Safe, efficient and selective synthesis of dinitro herbicides via a multifunctional continuous-flow microreactor: one-step dinitration with nitric acid as agent†
Yizheng
Chen
ab,
Yuchao
Zhao
a,
Mei
Han
a,
Chunbo
Ye
ab,
Minhui
Dang
ab and
Guangwen
Chen
*a
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: gwchen@dicp.ac.cn; Fax: +86-411-84379327; Tel: +86-411-84379031
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 13th November 2012
Abstract
A continuous and selective method for synthesis of dinitro herbicides through one-step dinitration approach has been successfully developed in microreactor systems. Compared to a conventional two-step batch process, reaction can be conducted without the need to separate intermediates and much less solvent or solvent-free condition has been employed.
Dinitroaniline derivatives are an important class of compounds with wide applications in herbicides and pesticides.1 For example, pendimethalin, N-(1-ethylpropyl)-3,4-dimethyl-2,6-dinitroaniline, is a widely-used selective herbicide in agricultural industry.2 Dinitroaniline derivatives are generally produced by nitration of aniline derivatives. These nitration reactions are extremely hazardous due to highly exothermic thermodynamics as well as possible decomposition and explosion of nitro compounds.3 The reaction heat of direct dinitration can be twice as much as that of mononitration. In the chemical industry, to carry out the reaction in a mild way, a two-step process is generally conducted, in which one mononitration step is followed by subsequent mononitration.4 Moreover, large quantities of solvents or diluents are used in each nitration step since nitration reactions are usually fast for reactive species such as phenol and aniline derivatives. Thus, it requires long reaction time and large amounts of waste solvents. Also, selectivity is not satisfying due to easy oxidation or over-nitration of aniline derivatives when reagents and products are exposed to the reaction conditions over a long residence time, and protection of the amino group by acetylation may be required prior to nitration.5 Still, the safety risk is not eliminated owing to mass and heat transfer limitations in conventional batch reactors.
The broad applications of dinitroaniline herbicides provide enormous impetus for extensive study of nitration reactions. The well-established mechanisms for nitration of reactive species, such as aniline and phenol compounds, include both electrophilic and charge-transfer processes.6 Recently, one solution complementary to the conventional batch or semi-batch mode is to employ a continuous and selective nitration mode. The continuous process can offer much small reaction volume and relatively large surface-to-volume ratio; thus process safety is improved and both temperature and residence time can be precisely controlled, which may improve selectivity by suppression of side reactions.7 Microreactors have been demonstrated as an effective tool in continuous processes for mononitration of some aromatic compounds, for instance benzene, toluene and chlorobenzene.8 Beneficial for its characteristic submillimeter dimensions, microreactors have much short diffusion paths and large surface-to-volume ratios compared with batch reactors, which are beneficial to realize excellent heat and mass transfer. So far, most academic and industrial attention has been focused on mononitration processes, and the straightforward one-step dinitration in continuous-flow reactors has been rarely reported.
To envisage that one-step dinitration of aniline derivatives may be carried out in a safe, efficient and selective way, a continuous-flow microreactor has been employed in this study. At the start of our investigation, we proposed the following criteria as the essential requirements of our microreaction system and method: one-step dinitration in a continuous-flow approach, precise reaction temperature control (the variation between reaction temperature and the fluid outlet temperature is less than 2 °C), avoidance of any pre-protection of the amino groups of aniline derivatives, obviation of sulfuric acid (this approach is beneficial to the downstream purification process), residence time <10 s, minimal solvent or solvent-free conditions, minimal HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aniline derivative molar ratio (although nitric acid can be separated by simple phase separation and reused after concentration). Other features include easily assembled and less-expensive peripherals,9 and a multi-functional microreactor integrated with mixing, heat exchange and reaction (see ESI†). The total reaction volume of the microreactor is ca. 0.2 ml. The heat transfer coefficient has been tested in the magnitude of 10 kW m−2 K−1 and mass transfer coefficient in the magnitude of 10 s−1 in the microreactor, which are two orders larger than those in a reported conventional reactor.10
:aniline derivative molar ratio (although nitric acid can be separated by simple phase separation and reused after concentration). Other features include easily assembled and less-expensive peripherals,9 and a multi-functional microreactor integrated with mixing, heat exchange and reaction (see ESI†). The total reaction volume of the microreactor is ca. 0.2 ml. The heat transfer coefficient has been tested in the magnitude of 10 kW m−2 K−1 and mass transfer coefficient in the magnitude of 10 s−1 in the microreactor, which are two orders larger than those in a reported conventional reactor.10
N-Alkylated anilines 1–4 have been prepared prior to use (see ESI†). Dinitration of aniline 1 in a microreactor has been chosen as a model reaction. Nitric acid acts as the nitrating agent and acid catalyst. The desired dinitrated product shown in Scheme 1 is pendimethalin (N-(1-ethylpropyl)-3,4-dimethyl-2,6-dinitroaniline, 1a). Another less-desired product is N-nitroso-pendimethalin (1b) which can be readily and quantitatively converted to pendimethalin 1a (see ESI†). It is a liquid–liquid biphasic reaction in which the reaction takes place in acid phase.
