Low temperature pretreatment of sugarcane bagasse at atmospheric pressure using mixtures of ethylene carbonate and ethylene glycol†
Zhanying
Zhang
*ab,
Ian M.
O'Hara
ab,
Darryn W.
Rackemann
b and
William O. S.
Doherty
b
aSyngenta Centre for Sugarcane Biofuels Development, Queensland University of Technology, Brisbane, Australia. E-mail: jan.zhang@qut.edu.au; Fax: +61 7 3138 4132; Tel: +61 7 3138 7792
bCentre for Tropical Crops and Biocommodities, Queensland University of Technology, Brisbane, Australia
First published on 13th November 2012
Abstract
A low temperature lignocellulose pretreatment process was developed using acid-catalysed mixtures of alkylene carbonate and alkylene glycol. Pretreatment of sugarcane bagasse with mixtures of ethylene carbonate (EC) and ethylene glycol (EG) was more effective than that with mixtures of propylene carbonate (PC) and propylene glycol (PG). These mixtures were more effective than the individual components in making bagasse cellulose more amenable to cellulase digestion. Glucan digestibilities of ≥87% could be achieved with a wide range of EC to EG ratios from 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1 (w/w). Pretreatment of bagasse by the EC–EG mixture with a ratio of 4:1 in the presence of 1.2% H2SO4 at 90 °C for 30 min led to the highest glucan enzymatic digestibility of 93%. The high glucan digestibilities obtained under these acidic conditions were due to (a) the ability of alkylene carbonate to cause significant biomass size reduction, (b) the ability of alkylene glycol to cause biomass defibrillation, (c) the ability of alkylene carbonate and alkylene glycol to remove xylan and lignin, and (d) the magnified above attributes in the mixtures of alkylene carbonate and alkylene glycol.
:1 to 1:1 (w/w). Pretreatment of bagasse by the EC–EG mixture with a ratio of 4:1 in the presence of 1.2% H2SO4 at 90 °C for 30 min led to the highest glucan enzymatic digestibility of 93%. The high glucan digestibilities obtained under these acidic conditions were due to (a) the ability of alkylene carbonate to cause significant biomass size reduction, (b) the ability of alkylene glycol to cause biomass defibrillation, (c) the ability of alkylene carbonate and alkylene glycol to remove xylan and lignin, and (d) the magnified above attributes in the mixtures of alkylene carbonate and alkylene glycol.
1. Introduction
Lignocellulose is the most abundant renewable biomass on the earth with annual production estimated to be 1 × 1010 million tonnes.1 Due to the recalcitrant structure of lignocellulosic biomass, pretreatment of lignocellulose is a prerequisite to make cellulose more accessible to cellulases for the release of fermentable sugars for subsequent production of chemicals (ethanol, lactic acid, succinic acid, 1,3-propanediol, etc.) in biological processes.2–5 The pretreatment cost has been described as the second most expensive unit cost in the cellulosic ethanol production process following the feedstock cost.6A large number of lignocellulose pretreatment approaches have been investigated on a wide variety of feedstock types and several recent review articles provide a general overview of the field.3,6–9 Pretreatment technologies can be categorized as chemical, physical, and biological, and have been used either singly or in combination. Chemical-based pretreatments are conducted at elevated temperatures using solvents (water, ionic liquids, organic solvents, etc.) with or without catalysts (acids, alkalis, etc.). The most widely studied chemical-based pretreatments are acid- and alkali-catalysed pretreatment using water as the solvent. However, pretreatment using water as the solvent typically requires temperatures of ≥150 °C and pressure-resistant reactors, which inevitably increase equipment and production costs.6 In the last decade, pretreatment/dissolution of lignocellulosic biomass by ionic liquid solvents has also attracted increasing interest worldwide as some ionic liquids are thermally stable, non-flammable, have low vapour pressures and have a tendency to remain liquid over a wide range of temperatures.3,10 Complete or nearly complete digestion of cellulose and hemicellulose can be achieved after pretreatment/dissolution of lignocellulose without producing significant amounts of sugar degradation products.11–14 However, ionic liquids are not bulk commodity chemicals and the costs relating to ionic liquids are currently prohibitive towards the development of commercial processes for the pretreatment of lignocellulosics.
Another type of chemical-based pretreatment uses organic solvents. Many organic solvents such as ethanol, formic acid, acetic acid, glycerol and ethylene glycol are relatively cheap compared to ionic liquids. However, pretreatment by low-boiling-point solvents (such as methanol and ethanol) is restricted to the laboratory or pilot scale due to challenges involving high pressure operation and highly volatile and flammable solvents.15 Hence, the use of high-boiling-point organic solvents for biomass pretreatment is an attractive proposition. Glycerol, ethylene glycol (EG) and propylene glycol (or 1,2-propanediol, PG) are the most studied high boiling point solvents.16–25 Researchers have found that pretreatment with acid-catalysed glycerol or EG solution was more effective than that with polyol alone or alkali-catalysed polyol solution for the pretreatment of lignocellulose.26–28 The disadvantage of these pretreatment systems is that they require relatively high pretreatment temperatures (158–225 °C) in order to achieve cellulose enzymatic digestibility of >90%.26–30
The initial work by Yamada and Ono established that lignocellulosic biomass can be liquefied in alkylene carbonates, e.g. ethylene carbonate (EC) and propylene carbonate (PC) in the presence of an acid catalyst.31 Satisfactory liquefaction of lignocellulosic materials was achieved at only 150 °C by blending EG with EC to improve lignin dissolution.31 It was found that the rate of EC-liquefaction of cellulose was 27.9 times faster than that of EG-liquefaction.31 Further studies on biomass liquefaction using EC or a mixture of EC and EG in the presence of acid-catalysts have been reported since this initial study.32–35
As lignocellulosic biomass can be liquefied at a temperature of 150 °C with mixtures of alkylene carbonate and alkylene glycol, it is likely that biomass can be destructed for enzymatic digestion of cellulose at lower temperatures with the same mixtures. Pretreatment of lignocellulosic biomass at a temperature of <100 °C can reduce the process energy consumption significantly. In this study, pretreatment of sugarcane bagasse, an abundant agricultural crop residue, at a low temperature range (60–90 °C) and atmospheric pressure using mixtures of alkylene carbonate and alkylene glycol was studied. Pretreatment by mixtures of alkylene carbonate and alkylene glycol was more effective than by alkylene carbonate or alkylene glycol alone. Higher glucan digestibility was achieved with bagasse pretreated by mixtures of EC and EG than by the mixtures of PC and PG. A high glucan digestibility of ≥93% was achieved with the pretreatment of sugarcane bagasse at only 90 °C for 30 min using a mixture of EC and EG (4:1, w/w) in the presence of 1.2% H2SO4. A wide ratio range of EC to EG could be used without significantly decreasing the glucan digestibility of bagasse. The effects of other operational conditions such as pretreatment temperature, pretreatment time, acid concentration and water content on pretreatment effectiveness were also investigated.
