 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge competition with oxygen molecules determines the growth of gold particles on doped CaO films
Yi
Cui
a,
Kai
Huang
a,
Niklas
Nilius
*ab and
Hans-Joachim
Freund
a
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, 14195 Berlin, Germany. E-mail: nilius@fhi-berlin.mpg.de
bCarl von Ossietzky Universität Oldenburg, Institut für Physik, D-26111 Oldenburg, Germany
First published on 8th January 2013
Abstract
The influence of gas-phase oxygen on the growth of Au nanoparticles on Mo-doped CaO films has been investigated by means of low temperature scanning tunnelling microscopy and X-ray photoelectron spectroscopy. Whereas at ideal vacuum conditions, only 2D Au islands develop on the oxide surface, the fraction of 3D deposits increases with increasing O2 pressure until they become the dominant species in 10−6 mbar oxygen. The morphology crossover arises from changes in the interfacial electron flow between Mo donors in the CaO lattice and different ad-species on the oxide surface. In the absence of O2 molecules, the donor electrons are predominately transferred to the Au ad-atoms, which consequently experience enhanced binding to the oxide surface and agglomerate into 2D islands. In an oxygen atmosphere, on the other hand, a substantial fraction of the excess electrons is trapped by adsorbed O2 molecules, while the Au atoms remain neutral and assemble into tall 3D particles that are typical for non-doped oxides. Our experiments demonstrate how the competition for charge between different adsorbates governs the physical and chemical properties of doped oxides, so widely used in heterogeneous catalysis.
Introduction
The ability of supported metal particles to promote certain catalytic reactions depends on their elemental composition and the electronic and chemical interplay with the oxide support. Also the particle shape has proven to be of relevance, as certain reactions require the availability of specific active sites on the catalyst surface. A prominent example is the CO → CO2 conversion on supported metal catalysts, where the metal deposits are responsible for capturing the CO molecules, while the oxide stabilizes and activates the O2 species. The actual reaction is then believed to occur at the metal–oxide perimeter, explaining why particles with 2D or raft-like shapes are particularly suited to catalyze this reaction.1–4The geometry of metal particles on oxide supports has been investigated down to the atomic scale by using a variety of microscopic and spectroscopic techniques.5–8 The metal–oxide interface, i.e. the atomic registry and interfacial bond length, has thereby received special attention, as it governs the morphology of the growing aggregates.9,10 A careful shape characterization is also required in order to rationalize the particles’ electronic properties that, to a certain extent, reflect the symmetry of the confining potential and hence the position of the metal ion cores.11 Whereas particle shapes could be analyzed only before and after a chemical reaction in the past,6,7,12in situ studies employing synchrotron-based EXAFS and environmental TEM now provide evidence for changes in coordination numbers and particle shapes directly during a catalytic process.13–17 In fact, the particles undergo substantial structural modifications at elevated pressure and temperature conditions, being explained with the different thermodynamic stability of bare and adsorbate-covered surfaces and the temporary formation of oxide or hydrate phases at reaction conditions.18,19
In this paper, we demonstrate how the presence of a gas phase influences the growth behaviour of supported metal particles and how this effect depends on the state of doping of the oxide beneath. The phenomenon described here is therefore different from a pure environmental effect on existing particles, and sheds light on the interplay between adsorbates and dopants inside the oxide matrix on the growth process of metals. The effect of gas-phase species on the morphology of metal surfaces and nanoparticles has been addressed in several experimental and theoretical studies before.20–23 The regular Pt(557) surface, for example, was found to develop a high density of kinks and corners in high-pressure CO,24 but exposes ultra-small Pt nanoclusters in an O2 environment.25 Electron micrographs of individual Pt particles, on the other hand, revealed the transformation of (111) and (100) facets into various high-index planes upon O2 exposure at 670 K.26,27 Similarly, the (100) facets of Au nanoclusters were shown to reconstruct in a CO environment in order to maximize the Au–CO interaction.28 The observed restructuring is driven by the enhanced binding of most adsorbates to open, corrugated surfaces that are, however, thermodynamically unfavourable at vacuum conditions due to their high surface free energy.
