Copper-catalyzed C–C bond cleavage and intramolecular cyclization: an approach toward acridones†
Wang
Zhou
*a,
Youqing
Yang
a,
Yong
Liu
b and
Guo-Jun
Deng
b
aCollege of Chemical Engineering, Xiangtan University, Xiangtan 411105, China. E-mail: wzhou@xtu.edu.cn
bCollege of Chemistry, Xiangtan University, Xiangtan 411105, China
First published on 23rd October 2012
Abstract
A copper-catalyzed approach for the synthesis of acridones via C–C bond cleavage and intramolecular cyclization using air as the oxidant under neutral conditions is described. This transformation offers an alternative method to prepare medicinally important acridones and a new strategy for C–C bond cleavage.
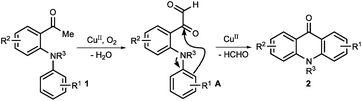
Because of its potential applications in organic synthesis and industry, transition-metal-catalyzed C–C and C–H bond cleavage has recently emerged as an active research topic in organic chemistry.1,2 Although the cleavage of C–C bonds is a challenging task, some elegant methods involved in employing the catalyst of noble metals, such as Rh,3 Ru,4 Pd,5 Pt,6 and others,7 have been developed. However, there are a limited number of strategies using the catalyst of cheap metals such as Cu8 and Fe.9 In the mid-1960s, Brackman and Volger reported the conversion of aliphatic aldehydes to aldehydes of one less carbon atom via a radical process.10 Later, Sayre and co-workers studied the mechanism of oxygenation α to carbonyl groups.11 To date, there are only a few reports employing the copper/O2 catalytic system for C–C bond cleavage.12 Recently, we reported a copper-catalyzed intramolecular direct amination of the sp2 C–H bond for the synthesis of N-aryl acridones.13 As part of our ongoing research, herein, we disclose an efficient copper-catalyzed approach for the synthesis of acridones via C–C bond cleavage and intramolecular cyclization using air as an oxidant under neutral conditions (Scheme 1).
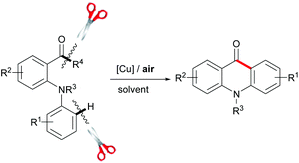 | ||
| Scheme 1 New route to synthesize acridones. | ||
Our initial studies focused on identifying the optimal conditions. 1-(2-(Phenylamino)phenyl)ethanone (1a) could be smoothly converted to the desired acridone 2a in 80% yield with 20 mol% CuI in dimethyl sulfoxide (DMSO) under O2 (1 atm) at 140 °C for 12 h (Table 1, entry 1). Carrying out the reaction under N2 or without CuI led to almost quantitative recovery of 1a, suggesting that the presence of oxygen and CuI are essential (Table 1, entries 2 and 3). Then, we began to screen solvents under O2, finding that polar solvents, such as N-methyl-2-pyrrolidone (NMP), N,N-dimethylacetamide (DMA), and N,N-dimethylformamide (DMF), gave products in lower yields (entries 4–6). Notably, none of 2a but unreacted 1a was detected in the presence of p-xylene as a solvent (entry 7). Gratifyingly, the desired acridone could be obtained in 85% yield under air by prolonging the reaction time (36 h, entry 8). Lower temperature or catalyst loading has a significant effect on the yield (entries 9–11). Other copper catalysts, such as CuBr, CuCl, CuBr2, Cu(OAc)2 and Cu(NO3)2·3H2O, showed sluggish catalytic activity (entries 12–16). Moreover, Pd(OAc)2 was not a suitable catalyst for this transformation (entry 17). On the basis of these observations, we tried to investigate the substrate scope under the reaction conditions as entry 8 listed (Table 1). However, the unsatisfying results spurred us to explore more conditions (ESI, Tables S1 and S2†). Finally, a mixed solvent furnished the best result unexpectedly (entry 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (20 mol%) | Oxidant (1 atm) | Solvent | 1 (h) | Yieldb (%) |
a Reaction conditions: 1a (0.3 mmol), catalyst, and solvent (1.6 mL) were stirred at 140 °C.
b Isolated yield.
c The reaction was carried out under N2.
d The reaction was carried out at 100 °C.
e The reaction was carried out at 120 °C.
f CuI (10 mol%) was used.
g Mixed solvent (vDMSO/vPhCl = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used. :1) was used.
|
|||||
| 1 | CuI | O2 | DMSO | 12 | 80 |
| 2c | CuI | None | DMSO | 12 | Trace |
| 3 | None | O2 | DMSO | 12 | Trace |
| 4 | CuI | O2 | NMP | 12 | 79 |
| 5 | CuI | O2 | DMA | 12 | 58 |
| 6 | CuI | O2 | DMF | 12 | 55 |
| 7 | CuI | O2 | p-Xylene | 12 | 0 |
| 8 | CuI | Air | DMSO | 36 | 85 |
| 9d | CuI | Air | DMSO | 36 | 17 |
| 10e | CuI | Air | DMSO | 36 | 75 |
| 11f | CuI | Air | DMSO | 36 | 17 |
| 12 | CuBr | Air | DMSO | 36 | 58 |
| 13 | CuBr2 | Air | DMSO | 36 | 70 |
| 14 | CuCl | Air | DMSO | 36 | 48 |
| 15 | Cu(OAc)2 | Air | DMSO | 36 | 17 |
| 16 | Cu(NO3)23H2O | Air | DMSO | 36 | 19 |
| 17 | Pd(OAc)2 | Air | DMSO | 36 | 58 |
| 18g | CuI | Air | DMSO/PhCl | 48 | 90 |