2. Experimental
2.1. Materials
Sugarcane bagasse was collected from the Racecourse sugar mill (Mackay Sugar Limited) in Mackay, Australia. Sugarcane bagasse was washed (hot water at 90 °C) to remove residual sugar to a negligible amount. The washed sugarcane bagasse was air-dried, depithed and ground to a powder by a cutter grinder (Retsch® SM100, Retsch GmbH, Germany). The milled bagasse was screened and bagasse having particle sizes of 250–500 μm was collected and stored for experiment. The moisture of the bagasse powder was 7.1%. The bagasse powder mainly consisted of 43.8% glucan, 20.2% xylan, 3.3% arabinan, 27.5% lignin, 2.5% acetyl and 2.1% ash. EC, EG, PC and PG were purchased from Sigma-Aldrich Company (Australia). Accellerase™ 1000 (Batch no. 1600877126), a Danisco product (Genencor Division, Danisco Inc., USA), was purchased through Enzymes Solutions Pty Ltd (Australia). The filter paper activity of Accellerase™ 1000 was approximately 40 FPU mL−1. All the chemicals used in this study were analytical standard reagents.2.2. Pretreatment experiment
A required amount of solvent solution was transferred into a 100 mL glass flask. A magnetic stirrer was placed in the flask. The flask was immersed in a silicone oil bath which was preheated to the required temperature. The flask was not sealed and the pretreatment was conducted at atmospheric pressure. The heating element was equipped with a magnetic stirring device (Ika Labortechnik, Germany) and the stirring speed was set at 500 rpm. The solvent in the flask was preheated for 5 min to reach the required temperature. A required amount of 72% (w/w) H2SO4 was added into the flask and mixed for another 0.5 min. After that, 4.31 g bagasse (4.0 g dry biomass) was transferred into the flask and the pretreatment was started. The ratio of liquid to solid was 10:1 (w/w). After a prescribed pretreatment time, 40 g of distilled water was added into the flask. The solution was mixed well and filtered (Whatman 541 filter paper) to collect the pretreated bagasse. The filtrate was collected and stored frozen for further analysis. The pretreated bagasse was washed with 800 mL distilled water (2 × 400 mL per wash). The washed pretreated bagasse was further washed with a 50 mM NaOH solution (4 × 40 mL per wash) followed by a further water wash (2 × 400 mL per wash). The washed pretreated bagasse was collected. A portion of the filtered pretreated bagasse was freeze-dried and stored for compositional analysis while the remaining filtered pretreated bagasse was stored at 4 °C for enzymatic hydrolysis. Compositional analysis of bagasse and pretreated bagasse samples was conducted according to a standard procedure developed by National Renewable Energy Laboratory (NREL, USA).36
2.3. Enzymatic hydrolysis
Enzymatic hydrolysis was carried out in a 20 mL glass vial containing a 5 g solution. A glucan loading of 2% was used based on the glucan content in the bagasse sample. The reaction solution contained 0.05 M citrate buffer to maintain pH 4.8 and 0.02% sodium azide to prevent the growth of microorganisms. The dosage of Accellerase for enzymatic hydrolysis was 0.5 mL Accellerase per g cellulose (approximately 20 FPU g−1 glucan) unless otherwise stated. The reaction was carried out at 50 °C in a rotary incubator (Ratek OM 11 Orbital Mixer, Australia) with a shaking speed of 150 rpm. The reaction solution was sampled at 0 h, 6 h, 12 h, 24 h, 48 h and 72 h. The sampling volume was 0.2 mL using a cut-off pipette tip. After sampling, the sample was sealed and incubated for 5 min in a boiling water bath to denature the cellulase. The sample was then centrifuged at 9000g for 5 min. 0.1 mL supernatant was diluted 10 times by de-ionized water. The diluted sample was filtered through a 0.22 μm disk filter prior to sugar analysis by a high performance liquid chromatography (HPLC) system. All the enzymatic hydrolysis experiments were conducted in duplicate with the mean data detailed in this study.2.4. HPLC analysis
A HPLC system with a Bio-Rad Aminex HPX-87H column and a Waters refractive index detector was used to detect and quantify sugar derivatives such as 5-hydroxymethylfurfural (HMF) and furfural in the pretreatment hydrolysate. The mobile phase was 5 mM H2SO4 at a flow rate of 0.6 mL min−1. The column temperature was 65 °C. A Phenominex RPM monosaccharide column was used to determine the sugars generated from compositional analysis, pretreatment hydrolysate and enzymatic hydrolysis. The pretreatment hydrolysate was neutralised with CaCO3 prior to sugar analysis. The column temperature was 85 °C and the mobile phase was water at a flow rate of 0.5 mL min−1.2.5. Characterisation of untreated bagasse and pretreated bagasse
Untreated bagasse and pretreated bagasse samples were characterised by X-ray powder diffraction (XRD), Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM).XRD analysis was used to estimate the crystallinity index (CrI) of the bagasse samples. The X-ray diffractometer (PANalytical, The Netherlands) with Cu Kα radiation (λ = 1.5418 nm) was operated at a voltage of 40 kV and a current of 40 mA. The 2θ range was from 4° to 30° in steps of 0.02° at a rate of 2.6° min−1. CrI was calculated by:
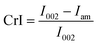 | (1) |
FTIR spectra of the samples were recorded between 4000 cm−1 and 500 cm−1 using a Thermo Nicolet Nexus 870 system (Thermo Nicolet, USA) with the processing software Omnic 7.3. SEM was used to record the surface morphological features of bagasse before and after pretreatment. The samples were coated with gold using a Leica EMS CD 005 system prior to analysis by an FEI scanning electron microscope (Quanta 200 3D, USA).