A second effect on the particle morphology, the presence of dopants in the oxide lattice, has moved into the focus of catalysis research only recently. In a combined scanning tunnelling microscopy (STM)/density functional theory (DFT) study, it was shown that small quantities of Mo embedded in a CaO matrix substantially alter the growth morphology of gold.29 Whereas mainly 3D deposits developed on the pristine oxide due to the weak metal–oxide adhesion in that case, monolayer Au islands occurred after doping with Mo. The morphology crossover was explained with an electron transfer from the Mo donors into the ad-gold, activating efficient Au–CaO interaction channels.30 The reverse effect was found upon inserting low-valence dopants, e.g. Li, into the CaO lattice. The Li ions generate acceptor-type hole-states in the oxide valence band, which effectively trap excess electrons and prevent their transfer into the ad-gold. The neutral gold follows a 3D growth behaviour, similar to the one on non-doped supports.31 In this paper, we demonstrate how a variable background of O2 alters the charge exchange between donors in the CaO lattice and adsorbed Au species. We show that gold grows into tall 3D deposits in the presence of oxygen and discuss possible implications of the underlying charge competition on the catalytic properties of doped metal–oxide systems.
Experimental
The studies were performed in an ultra-high vacuum (UHV) chamber equipped with a low-temperature STM (T = 9.5 K) and an X-ray photoelectron spectrometer (ESCALAB 200). The CaO films were prepared by Ca deposition onto a Mo(001) surface in 5 × 10−7 mbar O2 at room temperature, followed by an annealing step to 1000 K in UHV.32 The films contained a significant amount of Mo impurities, originating from atom diffusion out of the metal support into Ca substitutional sites of the CaO lattice. Due to a relatively large Mo diffusion barrier, the impurity concentration gradually diminished with film thickness from 25% directly at the interface to almost zero beyond the 20th monolayer (ML).33 For 20–25 ML thick films used in the experiment, the mean Mo concentration in the near-surface region was estimated to be 2–3%. Practically dopant-free films, being referred to as pristine in the following, could be produced by increasing the CaO thickness to around 50 ML. Both, the self-doped and pristine films displayed a sharp (1 × 1) square pattern in LEED, indicative for a fully relaxed, (001)-terminated rocksalt structure. XPS measurements confirmed the one-to-one stoichiometry of the CaO films and the presence of Mo dopants at low film thicknesses. The oxide surface consisted of atomically flat domains of up to 100 nm2 size, separated by predominately [100] oriented line defects, as derived from the STM data.32 The dislocation lines develop spontaneously in the oxide film in order to remove misfit-induced lattice strain with the metal substrate. The Au particles were prepared by depositing nominally 0.5 ML gold from a thermal evaporator using a deposition rate of 0.5 ML min−1. In order to investigate the impact of oxygen on the Au growth, the samples have been exposed to different O2 partial pressures at 300 K before, during or after gold exposure.Results
As discussed in earlier work,29 Au growth on pristine and Mo-doped CaO films proceeds in opposite manner (Fig. 1a, b). While on pristine films, the Au particles adopt large height-to-diameter ratios indicative for a small metal–oxide adhesion, flat, monolayer islands develop in the presence of Mo-dopants. The drastic change in particle morphology becomes evident in shape histograms, being produced by analysing a few hundred Au deposits on differently prepared oxide films (Fig. 2). Interestingly, the growth behaviour was found to be sensitive not only to the presence of dopants, but also to the gas atmosphere in which metal deposition was carried out. In 5 × 10−8 mbar O2, the Au growth starts diverging from the ideal 2D regime that is observed on doped films, and 3D deposits become visible in the STM (Fig. 1c and 2c). The alteration of the growth mode initially affects only particles around the oxide dislocation lines. However, with increasing O2 partial pressure, 3D particles appear even away from the surface defects. In 5 × 10−7 mbar of oxygen, already half of the Au deposits adopt 3D geometries, while the remaining entities still retain a monolayer nature (Fig. 1d). Surprisingly, no intermediate particle shapes are detected, which becomes evident in a bimodal shape distribution (Fig. 2d). At 5 × 10−6 mbar oxygen pressure, 3D shapes entirely dominate the Au particle