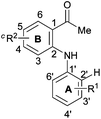
Based on the optimized conditions, the substrate scope was observed. The effect of different substituents on the aromatic rings A and B is listed in Table 2. Substrates bearing electron-donating substituents on the aromatic ring A could be successfully transformed into the desired products in high yields, such as 4′-Me (entry 2), 4′-OMe (entry 4) and 4′-NHAc (entry 5), but on the aromatic ring B in relatively low yields, such as 5-Me (entry 12) and 5-OMe (entry 13). The product with the phenyl substituent could be obtained in high yield (entry 6). It is worth noting that 3′-Me substituted substrate 1j led to a mixture of 3′-Me and 5′-Me products in 84% yield with the ratio of 1:4 (2j/2j′, entry 10). Electron-withdrawing functional groups, such as 4′-COOEt (entry 7) and 4′-F (entry 8), on the aromatic ring A or 5-OCF3 (entry 14) and 5-F (entry 15) on the aromatic ring B could be well-tolerated, giving products in good to excellent yields. Moreover, the reaction scope could be expanded to the substrates with substituents on both rings A and B at the same time (Table 2, entries 17–19).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) | ||||
| a Reaction conditions: 1 (0.30 mmol), CuI (20 mol%) in solvent (vDMSO/vPhCl = 1/1, 1.6 mL) were stirred at 140 °C under air (1 atm) for 48 h. b Isolated yield. c CuI (40 mol%) was used. d Determined by 1H NMR. | |||||||
| R1 | R2 | ||||||
| 1 |
|
H | H | 1a |

|
2a | 90 |
| 2 | 4′-Me | H | 1b | 2b | 91 | ||
| 3c | 6′-Me | H | 1c | 2c | 56 | ||
| 4 | 4′-OMe | H | 1d | 2d | 89 | ||
| 5 | 4′-NHAc | H | 1e | 2e | 86 | ||
| 6 | 4′-Ph | H | 1f | 2f | 85 | ||
| 7 | 4′-COOEt | H | 1g | 2g | 90 | ||
| 8 | 4′-F | H | 1h | 2h | 92 | ||
| 9 | 4′-Cl | H | 1i | 2i | 50 | ||
| 10d | 3′-Me(1j) | H |

|
84 | |||
| 11 | H | 3-Me | 1k | 2c | 38 | ||
| 12 | H | 5-Me | 1l | 2b | 65 | ||
| 13 | H | 5-OMe | 1m | 2d | 65 | ||
| 14 | H | 5-OCF3 | 1n | 2k | 84 | ||
| 15 | H | 5-F | 1o | 2h | 83 | ||
| 16 | H | 5-Cl | 1p | 2i | 84 | ||
| 17 | 4′-F | 5-F | 1q | 2l | 69 | ||
| 18 | 4′-Me | 5-OCF3 | 1r | 2m | 70 | ||
| 19 | 4′-OMe | 5-Cl | 1s | 2n | 53 | ||