2.6. Calculations
Glucan (xylan, lignin) recovery was calculated based on the following equation: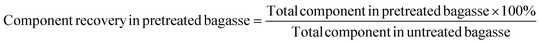 | (1a) |
Glucan digestibility was calculated based on the following equation:
 | (2) |
Total glucose yield after enzymatic hydrolysis was calculated based on the following equation:
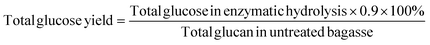 | (3) |
The yields of glucose (HMF, xylose and furfural) detected in pretreatment hydrolysate on total glucan (xylan) in untreated bagasse were calculated based on the following equations:
 | (4) |
 | (5) |
 | (6) |
 | (7) |
3. Results and discussion
3.1. Pretreatment by the mixtures of EC–EG and PC–PG at different ratios
Pretreatment of sugarcane bagasse was first conducted with mixtures of EC–EG and PC–PG at a ratio range from 1:0 to 0:1 at 90 °C for 30 min in the presence of 1.2% H2SO4.
| Ratio of EC to EG | Content in pretreated bagasse (%) | Biomass yield (%) | Recovery in pretreated bagasse (%) | 72 h glucan digestibility (%) | Total glucose yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Glucan | Xylan | Lignin | Glucan | Xylan | Lignin | ||||
| 1:0 |
67.7 | 7.4 | 16.0 | 60.0 | 92.7 | 22.0 | 34.9 | 13.4 | 12.4 |
| 9:1 |
78.2 | 8.4 | 6.7 | 53.5 | 95.5 | 22.2 | 13.0 | 91.2 | 87.1 |
| 4:1 |
77.0 | 8.9 | 6.1 | 54.9 | 96.5 | 24.2 | 12.2 | 93.4 | 90.1 |
| 2:1 |
75.5 | 11.6 | 7.3 | 56.2 | 96.8 | 32.3 | 14.9 | 91.1 | 88.2 |
| 1:1 |
73.0 | 12.2 | 8.0 | 58.2 | 97.1 | 35.2 | 16.9 | 86.7 | 84.2 |
| 0:1 |
67.7 | 13.0 | 14.6 | 62.3 | 96.3 | 40.1 | 33.1 | 65.0 | 62.6 |
| Untreated bagasse | 43.8 | 20.2 | 27.5 | 100.0 | 100.0 | 100.0 | 100.0 | 6.0 | 6.0 |
| Ratio of PC to PG | Content in pretreated bagasse (%) | Biomass yield (%) | Recovery in pretreated bagasse (%) | 72 h glucan digestibility (%) | Total glucose yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Glucan | Xylan | Lignin | Glucan | Xylan | Lignin | ||||
| 1:0 |
62.6 | 11.3 | 20.1 | 66.0 | 94.3 | 37.0 | 48.3 | 7.4 | 7.0 |
| 9:1 |
76.1 | 9.6 | 9.9 | 55.0 | 95.6 | 26.3 | 19.9 | 51.0 | 48.7 |
| 4:1 |
76.3 | 11.3 | 9.2 | 55.7 | 97.1 | 31.1 | 18.6 | 77.7 | 75.4 |
| 2:1 |
75.9 | 11.9 | 8.5 | 55.5 | 96.3 | 32.8 | 17.2 | 80.3 | 77.3 |
| 1:1 |
72.5 | 12.7 | 10.3 | 58.7 | 97.1 | 36.9 | 21.9 | 77.9 | 75.6 |
| 0:1 |
65.4 | 14.3 | 15.7 | 64.4 | 96.2 | 45.7 | 36.7 | 60.0 | 57.7 |
| Untreated bagasse | 43.8 | 20.2 | 27.5 | 100.0 | 100.0 | 100.0 | 100.0 | 6.0 | 6.0 |
When EG and PG were added into EC and PC respectively, delignification improved significantly (Tables 1 and 2). The recovery of glucan improved slightly compared to pretreatment by EC or PC alone indicating that EG and PG may protect cellulose from degradation. With increasing the EG or PG content in solution, the amount of xylan recovered in the pretreated bagasse increased.