ensemble, while 2D islands have vanished from the histogram. This growth situation does not change anymore at higher O2 partial pressures, only that the CaO surface starts degrading at such conditions (Fig. 1f). We relate the observed roughening effect to two factors. First, our O2 gas source might be contaminated with traces of water that interacts with the hydrophilic CaO surface and gradually transforms it into a disordered calcium hydroxide phase. Second, the excess oxygen might promote Mo segregation from the interior of the CaO film to its surface, where it aggregates into a rough Mo oxide overlayer. Such a segregation process is thermodynamically feasible due to the substantial energy gain when changing the oxidation state of Mo ions from +2 in an ideal rocksalt environment to +6 in saturated MoO3 surface units.33 As a consequence of the reduced surface quality, also the density of Au particles increases at high O2 partial pressures, as new nucleation sites becomes available for the incoming Au atoms (Fig. 2f). | ||
| Fig. 1 STM topographic images of 0.5 ML gold deposited on (a) 50 ML CaO without dopants in the surface region and on 25 ML thick, self-doped CaO films (b) in UHV and (c–f) in O2 pressures increasing from (c) 5 × 10−8 mbar, (d) 5 × 10−7 mbar, (e) 5 × 10−6 mbar to (f) 2 × 10−5 mbar. All images are 50 × 50 nm2 in size and have been obtained at 4.0 V sample bias. Note that 2D Au islands are imaged as depressions on the CaO due to their low conductivity.33 In (c,d), these islands are marked by dashed lines. | ||
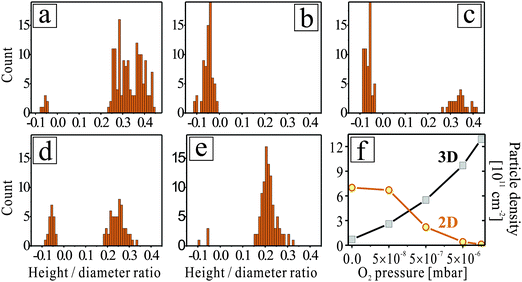 | ||
| Fig. 2 Histograms of Au-particle shapes for growth conditions identical to the ones shown in Fig. 1: (a) non-doped CaO in UHV, Mo-doped CaO in (b) UHV, (c) 5 × 10−8 mbar, (d) 5 × 10−7 mbar and (e) 5 × 10−6 mbar of oxygen. The occurrence of negative aspect ratios in the histograms relates to the negative contrast of monolayer Au islands in the respective STM images. Note the bimodal shape distributions in panels (c) and (d), reflecting the coexistence of 2D and 3D particles. (f) Evolution of the number density of 2D and 3D deposits as a function of the O2 pressure during deposition. | ||
In a second set of experiments, we have analysed the influence of pre-dosed oxygen on the growth morphology of gold. For this purpose, the CaO films were exposed to 20 L O2 prior to Au deposition, which produced large quantities of 0.5 Å-deep depressions in the surface (Fig. 3a). Following earlier DFT calculations,34,35 we assign those entities to O2 molecules that lie flat on the surface and therefore appear slightly elongated. This interpretation is supported by manipulation experiments, in which selected O2 molecules were split into their atomic constituents by applying a 3.0 V pulse with the STM tip36. Also pre-dosed oxygen was found to alter the Au growth morphology, although the actual deposition step was performed in UHV. The particle ensembles prepared on a Mo-doped, O2 pre-covered CaO have the same bimodal shape distribution as the ones produced in moderate O2 background pressure (Fig. 3c). Apparently, surface-bound and gas-phase molecules play a similar role, i.e. they promote the development of 3D instead of 2D particles even on doped oxide films. Only the efficiency of the geometry crossover is reduced, a trend that is easily explained with the smaller availability of surface oxygen at pre-dosing compared to continuous dosing.conditions
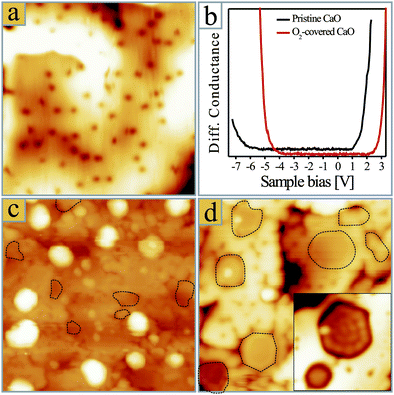 | ||
| Fig. 3 (a) STM topographic image of a Mo-doped CaO film after exposure to 20 L of oxygen (25 × 25 nm2). (b) Conductance spectra showing the onset of valence and conduction band on clean and O2-covered CaO films. (c) 0.5 ML Au deposited onto a Mo-doped CaO film pre-covered with 400 L O2 and (d) 0.5 ML Au post-oxidized with 100 L O2 (40 × 40 nm2). The 2D islands are marked with dashed lines. The inset in (d) shows two selected Au monolayer islands with an oxidized rim (10 × 10 nm2). | ||
Finally, we have explored the effect of post-oxidizing the Au particles. As discussed before, gold exposure in UHV produces 2D metal sheets on the Mo-doped CaO surface.29 Exposing such an ensemble to 100 L O2 does not affect the original 2D particle shapes (Fig. 3d). In fact, the main difference between as-grown and post-oxidized Au islands is a characteristic rim in the latter case, being assigned to adsorbed molecules along the low-coordinated island perimeter.37 On thick, non-doped CaO films, the Au growth morphology was found to be independent of oxygen, as the molecules promote the 3D growth that is already intrinsic to the pristine oxide.