In addition, a series of substrates with different substituents on both the nitrogen atom and the ketone were examined (Table 3). The substituting group R3 could be phenyl (Table 3, entry 1). R4 could be alkyl (entries 2–5) or alkenyl (entry 6). Notably, the substrate with tert-butyl only gave the product in 7% yield (entry 3). Moreover, we could detect p-methyl and p-methoxyl benzaldehydes as by-products when 1w and 1x were employed respectively (Table 3, entries 4 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) | |||
| a Reaction conditions: 1 (0.30 mmol), CuI (20 mol%) in solvent (vDMSO/vPhCl = 1/1, 1.6 mL) were stirred at 140 °C under air (1 atm) for 48 h. b Isolated yield. | ||||||
| R3 | R4 | |||||
| 1 |

|
Ph | Me | 1t | 2o | 82 |
| 2 | H | Et | 1u | 2a | 80 | |
| 3 | H | t Bu | 1v | 2a | 7 | |
| 4 | H | 4-Methylbenzyl | 1w | 2a | 60 | |
| 5 | H | 4-Methoxybenzyl | 1x | 2a | 65 | |
| 6 | H | Styryl | 1y | 2a | 19 | |

When 1-(2-(benzylamino)phenyl)ethanone (1z) was employed, we could only observe N-benzyl-indoline-2,3-dione (3z) as the product (Table 4, entry 1). Meanwhile, similar species could be detected by GC-MS analysis when 1a was used (ESI, Fig. S1†). To probe the reaction mechanism, indoline-2,3-dione 3a was synthesized and subjected to control experiments, giving 2a in 79% yield under the optimized conditions (Table 4, entry 3). When CuI or CuBr2 was used as the catalyst under N2, acridone 2a was obtained in 17% and 65% yields respectively (Table 4, entries 4 and 5). Furthermore, other possible intermediates also have been investigated (Table 4, entries 6–10). Although N-arylanthranilic aldehyde 7 could be successfully converted into the desired product in excellent yield (Table 4, entry 9), further studies ruled out the possibility of compound 7 working as a key intermediate in this transformation (see ESI, Table S3† for details). Moreover, radical mechanism studies indicate that a radical pathway may be not involved in this reaction (see ESI, Table S4† for details).
| Entry | Substrate | Productb (%) | Othersb (%) | ||
|---|---|---|---|---|---|
| a Reaction conditions: substrate (0.15 mmol), CuI (20 mol%) in solvent (vDMSO/vPhCl = 1/1, 0.8 mL) stirred at 140 °C under air (1 atm) for 48 h. b Isolated yield. c The reaction was carried out without CuI. d The reaction was carried out under N2. e CuBr2 was used instead of CuI. | |||||
| 1 |

|

|

|
||
| 2c |
|
2a | Trace | 3a | 66 |
| 3 | 2a | 79 | 3a | 0 | |
| 4d | 2a | 17 | 3a | 60 | |
| 5d,e | 2a | 65 | 3a | 35 | |
| 6 |

|
2a | Trace | ||
| 7 | 2a | 17 | |||
| 8 | 2a | Trace | |||
| 9 | 2a | 92 | |||
| 10 |