Although pretreatment by EC and PC removed 65% and 52% of the total lignin component from sugarcane bagasse (100% – recovery), it had a very limited effect on the improvement of glucan digestibility (13% for EC pretreatment and 7% for PC pretreatment compared to 6% for untreated bagasse). Pretreatment with EG or PG alone did improve glucan digestibility (65% for EG and 60% for PG) significantly compared to untreated bagasse and bagasse pretreated by EC or PC alone but there was still a large portion of the glucan component unable to be digested. When only 10% of the corresponding glycol was added to the carbonate, the glucan digestibility of pretreated bagasse markedly improved. Pretreatment with EC–EG ratios from 9:1 to 2:1 resulted in glucan digestibilities of ≥90%. The glucan digestibility dropped to 86.7% with an EC–EG ratio of 1:1. For pretreatment with mixtures of PC–PG, the maximum glucan digestibilities were achieved in the PC–PG ratio range of 4:1 to 1:1. The glucan digestibilities of bagasse pretreated with the EC–EG system were generally higher than those of the corresponding PC–PG system, indicating that the former system is more effective in the pretreatment of bagasse. Differences in pretreatment effectiveness between EC–EG and PC–PG systems may be related to differences in acidity potential. It is also reported that the PC–PG system is prone to degradation and generation of volatile and toxic compounds such as dioxolanes during prolonged heating.31 Thus, the EC–EG system is the preferred system for deconstructing bagasse.
SEM images show the morphological changes of bagasse samples before and after pretreatment by EC–EG and PC–PG solvents. The average diameter of untreated bagasse was ∼250–500 μm (Fig. 1a). Significant size reduction occurred with pretreatment by EC or PC alone (Fig. 1b and 1g). However, the extent of size reduction by EC pretreatment was higher than that by PC pretreatment. The average diameter was ∼40–150 μm for bagasse pretreated by EC whereas it was ∼60–230 μm for bagasse pretreated by PC. Despite the size reduction, the fibres were still compact and rigid, and defibrillation was not observed (Fig. 1b and 1g). Defibrillation and further fibre diameter size reduction occurred with blending EC with EG or PC with PG (Fig. 1c–1e, 1h and 1i). The average diameter of defibrillated fibres was ∼20–30 μm. Meanwhile, the defibrillated fibres were longer when the pretreatment contained a larger amount of EG than those with less EG (Fig. 1c–1e). The length of defibrillated fibres with an EC–EG ratio of 1:1 was in the range of ∼100–1000 μm whereas it was in the range of ∼100–400 μm with an EC–EG ratio of 9:1. There was still a large portion of fibre bundles not being defibrillated from pretreatment by EG alone (Fig. 1f). These SEM images indicate that the role of EC or PC is to reduce the fibre size, possibly through depolymerisation of cellulose whereas EG or PG breaks down the lignin and carbohydrate linkages. Hence the combination of an alkylene carbonate with the corresponding glycol enhances fibre defibrillation and delignification. Bagasse fibre size reduction itself did not significantly increase the glucan digestibility whereas defibrillation did as shown in Tables 1 and 2. Therefore, defibrillation of fibres makes cellulose more accessible to cellulases and enhances enzymatic hydrolysis.
 | ||
| Fig. 1 SEM images of (a) untreated bagasse, and bagasse samples pretreated by EC–EG solvents with the ratio of (b) 1:0, (c) 9:1, (d) 4:1, (e) 1:1 and (f) 0:1 and by PC–PG solvents with the ratio of (g) 1:0, (h) 9:1 and (i) 4:1. | ||
The bagasse samples pretreated with EC–EG solvents were also characterised using FTIR. A number of characteristic bands were used to monitor the chemical changes that occurred in lignin and carbohydrates. As shown in Fig. 2, the ester bond (i.e., lignin–hemicellulose) signal at about 1732 cm−1 diminished in the bagasse samples pretreated by EC–EG (4:1) but was still observable in the bagasse samples pretreated by EC or EG alone. This peak may be related to the uronic acid ester bonds formed between the carboxylic acid group in hemicellulose and the phenolic hydroxyl group in lignin, and/or the carboxylic acid group from lignin hydroxycinnamic acid and the hydroxyl group from the arabinofuranose unit.39,40 The reduction of this peak in the bagasse pretreated by EC–EG (4:1) confirmed the results of Table 1 with removal of 87% of the total amount of lignin and 74% of the total amount of xylan from bagasse. The peaks at 1605 cm−1 and 1515 cm−1, which are related to the aromatic skeleton vibrations in lignin,41 were prominent in the bagasse samples pretreated with EC or EG alone compared to those in the bagasse samples pretreated by the EC–EG mixture. The band intensity at 1460 cm−1, possibly associated with the methoxy group in lignin,42 was weak in the bagasse samples pretreated by EC–EG mixtures compared to pretreatment by EC or EG alone.
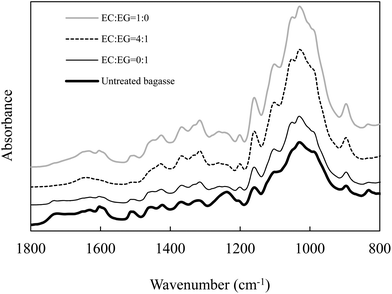 | ||
| Fig. 2 FTIR spectra of pretreated bagasse samples. | ||
The peak at 1318 cm−1 may be assigned to C–H vibration in cellulose and/or C–O vibration in syringyl derivatives according to previous reports.43,44 In this study, this peak is likely associated with C–H vibration in cellulose because the intensity of this peak increased slightly in all the pretreated bagasse samples, corresponding to increased cellulose content in pretreated bagasse samples. The peak at 1240 cm−1 is assigned to β-ether bonds.41 This peak diminished in the bagasse samples pretreated by EG and EC–EG, indicating the cleavage of ether linkages between lignin and carbohydrates.