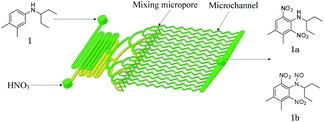 | ||
| Scheme 1 One-step dinitration of N-(1-ethylpropyl)-3,4-xylidine (1) in a continuous-flow microreactor system. | ||
In the industrial batch production of pendimethalin in chlorinated solvent, the aniline solution (30 wt%) is treated with dilute nitric acid as the first step. After separation of the organic phase and spent acid phase, the organic phase is treated with more concentrated nitric acid as the second step, obtaining 89% yield of 1a and 1b with a molar ratio for 1a:1b of 7:3. The total reaction time is 4 h, and 1e is usually produced in a significant amount since 1b can be further converted into 1e11 under long reaction time (Scheme 2).
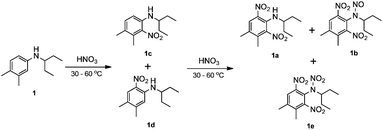 | ||
| Scheme 2 Conventional two-step dinitration of N-(1-ethylpropyl)-3,4-xylidine (1) in the batch system. | ||
The batch method threw some light on the development of our one-step dinitration process. From the beginning of our investigation, 1,2-dichloroethane was selected as solvent and concentration of aniline 1 was fixed at 30 wt% in the organic tank. Two commercial nitric acids, 65 wt% and 98 wt% HNO3, were prepared in the acid tank. Then the organic stream and the acid stream were pumped into the microreactor to carry out the reaction. Benefiting from the short residence time, 1e was not determined in all cases (Table 1), and the fluid outlet temperatures were almost the same as the reaction temperature, the variations of temperature were always within 2 °C. We observed that 85% conversion was obtained with 98% HNO3, while only 24% conversion was achieved with 65% HNO3 (entries 1–3, Table 1). Bearing in mind that increase in substrate concentration in microreactors may result in improved conversion, the concentration of aniline 1 was increased to 80% (entries 4–6, Table 1). We found complete conversion was achieved with a residence time of 9 s and temperature of 60 °C (entry 6, Table 1). However, the molar ratio of 1a:1b in the products distribution was very low, which meant that large amounts of 1b were required to be converted into 1a in the downstream process. When the concentration of nitric acid was changed to 65%, 100% conversion and 98% selectivity were obtained and the molar ratio of 1a:1b was as high as 3.6 (entry 7, Table 1).