|
2a | 0 | ||
Intermolecular kinetic isotope effects (kH/kD = 1.23) and intramolecular kinetic isotope effects (kH/kD = 1.69) indicate that aromatic C–H bond cleavage may be not turnover-limiting and does not occur via a chelation-assisted, SEAr or a free radical mechanism (see ESI† for details).14,15 Interestingly, 13C labeling experiments unveil that only about 86% of carbonyl carbon in the product originate from the carbonyl carbon of the substrate (Scheme 2).
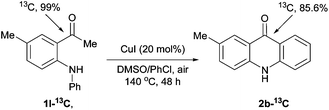 | ||
| Scheme 2 13C labeling experiments. | ||
On the basis of these preliminary results, we still can not speculate on a reasonable mechanistic pathway.16 Multiple pathways may be involved in this transformation. A thorough mechanistic study is needed to unravel the mechanistic intricacies of this process.
In conclusion, we have demonstrated a copper-catalyzed approach for the synthesis of acridones via C–C bond cleavage and intramolecular cyclization using air as the oxidant under neutral conditions. This reaction not only provides an efficient method for constructing medicinally important acridones, but also offers a new strategy for C–C bond cleavage.
Acknowledgements
Financial support from National Science Foundation of China (No. 21102123), Hunan Province Department of Education (No. 11C1208) and Xiangtan University (Nos. KZ08018 and KZ03011) is greatly appreciated.Notes and references
- For reviews on C–C bond cleavage, see: (a) K. C. Bishop III, Chem. Rev., 1976, 76, 461 CrossRef; (b) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS; (c) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (d) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (e) C.-H. Jun and J.-W. Park, Top. Organomet. Chem., 2007, 24, 117 Search PubMed; (f) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS; (g) C. Nájera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 48, 2452 CrossRef CAS.
- For reviews on C–H activation in recent years, see: (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS; (c) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS; (d) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (e) C. Copéret, Chem. Rev., 2010, 110, 656 CrossRef CAS; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (g) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS; (h) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS; (i) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060 CrossRef CAS; (j) D. Balcells, E. Colt and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS; (k) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS; (l) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS; (m) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS; (n) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS; (o) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS.
- For some examples on the rhodium-catalyzed cleavage of C–C bonds, see: (a) J. W. Suggs and C.-H. Jun, J. Am. Chem. Soc., 1984, 106, 3054 CrossRef CAS; (b) S.-Y. Liou, M. E. van der Boom and D. Milstein, Chem. Commun., 1998, 687 RSC; (c) M. Murakami, K. Takahashi, H. Amii and Y. Ito, J. Am. Chem. Soc., 1997, 119, 9307 CrossRef CAS; (d) S. C. Bart and P. J. Chirik, J. Am. Chem. Soc., 2003, 125, 886 CrossRef CAS; (e) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS; (f) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244 CrossRef CAS; (g) Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690 Search PubMed.
- For some examples of the ruthenium-catalyzed cleavage of C–C bonds, see: (a) N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 1999, 121, 8645 CrossRef CAS; (b) T. Kondo, A. Nakamura, T. Okada, N. Suzuki, K. Wada and T. Mitsudo, J. Am. Chem. Soc., 2000, 122, 6319 CrossRef CAS; (c) T. Kondo, K. Kaneko, Y. Taguchi, A. Nakamura, T. Okada, M. Shiotsuki, Y. Ura, K. Wada and T. Mitsudo, J. Am. Chem. Soc., 2002, 124, 6824 CrossRef CAS; (d) T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 6646 CrossRef CAS; (e) D. Nečas, M. Turský and M. Kotora, J. Am. Chem. Soc., 2004, 126, 10222 CrossRef CAS; (f) T. Kondo, Y. Taguchi, Y. Kaneko, M. Niimi and T. Mitsudo, Angew. Chem., Int. Ed., 2004, 43, 5369 CrossRef CAS; (g) J.-J. Lian, A. Odedra, C.-J. Wu and R.-S. Liu, J. Am. Chem. Soc., 2005, 127, 4186 CrossRef CAS.