The region of 1200–1000 cm−1 represents C–O stretch and deformation bands in cellulose, lignin and residual hemicellulose.45 The increase in band intensity at 1200 cm−1 of pretreated bagasse may be related to the increase in glucan content. The band intensity at 1105 cm−1, which corresponds to crystalline cellulose,46 increased in all the pretreated bagasse samples compared to the untreated bagasse sample, indicating that the pretreatment removed disproportionate amounts of amorphous components in the pretreated bagasse samples. XRD results confirmed the removal of amorphous content. As shown in Fig. S1,† the peak (2θ at ∼15.5°) which corresponds to crystalline cellulose remained significant in the pretreated bagasse. The estimated CrI increased from 0.70 for the untreated bagasse sample to 0.75–0.79 for the pretreated bagasse samples. The slight increase of CrI after pretreatment can be attributed to the removal/dissolution of amorphous components such as amorphous cellulose, hemicellulose and lignin. The highest CrI was 0.79 for the bagasse samples pretreated with the EC–EG ratios of 9:1 and 4:1 whereas it was 0.74–0.76 for the bagasse samples pretreated under other conditions.
The peak at 1050 cm−1 is associated with the C–O stretch in cellulose and hemicellulose.43 It was more prominent in the pretreated bagasse samples, corresponding to the increase in glucan content. The peak at 898 cm−1 is characteristic of β-glycosidic linkages and demonstrates the dominance of these linkages between the sugar units in cellulose and hemicellulose.41 The peak at 835 cm−1 belongs to a C–H out of plane vibration in lignin.43 The peak at 835 cm−1 was still prominent with EC pretreatment, diminished with EG pretreatment, and disappeared with EC–EG pretreatment, indicating removal of a higher proportion of lignin component.
| Ratio of EC to EG | Glucose yield on glucan (%) | Yield on xylan (%) | |
|---|---|---|---|
| Xylose | Furfurala | ||
|
a Furfural yield is based on the total xylan and arabinan components in untreated bagasse.
b Hydrolysate from pretreatment using the EC–EG (4:1) mixture was further diluted four times with water and incubated at 130 °C for 30 min.
|
|||
| 1:0 |
0.0 | 4.6 | 8.0 |
| 9:1 |
0.0 | 4.6 | 3.7 |
| 4:1 |
0.0 | 3.9 | 0.0 |
| 2:1 |
0.0 | 3.2 | 0.0 |
| 1:1 |
0.0 | 2.8 | 0.0 |
| 0:1 |
0.0 | 0.0 | 0.0 |
| 4:1b |
1.3 | 57.5 | 1.5 |
Studies on the liquefaction of lignocellulosics with the EC–EG system have reported the formation of EG-glucosides (Fig. 3a) during the early stages of the liquefaction.31,47,48 When the time of liquefaction was prolonged, EG-glucosides were decomposed, leading to the formation of levulinic acid–EG esters.31 EG-glucosides or levulinic acid–EG esters could be hydrolysed to EG, glucose or levulinic acid if water is added to the liquefaction process.47,48 The formation of EG-glucosides may explain why free glucose was not detected in the solution. Production of EG-xylosides from biomass liquefaction by the EC–EG system was not reported. However, it is probable that xylan is degraded in a similar way with the formation of EG-xylosides as shown in Fig. 3b. To confirm our hypothesis, the hydrolysate originating from pretreatment by the EC–EG (4:1) mixture was further hydrolysed at 130 °C for 30 min after adding water (final water content of ∼75% and acid concentration of ∼0.3%). As shown in Table 3, the xylose yield increased from 3.9% to 57.5%, and a small amount of glucose was also detected in the hydrolysate. In another experiment, an EG solution containing pure xylose and 1.2% H2SO4 was incubated at 130 °C for 30 min. After incubation, ∼6% of the total xylose was detected. However, when water (∼75% of the total solution) was added into the incubated solution and the solution was further hydrolysed at 130 °C for 30 min, the yield of xylose increased to 35%. These experimental results therefore indicated that pretreatment of sugarcane bagasse with the acid-catalysed EC–EG system produced EG-xylosides. It was also likely that the formation of PG-xylosides occurred in the acid-catalysed PC–PG system. The low temperature EC–EG pretreatment process developed in our study retained the majority of cellulose components for enzymatic production of fermentable sugars. The hydroxyl-rich hydrolysates (containing liquefied lignin and xylan components) from pretreatment with the EC–EG system may be useful in the production of polymers as has been shown previously with liquefied biomass.33,49,50
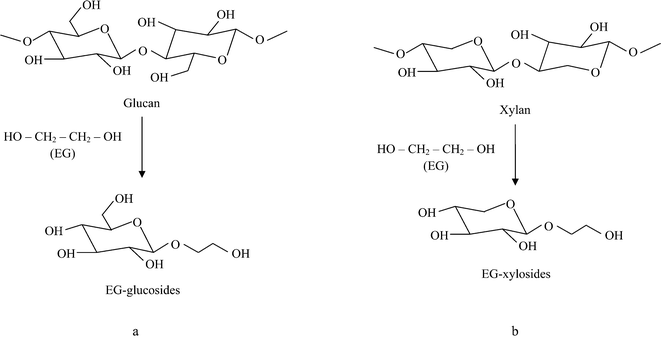 | ||
| Fig. 3 Schemes of (a) the reported glucan degradation mechanism31 and (b) the proposed xylan degradation mechanism in an acidified EG solution. | ||
Although the EC–EG system is an effective pretreatment/liquefaction system, EC itself is not stable under either acidic or basic conditions.51 Decomposition of EC produces EG and CO2.51 We observed that CO2 bubbles emerged in all pretreatments using EC–EG mixtures and ∼3% EC was converted into EG in the pretreatment by EC alone. Given that pretreatment effectiveness (in terms of glucan digestibility) remains high in a wide EC–EG ratio range (from 9:1 to 2:1), the EC–EG solvent can be reused several times. Thereafter, the hydrolysate may be processed by size exclusion chromatography to separate the free solvents (EC and EG) from EG-glycosides and lignin.24 The EG-glycosides and lignin have the potential for the production of polyesters and resins.33 EG also can be converted into EC to compensate for the loss of EC during decomposition.52 The low temperature pretreatment process with the use of acidified EC–EG solutions is shown in Fig. 4.