Before discussing the effect of oxygen on the Au growth, we want to highlight another interesting phenomenon. Although 3D particles develop on both, non-doped CaO in vacuum and Mo-doped CaO in an O2 environment, the resulting particle shapes are rather different. Vacuum-grown particles are mostly hemispherical and do not exhibit well-shaped basal planes or side facets (Fig. 1a). We are nonetheless assured of their crystalline nature and assign them to fcc-semispheres, densely covered with step edges. In contrast, particles grown in an oxygen environment have distinct geometries, as depicted in Fig. 4. The most abundant particle type has a quasi-rectangular basal plane and two roof-like side facets extending along the main axis (Fig. 4b). The particle orientation always follows a [100] or [010] direction of the CaO(001) lattice, indicating an epitaxial relationship between gold and the CaO surface. A second, frequently observed particle type has a truncated pyramidal shape with three large and three small side facets (Fig. 4c). Finally, particles with hexagonal or octahedral base planes and pyramidal appearance are found at oxygen-rich conditions (Fig. 4e). The latter two types have a random orientation with respect to the CaO lattice and are less abundant than the roof-like deposits. In general, the 3D deposits produced in an O2 environment feature higher crystallinity than the vacuum-grown species, suggesting that the surface oxygen increases the mobility of the Au atoms and facilitates their aggregation.12
 | ||
Fig. 4 (a) Pseudo-3D representation of crystalline Au particles grown on a Mo-doped CaO film in 5 × 10−6 mbar O2 (50 × 50 nm2). Enlarged images of four particles with (b) roof-like, (c) truncated pyramidal (8 × 8 nm2) and (e) hexagonal and octahedral shapes (8 × 12 nm2). The particle edges are retraced with dashed lines. (d) Ball model showing the hcp-Au (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0)/CaO(001) interface that gets pseudomorphic upon slight distortion of the Au bulk lattice. The A and B planes of the hcp lattice are indicated with arrows. 0)/CaO(001) interface that gets pseudomorphic upon slight distortion of the Au bulk lattice. The A and B planes of the hcp lattice are indicated with arrows. | ||
Discussion
According to the main experimental observations, we divide our discussion into two parts that are (i) the transition from 2D to 3D deposits upon gold deposition at increasing O2 pressures and (ii) the improved crystallinity of particles grown in oxygen excess.The fundamental reason for the development of 2D Au islands on Mo-doped CaO films is an electron transfer from the Mo donors into the Au ad-species.29,38 The process is energetically favourable as the filled Mo 4d states in the CaO band gap are higher in energy than the Au 6s affinity levels. Thus produced Au− species experience effective interaction schemes with the CaO support, namely Coulomb attraction to the oxide cations and polaronic stabilization of the Au− on the ionic surface,30 both promoting a 2D growth regime on the doped oxide film (see Fig. 1b). The impact of oxygen on the Mo–Au charge exchange now provides a plausible explanation for the modified Au growth behaviour at non-vacuum conditions. Similar to gold, the O2 molecules are electron acceptors and able to trap Mo excess electrons in their low-lying antibonding orbitals. Taking the electron affinity of the gas-phase species as reference (EA = 0.5 eV),39 the lowest acceptor level of the molecule lies below the highest occupied Mo 4d gap state, which renders formation of surface O2− species feasible. Co-deposition of oxygen and gold leads therefore to a situation in which both adsorbates compete for charges from the Mo impurities. For the experimental Au deposition rate of 0.5 ML min−1 and different O2 partial pressures, the following interplay occurs between the ad-species on the CaO surface. At the lowest O2 pressure (10−8 mbar), the number of both, Au atoms and O2 molecules increases by roughly 1 × 1013 cm−2 per second, assuming a sticking of one. However, as gold is the much stronger electron acceptor (EA = 2.3 eV)39 most extra charges get trapped by the Au atoms and their growth behaviour changes only moderately (Fig. 1c). Deviations from the 2D growth mode are only observed around the CaO dislocation lines, where empty defect states in the band gap emerge as third opponent in the charge competition.40 At 10−7 mbar O2, every incoming