| Entry | T (°C) | t (s) | LHSV (103 h−1) | [Aniline 1] (wt%) | [HNO3] (wt%) | HNO3:1 (mol/mol) |
Conv.c (%) | Sel.c (%) |
1a:1bd (wt/wt) |
|---|---|---|---|---|---|---|---|---|---|
| a See ESI. b Residence time. c Determined by HPLC. d In all cases, 1e was not determined. | |||||||||
| 1 | 80 | 6.1 | 0.6 | 30 | 65 | 3.0 | 24 | 72 | 1.1 |
| 2 | 80 | 9.0 | 0.4 | 30 | 98 | 3.0 | 85 | 64 | 0.4 |
| 3 | 80 | 40 | 0.1 | 30 | 98 | 3.0 | 40 | 57 | 0.3 |
| 4 | 40 | 7.2 | 0.5 | 80 | 98 | 3.1 | 61 | 89 | 0.5 |
| 5 | 50 | 7.2 | 0.5 | 80 | 98 | 3.1 | 74 | 90 | 0.4 |
| 6 | 60 | 9.0 | 0.4 | 80 | 98 | 4.3 | 100 | 92 | 0.2 |
| 7 | 70 | 6.1 | 0.6 | 80 | 65 | 3.9 | 100 | 98 | 3.6 |
| 8 | 60 | 0.8 | 4.1 | 80 | 65 | 3.0 | 100 | 97 | 4.2 |
The success with 65% nitric acid and 80% aniline 1 solution prompted us to study the influence of other process parameters, such as LHSV (liquid hourly space velocity), temperature and initial HNO3:aniline 1 molar ratio. Fig. 1 demonstrates that the conversion of aniline 1 first increased with the increase of LHSV (600–1800 h−1), then a stable conversion of 100% could be obtained when LHSV was higher than 1800 h−1 at a temperature of 60 °C. This phenomenon was a direct evidence that the reaction was controlled by mass transfer, for an increase in LHSV could lead to an increase in Reynolds numbers, which finally improved the mass transport on the reaction process.12 This trend became less apparent when temperature was raised to 70 °C and 80 °C, for the conversion of aniline 1 was slightly changed.
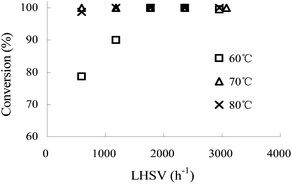 | ||
| Fig. 1 The influence of LHSV on the conversion of aniline 1 at different reaction temperatures. HNO3:aniline 1 = 3.9 (mol/mol). Concentration of HNO3, 65 wt%; concentration of aniline 1, 80 wt%. | ||
Fig. 2 shows the variation of molar ratio of 1a:1b with LHSV and temperature. Surprisingly, the molar ratio of 1a:1b turned out to be 1.4–5 with 65 wt% nitric acid, whereas it was 0–0.5 with 98 wt% nitric acid in the previous study. Thus, 65 wt% nitric acid was advantageous over 98 wt% nitric acid. The results indicated both low temperature and low LHSV were advantageous to obtain a high molar ratio of 1a:1b. However, low temperature or LHSV may lead to an incomplete conversion of aniline 1 as profiled in Fig. 1. Based on the total effect of reaction temperature and LHSV, temperature of 70 °C or 60 °C and LHSV > 1800 h−1 were appropriate. Further effort was concentrated on studying the influence of initial molar ratio of HNO3:aniline 1 to choose an optimal value so as to minimize the quantities of spent acid. Fig. 3 demonstrates that at LHSV of 800–4200 h−1 and temperature of 70 °C, quantitative conversion was achieved at a molar ratio of 3.1 or 3.5, while <90% conversion was obtained with a low molar ratio of 2.7.
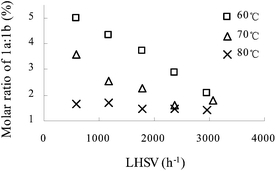 | ||
| Fig. 2 The influence of LHSV on the molar ratio of 1a:1b at different reaction temperatures. HNO3:aniline 1 = 3.9 (mol/mol). Concentration of HNO3, 65 wt%; concentration of aniline 1, 80 wt%. | ||
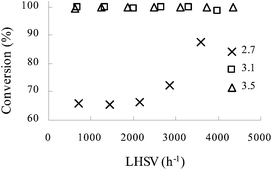 | ||
| Fig. 3 The influence of LHSV on the conversion of aniline 1 at different initial HNO3:aniline 1 molar ratios. T = 70 °C. Concentration of HNO3, 65 wt%; concentration of aniline 1, 80 wt%. | ||
Based on study of the process parameters, the reaction conditions were finally selected as HNO3:aniline 1 molar ratio of 3.0 (HNO3: 6.0 ml min−1, aniline solution: 7.8 ml min−1), temperature of 60 °C and LHSV of 4100 h−1, and 100% conversion and 97% were obtained (entry 8, Table 1). To test the stability of our microreactor system under these conditions, the flow process was run continuously for 1 h, obtaining 0.54 kg of target products of 1a and 1b in 97% yield as expected (isolated yield). This corresponds to an output of 12.96 kg per day or 4.32 t per year (considering 8000 h running time in a year) for the target products. Besides, the reaction proceeded smoothly without evaporation of solvent or clogging of channels. Scale-up can be realized13 by a factor of 100 (plate to stack, see in ESI†), which corresponds to an output of 432 t per year, thus approaching factory-scale. Actually, the 16-channel microreactor in this study can be considered as 16× scale-up compared with a single-channel microreactor.