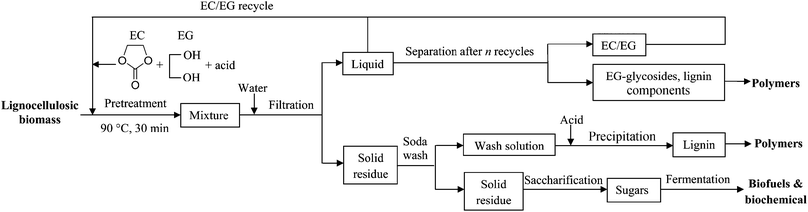 | ||
| Fig. 4 An EC–EG-based lignocellulosic biomass pretreatment process. | ||
3.2. Effects of operational conditions
:1) in the presence of 1.2% H2SO4. As shown in Table 4, pretreatment with EC alone was not effective at the temperatures used in the present study. However, pretreatment with EG alone led to glucan digestibilities increasing from 12.0% to 65.0% with increasing temperature from 60 °C to 90 °C. When the mixture of EC–EG was used, the glucan digestibilities were 41.1%, 68.9%, 87.2% and 93.4% at 60 °C, 70 °C, 80 °C and 90 °C respectively. The glucan digestibility of the bagasse pretreated with the mixture of EC–EG at 60 °C was 240% higher than that from pretreatment with EG alone. These results show that the improvement in glucan digestibility with the use of the EC–EG mixture at lower temperatures is more significant than that at higher temperatures.
:1) solvents at different temperatures
| Pretreatment temperaturea (°C) | Content in pretreated bagasse (%) | Biomass yield (%) | Recovery in pretreated bagasse (%) | 72 h glucan digestibility (%) | Total glucose yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Glucan | Xylan | Lignin | Glucan | Xylan | Lignin | ||||
| a Pretreatment was conducted in the presence of 1.2% H2SO4. | |||||||||
| 90 (EC:EG = 1:0) |
67.7 | 7.4 | 16.0 | 60.0 | 92.7 | 22.0 | 34.9 | 13.4 | 12.4 |
| 80 (EC:EG = 1:0) |
65.8 | 8.9 | 17.8 | 62.9 | 94.5 | 27.7 | 40.7 | 12.3 | 11.6 |
| 70 (EC:EG = 1:0) |
61.3 | 12.1 | 18.3 | 68.4 | 95.7 | 41.0 | 45.5 | 11.9 | 11.4 |
| 60 (EC:EG = 1:0) |
54.3 | 15.6 | 21.2 | 78.3 | 97.0 | 60.4 | 60.3 | 11.6 | 11.3 |
| 90 (EC:EG = 4:1) |
77.0 | 8.9 | 6.1 | 54.9 | 96.5 | 24.2 | 12.2 | 93.4 | 90.1 |
| 80 (EC:EG = 4:1) |
74.9 | 11.8 | 8.6 | 56.5 | 96.6 | 33.0 | 17.6 | 87.2 | 84.2 |
| 70 (EC:EG = 4:1) |
67.5 | 14.5 | 14.0 | 62.9 | 96.9 | 45.1 | 32.0 | 68.9 | 66.8 |
| 60 (EC:EG = 4:1) |
59.5 | 18.0 | 19.3 | 72.5 | 98.5 | 64.6 | 50.9 | 41.1 | 40.5 |
| 90 (EC:EG = 0:1) |
67.7 | 13.0 | 14.6 | 62.3 | 96.3 | 40.1 | 33.1 | 65.0 | 62.6 |
| 80 (EC:EG = 0:1) |
62.5 | 16.8 | 16.4 | 68.5 | 97.7 | 56.9 | 40.8 | 52.8 | 51.6 |
| 70 (EC:EG = 0:1) |
56.2 | 19.5 | 20.4 | 76.1 | 97.7 | 73.5 | 56.5 | 35.5 | 34.7 |
| 60 (EC:EG = 0:1) |
48.6 | 20.5 | 24.4 | 88.9 | 98.6 | 90.2 | 78.8 | 12.0 | 11.8 |
| Untreated bagasse | 43.8 | 20.2 | 27.5 | 100.0 | 100.0 | 100.0 | 100.0 | 6.0 | 6.0 |
Fig. 5 shows the kinetics of enzymatic hydrolysis of bagasse pretreated with the mixture of EC–EG (4:1) at different temperatures. Enzymatic hydrolysis was fast in the first 24 h and thereafter it increased only slightly. The maximum glucan digestibility obtained was from the bagasse pretreated at 90 °C. It was 89.6% at 24 h and 93.4% at 72 h.
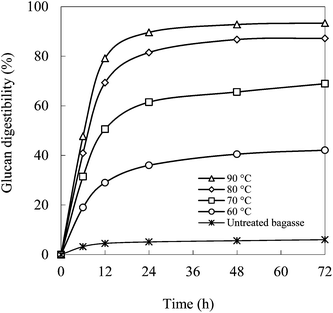 | ||
| Fig. 5 Kinetics of enzymatic hydrolysis of bagasse pretreated with EC–EG (4:1) solvents at different temperatures. | ||
:1. As shown in Table 5, increasing the acid content from 0.4% to 1.2% improved the glucan digestibility from 79% to 93%. Increasing the pretreatment time also had a positive effect on glucan digestibility of bagasse. However, increases in acid content and pretreatment time may also result in the decomposition of more EC to EG. Increasing water content from 1.2% to 10% decreased glucan digestibility significantly from 93.4% to 68.2%. Adding water possibly decreased the acidity potential. This is reflected by the decreased xylan removal in Table 5. The presence of water also decreased the delignification process (Table 5). Another disadvantage of increasing the water content is that it may lead to an increased conversion of EC to EG as EC decomposition is a hydrolysis process.