Au atom already faces 10 O2 molecules on the surface. Moreover, the charge reservoir established by near-surface Mo donors quickly depletes due to the high number of oxygen acceptors on the surface. However, as not every O2 molecule immediately sticks and takes an electron, a dynamical situation arises in which the Au atoms are still able to attract a substantial charge fraction from the Mo dopants. This new balance gives rise to a bimodal shape distribution in which 2D and 3D deposits coexist on the CaO surface (Fig. 2c). At 10−6 mbar O2, finally, the ratio between O2 and Au species approaches 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and acquisition of relevant charge quantities becomes difficult for the gold. Consequently, most Au atoms remain neutral and grow into tall 3D deposits right from the beginning. The resulting small interface area with the CaO support hampers the electron transfer also in a later stage, further stabilizing the 3D growth regime of Au in this stage (Fig. 1e).
:1 and acquisition of relevant charge quantities becomes difficult for the gold. Consequently, most Au atoms remain neutral and grow into tall 3D deposits right from the beginning. The resulting small interface area with the CaO support hampers the electron transfer also in a later stage, further stabilizing the 3D growth regime of Au in this stage (Fig. 1e).
To support the scenario of charge competition, we give circumstantial evidence for a negative charging of Au and O2 species on the CaO surface. The charge donation from the Mo impurities can be derived indirectly from changes in the CaO electronic structure. In STM conductance spectra, an up-shift of the oxide valence and conduction band by around 1.0 V is revealed upon oxygen exposure (Fig. 3b), an effect that was also observed after Au deposition onto the doped CaO films.33 XP-spectra show a similar trend and both, the O1s and Ca2p core levels lower their binding energy upon dosing the electron-accepting species (0.3 and 0.2 eV for Au and O2, respectively, Fig. 5). We relate this shift to an upward bending of the oxide bands, induced by the enhanced e–e repulsion between adsorbates and oxide electronic states in response to the electron transfer from the Mo donors.31 The magnitude of the effect is smaller in XPS than in STM due to the extreme surface sensitivity of the latter technique. Note that the presence of neutral or positively-charged adsorbates would lead to a constant or down-shifted state-density, in disagreement with the experimental data. The corresponding shift of the Mo 3d core levels could not be resolved in our XP data because of the low dopant concentration and the poor signal-to-noise ratio. This information needs to be obtained with a synchrotron-based experiment in future.
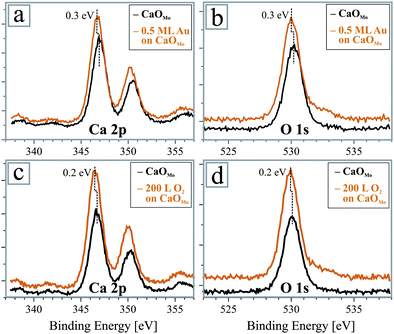 | ||
| Fig. 5 XP spectra of Ca 2p and O 1s peaks of a Mo-doped CaO film before and after (a,b) Au deposition and (c,d) O2 exposure. In both cases, the oxide states experience a shift to lower binding energies as a response to the charge transfer into the adsorbates. | ||
A charge transfer into oxygen and mediated by this, a strengthening of the O2–CaO interaction, is also in line with the observed thermal stability of O2 at room temperature and the easiness to image the molecules in the STM even on thick oxide films. On non-doped wide-gap insulators, such as MgO and Al2O3, molecular O2 typically desorbs well below 300 K and is hard to detect in STM even at cryogenic temperature.41,42 Moreover, recent electron paramagnetic resonance studies indicated the formation of O2− species on ultrathin MgO/Ag films, with the only difference that the extra electrons originated from the metal support below and not from donors inside the oxide film.43 The maximum density of O2− species was determined to be 1013 cm−2, a value that is likely higher on the more basic CaO surface in our case.43 We finally note that surface-bound oxygen may not only alter the charge transfer into the gold, but also reduce the Au–CaO adhesion directly, e.g. by forming a chemisorbed oxygen layer just below the growing ad-islands.19,44 The reduced interface interaction in this case would shift the Au equilibrium shape towards 3D, reflecting Wulff's principle that a smaller adhesion leads to larger particle heights.6,45 We expect, however, that this effect is less relevant than the charge competition between electron-accepting species in our experiment.