Two other N-alkyl-substituted derivatives underwent complete conversion and high selectivity even under solvent-free conditions (entries 2 and 3, Table 2). The reaction temperature for anilines 2 and 3 must be increased to 90 °C and 80 °C, respectively; presumably anilines 2 and 3 are less reactive than aniline 1. One sterically hindered aniline 4 was also tested and its initial concentration was limited by its solubility in 1,2-dichloroethane. However, its conversion was slightly reduced (entry 4, Table 2).

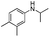

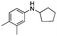
In conclusion, one-step dinitration method has been successfully developed in a continuous-flow microreactor system without the need for separation of intermediates. The risks associated with highly exothermic dinitration are eliminated. Pendimethalin and two other dinitro herbicides were synthesized in high yields when the residence time was less than 2 seconds, and no pre-protection of the amino group is needed. The undesired by-product 1e has been eliminated by virtue of short residence time. When the HNO3 concentration is changed from 98% to 65%, the molar ratio of 1a:1b in the products can switch from 0.2 to 4.2, which greatly benefit downstream processing. It is worth noting that pendimethalin can be produced at 4.32 t per year via this microreactor, and industrial-scale production can be realized by numbering-up principle.
Finally, it should be emphasized that the most important features of this method (high yield, high output, seconds-level residence time, HNO3 as the only agent, no pre-protection of amino group) are collectively possible only because the microreactor has very effective heat and mass transfer rate and the reaction temperature and aniline concentration have been improved to 60–90 °C and over 80%, respectively. A batch reactor performing one-step dinitration under identical conditions would be impossible or very hazardous because of its low heat and mass transfer ability and relatively large reaction volume. Microreactors thus offer great opportunities for one-step dinitration of aniline derivatives in a continuous and selective way. Studies on one-step dinitration of more aniline compounds and other aromatics are under way.
The work reported in this article was financially supported by research grants from the National Natural Science Foundation of China (No. 21225627), Ministry of Science and Technology of China (Nos. 2009CB219903, 2012BAA08B02), and the frame work of the Sino-French project MIGALI via the National Natural Science Foundation of China (No. 20911130358).
Notes and references
- (a) D. Moreland, F. Farmer and G. Hussey, Pestic. Biochem. Physiol., 1972, 2, 342–353 CrossRef CAS; (b) S. Traore and J. J. Aaron, Analyst, 1989, 114, 609–613 RSC; (c) J. W. Benbow, E. L. Bernberg, A. Korda and J. R. Mead, Antimicrob. Agents Chemother., 1998, 42, 339–343 CAS.
- (a) A. Walker and W. Bond, Pestic. Sci., 1977, 8, 359–365 CrossRef CAS; (b) S. Piutti, A. L. Marchand, B. Lagacherie, F. Martin-Laurent and G. Soulas, Pest Manage. Sci., 2002, 58, 303–312 CrossRef CAS.
- (a) C. Bunton, E. Hughes, G. Minkoff and R. Reed, Nature, 1946, 158, 514–515 CrossRef CAS; (b) D. E. O'Reilly and E. M. Peterson, J. Chem. Phys., 1972, 56, 2262 CrossRef CAS; (c) J. Antes, D. Boskovic, H. Krause, S. Loebbecke, N. Lutz, T. Tuercke and W. Schweikert, Chem. Eng. Res. Des., 2003, 81, 760–765 CrossRef CAS; (d) L. Ducry and D. M. Roberge, Angew. Chem., 2005, 117, 8186–8189 CrossRef; L. Ducry and D. M. Roberge, Angew. Chem., Int. Ed., 2005, 44, 7972–7975 Search PubMed; (e) R. Andreozzi, V. Caprio, I. Di Somma and R. Sanchirico, J. Hazard. Mater., 2006, 134, 1–7 CrossRef CAS; (f) J. N. Shen, Y. C. Zhao, G. W. Chen and Q. Yuan, Chin. J. Chem. Eng., 2009, 17, 412–418 CrossRef CAS.