- For some examples on the palladium-catalyzed cleavage of C–C bonds, see: (a) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2000, 122, 12049 CrossRef CAS; (b) S. Kim, D. Takeuchi and K. Osakada, J. Am. Chem. Soc., 2002, 124, 762 CrossRef CAS; (c) S. Matsumura, Y. Maeda, T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2003, 125, 8862 CrossRef; (d) S.-M. Ma and J.-L. Zhang, Angew. Chem., Int. Ed., 2003, 42, 183 CrossRef CAS; (e) S. Chiba, Y.-J. Xu and Y.-F. Wang, J. Am. Chem. Soc., 2009, 131, 12886 CrossRef CAS; (f) A. J. Grenning and J. A. Tunge, J. Am. Chem. Soc., 2011, 133, 14785 Search PubMed; (g) A. J. Grenning and J. A. Tunge, Angew. Chem., Int. Ed., 2011, 50, 1688 Search PubMed.
- For some examples on the platinum-catalyzed cleavage of C–C bonds, see: (a) C. Müller, C. N. Iverson, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 9718 CrossRef CAS; (b) A. Gunay and W. D. Jones, J. Am. Chem. Soc., 2007, 129, 8729 CrossRef CAS.
- (a) N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921 CrossRef CAS; (b) C. Zhang, C. Xu, T. Shen, G. Wu, L. Zhang and N. Jiao, Org. Lett., 2012, 14, 2362 Search PubMed.
- For copper-catalyzed cleavage of C–C bonds, see: (a) T. Sugiishi, A. Kimura and H. Nakamura, J. Am. Chem. Soc., 2010, 132, 5332 CrossRef CAS; (b) C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273 CrossRef CAS; (c) M. Sai, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2011, 50, 3294 CrossRef CAS; (d) F. Chen, C. Qin, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11487 CrossRef CAS.
- For iron-catalyzed cleavage of C–C bonds, see: (a) H. Li, W. Li, W. Liu, Z. He and Z. Li, Angew. Chem., Int. Ed., 2011, 50, 2975 CrossRef CAS; (b) C. Qin, W. Zhou, F. Chen, Y. Ou and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 12595 CrossRef CAS; (c) C. Qin, T. Shen, C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 6971 Search PubMed.
- (a) H. C. Volger, W. Brackman and J. W. F. M. Lemmers, Recl. Trav. Chim. Pays-Bas, 1965, 84, 1203 Search PubMed; (b) W. Brackman, C. J. Gaasbeek and P. J. Smit, Recl. Trav. Chim. Pays-Bas, 1966, 85, 437 Search PubMed; (c) W. Brackman and H. C. Volger, Recl. Trav. Chim. Pays-Bas, 1966, 85, 446 Search PubMed.
- (a) L. M. Sayre and S.-J. Jin, J. Org. Chem., 1984, 49, 3498 CrossRef CAS; (b) S.-J. Jin, P. K. Arora and L. M. Sayre, J. Org. Chem., 1990, 55, 3011 CrossRef CAS.
- (a) J. Cossy, D. Belotti, V. Bellosta and D. Brocca, Tetrahedron Lett., 1994, 35, 6089 CrossRef CAS; (b) K. M. Steward and J. S. Johnson, Org. Lett., 2011, 13, 2426 Search PubMed.
- W. Zhou, Y. Liu, Y. Yang and G.-J. Deng, Chem. Commun., 2012, 48, 10678 RSC.
- (a) W. D. Jones and F. J. Feher, J. Am. Chem. Soc., 1986, 108, 4814 CrossRef CAS; (b) W. D. Jones and F. J. Feher, Acc. Chem. Res., 1989, 22, 91 CrossRef CAS; (c) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS; (d) A. Pinto, L. Neuville, P. Retailleau and J. Zhu, Org. Lett., 2006, 8, 4927 CrossRef CAS; (e) B.-X. Tang, R.-J. Song, C.-Y. Wu, Y. Liu, M.-B. Zhou, W.-T. Wei, G.-B. Deng, D.-L. Yin and J.-H. Li, J. Am. Chem. Soc., 2010, 132, 8900 CrossRef CAS.
- (a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS; (b) X. Chen, G. Dobereiner, X.-S. Hao, R. Giri, N. Maugel and J.-Q. Yu, Tetrahedron, 2009, 65, 3085 CrossRef CAS.
- At the beginning, we speculated that oxygenation α to the carbonyl group of 1-(2-(arylamino)phenyl)ethanone 1 gave α-keto aldehyde A, which undergoes a copper-catalyzed intramolecular Friedel–Crafts type reaction to give product 2. However, we disfavor this pathway because (1) the electron-deficient aromatic ring gave good or better results, clearly contradictory to this Friedel–Crafts type pathway. If the reaction does work in this manner, ester 6 should give product 2a under the optimized conditions more likely (Table 4, entry 8); (2) it contradicts with the result of the 13C labeling experiment (Scheme 2).
.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36502b |
| This journal is © The Royal Society of Chemistry 2013 |