In the last paragraph, we discuss the improved crystallinity of Au deposits grown at O2-rich conditions and try to rationalize the observed particle geometries. The most abundant particle type is a roof-like crystallite oriented along a CaO[100] direction (Fig. 4b). Its elongated shape indicates a strong unidirectional growth and a pseudomorphic relationship with the CaO(001) lattice beneath. As a hexagonal Au(111) interface, being the thermodynamically favoured termination of fcc-gold, is incompatible with an epitaxial growth on the square oxide lattice, we suggest that gold crystallizes in an hcp lattice and develops (110) interface planes. This growth motive has recently been identified for Ni and Mg deposits grown on the iso-structural MgO(001) surface.10,46 Due to the two-fold symmetry of the hcp (110) plane, a dense array of metal–oxygen bonds is formed across the interface that substantially contributes to the metal–oxide adhesion. A similar interface geometry can now be realized for hcp Au (110) planes on the slightly larger CaO(001) lattice. The required distortion of the Au lattice amounts to 2% and −4% along the two orthogonal CaO[100] directions, respectively, values that are in reach for metal nanoclusters with their high structural flexibility (Fig. 4d). A (110) interface would also explain the unidirectional growth of gold, as the particles primarily expand along the low-strain direction on the CaO support. Even the side-facets of the roof-shaped Au deposits can be assigned with this model. The most plausible candidates are the hcp (0001) planes, which are tilted by 45° against the surface plane and characterized by particularly low surface free energies due to their closed-packed nature. We finally note that DFT calculations found similar total energies for Au40 clusters in either hcp or fcc configuration on the CaO(001), suggesting that both growth modes are energetically accesible.46
Apart from the roof-shaped crystallites, none of the other particle types develop an epitaxial relationship with the square CaO(001) lattice (Fig. 4c,e). Evidently, those deposits have other means to interact with the oxide support and are not forced to grow in registry with the oxide lattice. Icosdahedral and pyramidal particle shapes have been observed for various metals (Au, Ag, Pd and Cu) on different oxide supports before, e.g. on hexagonal Al2O3(0001),5 square MgO(001)10 and square SrTiO3(001),47,48 especially at high deposition temperature. This universality suggests that such geometries are mainly determined by the low formation energy of (111) facets of fcc metals and follow the classical Wulff construction principle for metal crystallites on weakly interacting supports.45,49 The ratio between epitaxial-hcp and symmetry-mismatched fcc deposits is again connected to the role of oxygen in the particle growth. At O2-rich conditions (1 × 10−6 mbar), the ad-gold is unable to acquire charges from the Mo donors and remains neutral. Interfacial Au–OCaO bonds are thus a crucial means to anchor the particles on the CaO surface, a fact that promotes the formation of epitaxial hcp(110) interface planes. With an enhanced electron flow to the gold at lower O2 pressures, charge-mediated interaction schemes become available. As a result, the particles gain structural flexibility and adopt growth shapes that are energetically more favorable than the hcp configuration. In the limit of zero oxygen, the Mo excess charges entirely govern the growth on the CaO surface and the gold spreads into large, monolayer islands.29
We note that other oxygen-induced effects may contribute to the particle shapes observed in our experiment. Oxygen is a known surfactant50 and increases the mobility of Au adatoms by forming intermediate metal–oxygen units. A higher diffusivity of the incoming species naturally improves the crystallinity of the ad-particles. In addition, O2 adsorption may selectively stabilize certain Au facets,44 modifying the particle equilibrium shape as compared to UHV conditions.25,26 Given the weakness of the oxygen–Au bond,39 we believe however that direct adsorption of O2 species is only of minor importance for the growth morphology of the Au particles.
