements.
Procedure for catalyst preparation
Aluminium and copper were first alloyed using an AM arc melter (Edmund Bühler GmbH, Germany) with a melt stream of 300 A. The reactor was first evacuated to 10−5 mbar and then pressurised to 500 mbar argon. For homogenization, the sample was overturned three times and fused each time. The bulk material was cut into pieces and ground using a rotary mill (Pulverisette 14, Fritsch GmbH, Germany) with a sieve ring of 0.12 mm. The resulting powder (typically 30 g alloy, 25.0 wt% Cu) was then sieved by a 150 μm sieve.Five grams of this Cu/Al alloy were then dispersed in 50 ml purified water and sonicated for 60 min with an ultrasound tip (VIP1000hd, Hielscher Ultrasonics GmbH, Germany). The device was operated at 20 kHz with a maximum output power of 1000 W by an ultrasonic horn BS2d22 (head area of 3.8 cm2). It was equipped with a booster B2–1.8 for 60 min. The maximum intensity was calculated to be 140 W cm−2 at a mechanical amplitude of 106 μm. To avoid the temperature increase during sonication the experiment was performed in a thermostatic cell. The resulting powder was then dried at 120 °C for 5 h.
General procedure for Cu/Al-catalysed A3 coupling
The amine (1.2 mmol), aldehyde (1.0 mmol), alkyne (1.3 mmol), Cu/Al (0.12 mmol based on Cu) and toluene (1.7 ml) were added to a 50 ml round-bottom flask equipped with a magnetic stirring bar and connected to a water-cooled condenser. The mixture was degassed and backfilled with nitrogen, and then stirred for 22 h at 100 °C (oil bath). After the reaction, the mixture was cooled to ambient temperature and the catalyst was filtered. The residue was washed with an additional 5 ml toluene, which was then combined with the filtrate. The samples were analyzed by gas chromatography (VB-1 column, FID), and/or purified by column chromatography (silica gel; hexane:EtOAc 4:1) and analysed using 1H NMR. Product identity and purity was confirmed by 1H NMR spectroscopy and mass spectrometry.
Example: N-(1,3-Diphenyl-2-propynyl)piperidine (Table 1, entry 1): Benzaldehyde (1.0 mmol, 106.1 mg), piperidine (1.2 mmol, 102.2 mg), phenylacetylene (1.3 mmol, 132.8 mg), Cu/Al catalyst (0.12 mmol based on Cu, 30 mg), and toluene (1.7 ml) were reacted and analysed as above (258 mg, 94% yield). 1H NMR δ = 7.00–7.65 (m, 10H), 4.80 (s, 1H), 2.40–2.60 (m, 4H), 1.56–1.69 (m, 4H), 1.30–1.50 (m, 2H); MS m/z (%) 275 (M+, 20), 274 (10), 191 (100), 198 (79), 192 (24), 189 (24), 115 (14), 165 (10), 232 (8).
Acknowledgements
We thank Dr C.S. Gopinath (National Chemical Laboratory, Pune) for his suggestions on XPS measurements and B. Putz (University of Bayreuth) for the PXRD measurements. J. D. and D. A. thank SFB 840 for financial support.Notes and references
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS.
- M. K. Singh and M. K. Lakshman, J. Org. Chem., 2009, 74, 3079–3084 CrossRef CAS.
- J. W. Lee, T. Y. Kim, Y.-S. Jang, S. Choi and S. Y. Lee, Trends Biotechnol., 2011, 29, 370–378 Search PubMed.
- A. A. Boulton, B. A. Davis, D. A. Durden, L. E. Dyck, A. V. Juorio, X.-M. Li, I. A. Paterson and P. H. Yu, Drug Dev. Res., 1997, 42, 150–156 CrossRef CAS.
- M. Miura, M. Enna, K. Okuro and M. Nomura, J. Org. Chem., 1995, 60, 4999–5004 CrossRef CAS.