:1) solvents at other conditions
| Pretreatment conditions | Content in pretreated bagasse (%) | Biomass yield (%) | Recovery in pretreated bagasse (%) | 72 h glucan digestibility (%) | Total glucose yield (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acid content (%) | Water content (%) | T (°C) | Time (min) | Glucan | Xylan | Lignin | Glucan | Xylan | Lignin | |||
| 0.4 | 1.2 | 90 | 30 | 72.2 | 12.7 | 9.7 | 58.8 | 97.0 | 37.1 | 20.7 | 79.0 | 76.6 |
| 0.8 | 1.2 | 90 | 30 | 76.0 | 11.3 | 7.7 | 56.0 | 97.2 | 31.4 | 15.7 | 87.2 | 84.8 |
| 1.2 | 1.2 | 90 | 30 | 77.0 | 8.9 | 6.1 | 54.9 | 96.5 | 24.2 | 12.2 | 93.4 | 90.1 |
| 1.2 | 1.2 | 90 | 15 | 73.7 | 12.3 | 8.9 | 57.7 | 97.1 | 35.2 | 18.7 | 82.0 | 79.6 |
| 1.2 | 1.2 | 90 | 45 | 79.5 | 9.2 | 5.3 | 52.4 | 95.0 | 23.9 | 10.1 | 95.0 | 90.3 |
| 1.2 | 5 | 90 | 30 | 74.1 | 13.2 | 8.4 | 57.3 | 97.0 | 37.5 | 17.6 | 85.9 | 83.3 |
| 1.2 | 10 | 90 | 30 | 66.7 | 14.8 | 13.5 | 63.8 | 97.2 | 46.8 | 31.3 | 68.2 | 66.3 |
| Untreated bagasse | 43.8 | 20.2 | 27.5 | 100.0 | 100.0 | 100.0 | 100.0 | 6.0 | 6.0 | |||
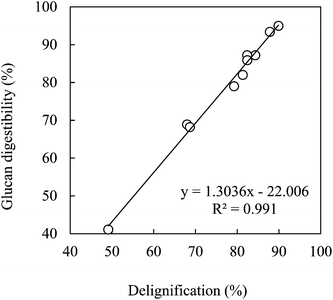
Fig. 6 shows the correlation of glucan digestibility with delignification for bagasse pretreated with the mixtures of EC–EG (4:1) for all the conditions tested (temperature, acid content, water content and time). A very good linear relationship between glucan digestibility and delignification was observed (r2 = 0.991) for this particular system.
4. Conclusions
Both EC and EG are bulk commodity chemicals. These chemicals have low toxicity and high boiling points (260 °C for EC and 197 °C for EG). EC and EG can be converted to each other under certain conditions.51,52 EC and EG are also renewable chemicals as technologies on the conversion of lignocellulose to EG have been demonstrated.53,54 A low energy consumption lignocellulose pretreatment process was developed in the present study with the use of an acid-catalysed EC–EG system. Pretreatment of bagasse at atmospheric pressure with a low temperature of 90 °C by the EC–EG system retained the majority of the glucan component and removed the majority of xylan and lignin components. Over 90% of the retained glucan component was hydrolysed to glucose by cellulases.Besides the benefit of low energy consumption, the low temperature pretreatment strategy also brings the possibility for the development of cellulase-compatible pretreatment technologies which will be welcomed by researchers focusing on expressing cellulases in plants. Production of plant-expressed cellulases has the potential to reduce cellulase costs for the saccharification of cellulose. This developed pretreatment process may be compatible with some thermo- and acid-stable cellulases as such cellulases have the ability to perform under very acidic conditions (pH < 2.0) and temperatures of 70–90 °C.55 Currently, the cellulase-compatibility of this pretreatment system is being investigated with the use of commercially available cellulases and sugarcane-expressed fungal cellulases.
References
- O. J. Sanchez and C. A. Cardona, Bioresour. Technol., 2008, 99, 5270–5295 CrossRef CAS.
- R. K. Saxena, P. Anand, S. Saran and J. Isar, Biotechnol. Adv., 2009, 27, 895–913 CrossRef CAS.
- P. Alvira, E. Tomas-Pejo, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS.
- Z. Y. Zhang, B. Jin and J. M. Kelly, Biochem. Eng. J., 2007, 35, 251–263 CrossRef CAS.
- P. Zheng, J. J. Dong, Z. H. Sun, Y. Ni and L. Fang, Bioresour. Technol., 2009, 100, 2425–2429 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS.
- B. Yang and C. E. Wyman, Biofuels Bioprod. Bioref., 2008, 2, 26–40 CrossRef CAS.
- M. FitzPatrick, P. Champagne, M. F. Cunningham and R. A. Whitney, Bioresour. Technol., 2010, 101, 8915–8922 CrossRef CAS.
- A. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- K. S. Kimon, E. L. Alan and D. W. O. Sinclair, Bioresour. Technol., 2011, 102, 9325–9329 CrossRef CAS.
- T. V. Doherty, M. Mora-Pale, S. E. Foley, R. J. Linhardt and J. S. Dordick, Green Chem., 2010, 12, 1967–1975 RSC.
- A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS.
- W. Y. Li, N. Sun, B. Stoner, X. Y. Jiang, X. M. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038–2047 RSC.
- P. Rezayati-Charani, J. Mohammadi-Rovshandeh, S. J. Hashemi and S. Kazemi-Najafi, Bioresour. Technol., 2006, 97, 2435–2442 CrossRef CAS.