- L. A. Daniel, US4621157, 1986 Search PubMed.
- (a) J. B. Tingle and F. Blanck, J. Am. Chem. Soc., 1908, 30, 1395–1412 CrossRef CAS; (b) E. R. Riegel, H. W. Post and E. E. Reid, J. Am. Chem. Soc., 1929, 51, 505–508 CrossRef CAS; (c) L. C. Raiford and J. N. Wickert, J. Am. Chem. Soc., 1931, 53, 3143–3147 CrossRef CAS; (d) B. Gigante, A. O. Prazeres, M. J. Marcelo-Curto, A. Cornelis and P. Laszlo, J. Org. Chem., 1995, 60, 3445–3447 CrossRef CAS.
- (a) J. Ridd, Chem. Soc. Rev., 1991, 20, 149–165 RSC; (b) R. N. Loeppky, S. P. Singh, S. Elomari, R. Hastings and T. E. Theiss, J. Am. Chem. Soc., 1998, 120, 5193–5202 CrossRef CAS; (c) M. Tanaka, E. Muro, H. Ando, Q. Xu, M. Fujiwara, Y. Souma and Y. Yamaguchi, J. Org. Chem., 2000, 65, 2972–2978 CrossRef CAS; (d) S. R. Gwaltney, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 3273–3283 CrossRef CAS.
- (a) K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Angew. Chem., 2004, 116, 410–451 CrossRef; K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 Search PubMed; (b) D. R. J. Acke and C. V. Stevens, Green Chem., 2007, 9, 386 RSC; (c) T. H. Yoon, S. H. Park, K. I. Min, X. Zhang, S. J. Haswell and D. P. Kim, Lab Chip, 2008, 8, 1454–1459 RSC; (d) F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., 2010, 122, 2530–2537 CrossRef; F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 Search PubMed; (e) R. A. Maurya, C. P. Park, J. H. Lee and D. P. Kim, Angew. Chem., 2011, 123, 6074–6077 CrossRef; R. A. Maurya, C. P. Park, J. H. Lee and D. P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952–5955 Search PubMed; (f) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC; (g) A. Nagaki, K. Imai, H. Kim and J.-i. Yoshida, RSC Adv., 2011, 1, 758–760 RSC; (h) T. Illg, V. Hessel, J. C. Schouten and P. Loeb, Green Chem., 2012, 14, 1420–1433 RSC.
- (a) J. R. Burns and C. Ramshaw, Chem. Eng. Commun., 2002, 189, 1611–1628 CrossRef CAS; (b) R. Halder, A. Lawal and R. Damavarapu, Catal. Today, 2007, 125, 74–80 CrossRef CAS.
- P. B. Palde and T. F. Jamison, Angew. Chem., 2011, 123, 3587–3590 CrossRef CAS; P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525–3528 CrossRef.
- (a) Y. C. Zhao, G. W. Chen and Q. Yuan, AIChE J., 2007, 53, 3042–3053 CrossRef CAS; (b) H. S. Cao, G. W. Chen and Q. Yuan, Ind. Eng. Chem. Res., 2009, 48, 4535–4541 CrossRef CAS; (c) H. S. Cao, G. W. Chen and Q. Yuan, Ind. Eng. Chem. Res., 2010, 49, 6215–6220 CrossRef CAS; (d) Y. H. Su, Y. C. Zhao, G. W. Chen and Q. Yuan, Chem. Eng. Sci., 2010, 65, 3947–3956 CrossRef CAS.
- G. R. Clemo and J. M. Smith, J. Chem. Soc., 1928, 2414–2422 RSC.
- Y. Z. Chen, Y. H. Su, F. J. Jiao and G. W. Chen, RSC Adv., 2012, 2, 5637–5644 RSC.
- (a) R. Schenk, V. Hessel, C. Hofmann, J. Kiss, H. Löwe and A. Ziogas, Chem. Eng. J., 2004, 101, 421–429 CrossRef CAS; (b) V. Hessel, D. Kralisch and U. Krtschil, Energy Environ. Sci., 2008, 1, 467–478 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36652e |
| This journal is © The Royal Society of Chemistry 2013 |