- T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599–2660 CrossRef CAS.
- L. Zani and C. Bolm, Chem. Commun., 2006, 4263–4275 RSC.
- M. E. Jung and A. Huang, Org. Lett., 2000, 2, 2659–2661 CrossRef CAS.
- T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968–5969 CrossRef CAS.
- G. Dyker, Angew. Chem., Int. Ed., 1999, 111, 1808–1822 Search PubMed.
- C. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS.
- V. K.-Y. Lo, Y. Liu, M.-K. Wong and C.-M. Che, Org. Lett., 2006, 8, 1529–1532 CrossRef CAS.
- C. Wei, Z. Li and C.-J. Li, Org. Lett., 2003, 5, 4473–4475 CrossRef CAS.
- Z. Li, C. Wei, L. Chen, R. S. Varma and C.-J. Li, Tetrahedron Lett., 2004, 45, 2443–2446 CrossRef CAS.
- K. Mohan Reddy, N. Seshu Babu, I. Suryanarayana, P. S. Sai Prasad and N. Lingaiah, Tetrahedron Lett., 2006, 47, 7563–7566 CrossRef CAS.
- Y. Zhang, A. M. Santos, E. Herdtweck, J. Mink and F. E. Kuhn, New J. Chem., 2005, 29, 366–370 RSC.
- L. Pin-Hua and W. Lei, Chin. J. Chem., 2005, 23, 1076–1080 CrossRef.
- A. Hoffmann-Roder and N. Krause, Org. Biomol. Chem., 2005, 3, 387–391 RSC.
- X. Zhang and A. Corma, Chem. Commun., 2007, 3080–3082 RSC.
- X. Zhang and A. Corma, Angew. Chem., Int. Ed., 2008, 47, 4358–4361 CrossRef CAS.
- M. L. Kantam, B. V. Prakash, C. R. V. Reddy and B. Sreedhar, Synlett, 2005, 2329–2332 CrossRef CAS.
- W. Yan, R. Wang, Z. Xu, J. Xu, L. Lin, Z. Shen and Y. Zhou, J. Mol. Catal. A: Chem., 2006, 255, 81–85 CrossRef CAS.
- B. M. Choudary, C. Sridhar, M. L. Kantam and B. Sreedhar, Tetrahedron Lett., 2004, 45, 7319–7321 CrossRef CAS.
- A. Fodor, A. Kiss, N. Debreczeni, Z. Hell and I. Gresits, Org. Biomol. Chem., 2010, 8, 4575–4581 RSC.
- M. Kidwai, V. Bansal, A. Kumar and S. Mozumdar, Green Chem., 2007, 9, 742–745 RSC.
- A. S.-Y. Lee, G.-A. Chen, Y.-T. Chang and S.-F. Chu, Synlett, 2009, 441–446 Search PubMed.
- V. K. Y. Lo, Y. G. Liu, M. K. Wong and C. M. Che, Org. Lett., 2006, 8, 1529–1532 CrossRef CAS.
- J. V. Madhav, B. S. Kuarm, P. Someshwar, B. Rajitha, Y. T. Reddy and P. A. Crooks, Synth. Commun., 2008, 38, 3215–3223 CrossRef CAS.
- O. P. Pereshivko, V. A. Peshkov and E. V. Van der Eycken, Org. Lett., 2010, 12, 2638–2641 CrossRef CAS.
- C. Wetzel, P. C. Kunz, I. Thiel and B. Spingler, Inorg. Chem., 2011, 50, 7863–7870 CrossRef CAS.
- Z. Xu, X. Yu, X. Feng and M. Bao, J. Org. Chem., 2011, 76, 6901–6905 Search PubMed.
- Q. Zhang, J.-X. Chen, W.-X. Gao, J.-C. Ding and H.-Y. Wu, Appl. Organomet. Chem., 2010, 24, 809–812 CrossRef CAS.