- A. Demirbas, Energy Source Part A, 2010, 32, 689–696 CrossRef CAS.
- A. Demirbaş, Bioresour. Technol., 1998, 63, 179–185 CrossRef.
- A. Demirbas, Energy Source Part A, 2008, 30, 1120–1126 CrossRef CAS.
- M. M. Kucuk, Energy Sources, 2005, 27, 1245–1255 CrossRef.
- A. Demirbas and A. Celik, Energy Sources, 2005, 27, 1073–1084 CrossRef CAS.
- A. Johansson, O. Aaltonen and P. Ylinen, Biomass, 1987, 13, 45–65 CrossRef CAS.
- L. Jimenez, A. Perez, M. J. De la Torre, A. B. Rodriguez and V. Angulo, Bioresour. Technol., 2008, 99, 2170–2176 CrossRef CAS.
- E. Jasiukaityte, M. Kunaver and M. Strlic, Cellulose, 2009, 16, 393–405 CrossRef CAS.
- A. Kržan and E. Zagar, Bioresour. Technol., 2009, 100, 3143–3146 CrossRef.
- A. Kržan, M. Kunaver and V. Tišler, Acta Chim. Slov., 2005, 52, 253–258 Search PubMed.
- J. A. Liu, R. Takada, S. Karita, T. Watanabe, Y. Honda and T. Watanabe, Bioresour. Technol., 2010, 101, 9355–9360 CrossRef CAS.
- C. Martin, J. Puls, B. Saake and A. Schreiber, Cellul. Chem. Technol., 2011, 45, 487–494 CAS.
- D. H. Lee, E. Y. Cho, C. J. Kim and S. B. Kim, Biotechnol. Bioprocess Eng., 2010, 15, 1094–1101 CrossRef CAS.
- F. Sun and H. Chen, Bioresour. Technol., 2008, 99, 5474–5479 CrossRef CAS.
- F. Sun and H. Chen, Bioresour. Technol., 2008, 99, 6156–6161 CrossRef CAS.
- T. Yamada and H. Ono, Bioresour. Technol., 1999, 70, 61–67 CrossRef CAS.
- J. Yip, M. J. Chen, Y. S. Szeto and S. C. Yan, Bioresour. Technol., 2009, 100, 6674–6678 CrossRef CAS.
- F. Yu, Y. H. Liu, X. J. Pan, X. Y. Lin, C. M. Liu, P. Chen and R. Ruan, Appl. Biochem. Biotechnol., 2006, 130, 574–585 CrossRef.
- T. Xie and F. G. Chen, J. Appl. Polym. Sci., 2005, 98, 1961–1968 CrossRef CAS.
- L. Y. Liang, Z. H. Mao, Y. B. Li, C. X. Wan, T. P. Wang, L. H. Zhang and L. Y. Zhang, Bioresources, 2006, 1, 248–256 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, National Renewable Energy Laboratory Battelle, USA, 2008 Search PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3 DOI:10.1186/1754-6834-3-10.
- L. Paszner and P. C. Chang, US Patent4409032, 1983 Search PubMed.
- N. C. Carpita and D. M. Gibeaut, Plant J., 1993, 3, 1–30 CrossRef CAS.
- K. Iiyama, T. B. T. Lam and B. A. Stone, Plant Physiol., 1994, 104, 315–320 CAS.
- L. Liu, J. S. Sun, M. Li, S. H. Wang, H. S. Pei and J. S. Zhang, Bioresour. Technol., 2009, 100, 5853–5858 CrossRef CAS.
- G. L. Guo, W. H. Chen, W. H. Chen, L. C. Men and W. S. Hwang, Bioresour. Technol., 2008, 99, 6046–6053 CrossRef CAS.
- X. B. Zhao, L. Wang and D. H. Liu, J. Chem. Technol. Biotechnol., 2008, 83, 950–956 CrossRef CAS.
- T. H. Kim, J. S. Kim, C. Sunwoo and Y. Y. Lee, Bioresour. Technol., 2003, 90, 39–47 CrossRef CAS.
- X. F. Sun, F. Xu, R. C. Sun, P. Fowler and M. S. Baird, Carbohydr. Res., 2005, 340, 97–106 CrossRef CAS.
- C. L. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900–4906 CrossRef CAS.
- T. Yamada and H. Ono, J. Wood Sci., 2001, 47, 458–464 CrossRef CAS.
- T. Yamada, M. Aratani, S. Kubo and H. Ono, J. Wood Sci., 2007, 53, 487–493 CrossRef CAS.
- S. H. Lee, Y. Teramoto and N. Shiraishi, J. Appl. Polym. Sci., 2002, 83, 1482–1489 CrossRef CAS.
- Y. P. Wei, F. Cheng, H. P. Li and J. G. Yu, J. Appl. Polym. Sci., 2004, 92, 351–356 CrossRef CAS.
- W. J. Peppel, Ind. Eng. Chem., 1958, 50, 767–770 CrossRef CAS.
- S. Bhadauriaa, S. Saxena, R. Prasad, P. Sharma, R. Prasad and R. Dwivedi, Eur. J. Chem., 2012, 3, 235–240 CrossRef.
- J. Y. Sun and H. C. Liu, Green Chem., 2011, 13, 135–142 RSC.
- X. C. Wang, L. Q. Meng, F. Wu, Y. J. Jiang, L. Wang and X. D. Mu, Green Chem., 2012, 14, 758–765 RSC.
- Y. Huang, G. Krauss, S. Cottaz, H. Driguez and G. Lipps, Biochem. J., 2005, 385, 581–588 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36323b |
| This journal is © The Royal Society of Chemistry 2013 |