- L. Abahmane, J. M. Kohler and G. A. Groß, Chem.–Eur. J., 2011, 17, 3005–3010 Search PubMed.
- M. L. Kantam, S. Laha, J. Yadav and S. Bhargava, Tetrahedron Lett., 2008, 49, 3083 CrossRef CAS.
- G. Villaverde, A. Corma, M. Iglesias and F. Sanchez, ACS Catal., 2012, 2, 399–406 Search PubMed.
- K. K. R. Datta, B. V. S. Reddy, K. Ariga and A. Vinu, Angew. Chem., Int. Ed., 2010, 49, 5961–5965 CAS.
- K. Layek, R. Chakravarti, M. L. Kantam, H. Maheswaran and A. Vinu, Green Chem., 2011, 13, 2878–2887 RSC.
- K. M. Reddy, N. S. Babu, I. Suryanarayana, P. S. Sai Prasad and N. Lingaiah, Tetrahedron Lett., 2006, 47, 7563–7566 CrossRef CAS.
- G. Shore, W.-J. Yoo, C.-J. Li and M. G. Organ, Chem.–Eur. J., 2010, 16, 126–133 CrossRef CAS.
- L. Liu, X. Zhang, J. Gao and C. Xu, Green Chem., 2012, 14, 1710–1720 RSC.
- V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC.
- (a) J. Schäferhans, S. Gómez-Quero, D. V. Andreeva and G. Rothenberg, Chem.–Eur. J., 2011, 17, 12254–12256 CrossRef; (b) J. Dulle, S. Nemeth, E. V. Skorb, T. Irrgang, J. Senker, R. Kempe, A. Fery and D. V. Andreeva, Adv. Funct. Mater., 2012, 22, 3128–3135 Search PubMed.
- D. V. Andreeva, Int. J. Mater. Res., 2011, 102, 597–598 Search PubMed.
- N. Pazos-Perez, T. Borke, D. V. Andreeva and R. A. Alvarez-Puebla, Nanoscale, 2011, 3, 3265–3268 RSC.
- E. V. Skorb, D. Fix, D. G. Shchukin, H. Möhwald, D. V. Sviridov, R. Mousa, N. Wanderka, J. Schäferhans, N. Pazos-Pérez, A. Fery and D. V. Andreeva, Nanoscale, 2011, 3, 985–993 RSC.
- N. Pazos-Pérez, J. Schäferhans, E. V. Skorb, A. Fery and D. V. Andreeva, Microporous Mesoporous Mater., 2011, 154, 164–169 Search PubMed.
- C.-J. Li and C. Wei, Chem. Commun., 2002, 268 RSC.
- L. T. Shi, Y.-Q. Tu, M. Wang, F.-M. Zhang and C.-A. Fan, Org. Lett., 2004, 6, 1001 CrossRef CAS.
- S. H. H. Zaidi, R. Halder, K. S. Singh, J. Das and J. Iqbal, Tetrahedron Lett., 2002, 43, 6485 CrossRef CAS.
- G. W. Kabalka, L. Wang and R. M. Pagni, Synlett, 2001, 676 CAS.
- M. J. Albaladejo, F. Alonso, Y. Moglie and M. Yus, Eur. J. Org. Chem., 2012, 3093–3104 CrossRef CAS.
- (a) S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723 CrossRef CAS; (b) IUPAC Commission on Colloid and Surface Chemistry Including Catalysis, Pure Appl. Chem, 1985, 57, 603–619 CrossRef CAS.
- S. Velu, K. Suzuki, M. Vijayaraj, S. Barman and C. S. Gopinath, Appl. Catal., B, 2005, 55, 287 CrossRef.
- M. Vijayaraj and C. S. Gopinath, J. Catal., 2006, 241, 83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Pore-size distribution of catalyst and 1H NMR spectral data. See DOI: 10.1039/c3gc36607c |
| This journal is © The Royal Society of Chemistry 2013 |