Summary
Using a combined STM and XPS approach, we have demonstrated that the equilibrium shape of nanoparticles on doped CaO films results from a competition for electrons between Au atoms and oxygen species. At vacuum conditions, excess electrons provided by Mo ions in the oxide lattice are transferred to incoming Au atoms, which subsequently agglomerate into flat 2D islands in order to maximize the metal–oxide adhesion. Simultaneous oxygen supply from the gas phase interrupts this electron flow, as the surface O2 species are able to trap extra electrons as well. Once the majority of electrons are stabilized by O2 molecules, the Au growth changes from a 2D to a 3D mode, the latter being characteristic for non-doped oxides. The oxygen-induced morphology crossover is irreversible and the Au particles retain a 3D shape even after the oxygen has been removed from the system.Our paper highlights the complexity of charge-transfer processes between dielectric supports, ad-metals and molecules, a combination that typically occurs in heterogeneous catalysis. The direction of the charge flow is given by the actual potential situation at the catalyst surface, the relative energies of competing donor and acceptor levels and finally the electro-negativity of the involved molecules. The efficiency of the charge transfer therefore depends not only on intrinsic catalyst properties but also on the composition of the surrounding gas phase. The highly dynamic nature of charge-transfer processes calls for an exploration of catalytically-relevant systems directly at operation conditions. Our findings also disclose a possibility to alter the growth mode of metals simply by changing the gas atmosphere during deposition, which opens a new pathway to optimize structure and morphology of supported metal catalysts.
Acknowledgements
The authors are grateful for financial support from the DFG within the Cluster of Excellence ‘Unicat’. Y.C. thanks the Humboldt Foundation for a fellowship.References
- L. M. Molina and B. Hammer, Appl. Catal., A, 2005, 291, 21–31 CrossRef CAS.
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS.
- K. H. Hansen, T. Worren, S. Stempel, E. Lægsgaard, M. Bäumer, H. J. Freund, F. Besenbacher and I. Stensgaard, Phys. Rev. Lett., 1999, 83, 4120–4123 CrossRef CAS.
- M. Bäumer and H.-J. Freund, Prog. Surf. Sci., 1999, 61, 127–198 CrossRef.
- C. R. Henry, Prog. Surf. Sci., 2005, 80, 92–116 CrossRef CAS.
- G. J. Hutchings, Chem. Commun., 2008, 1148–1164 RSC.
- G. Pacchioni and N. Rosch, J. Chem. Phys., 1996, 104, 7329–7337 CrossRef CAS.
- S. Benedetti, P. Myrach, A. di Bona, S. Valeri, N. Nilius and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 125423 CrossRef.
- X. Lin, N. Nilius, H. J. Freund, M. Walter, P. Frondelius, K. Honkala and H. Häkkinen, Phys. Rev. Lett., 2009, 102, 206801–206804 CrossRef CAS.
- D. Starr, S. Shaikhutdinov and H.-J. Freund, Top. Catal., 2005, 36, 33–41 CrossRef CAS.
- J.-D. Grunwaldt and B. Clausen, Top. Catal., 2002, 18, 37–43 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS.
- G. Jacobs, J. A. Chaney, P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2004, 264, 203–212 CrossRef CAS.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS.
- M. W. Small, S. I. Sanchez, N. S. Marinkovic, A. I. Frenkel and R. G. Nuzzo, ACS Nano, 2012, 6, 5583–5595 CrossRef CAS.
- M. D. Ackermann, T. M. Pedersen, B. L. M. Hendriksen, O. Robach, S. C. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505–255508 CrossRef CAS.
- T. Schalow, M. Laurin, B. Brandt, S. Schauermann, S. Guimond, H. Kuhlenbeck, D. E. Starr, S. K. Shaikhutdinov, J. Libuda and H. J. Freund, Angew. Chem., Int. Ed., 2005, 44, 7601–7605 CrossRef CAS.
- S. Giorgio, M. Cabié and C. R. Henry, Gold Bull., 2008, 41, 167–173 CrossRef CAS.
- B. J. McIntyre, M. Salmeron and G. A. Somorjai, J. Vac. Sci. Technol., A, 1993, 11, 1964–1968 CAS.
- G. A. Somorjai and G. Rupprechter, J. Phys. Chem. B, 1999, 103, 1623–1638 CrossRef CAS.
- K. P. McKenna and A. L. Shluger, J. Phys. Chem. C, 2007, 111, 18848–18852 CAS.
- F. Tao, S. Dag, L.-W. Wang, Z. Liu, D. R. Butcher, H. Bluhm, M. Salmeron and G. A. Somorjai, Science, 2010, 327, 850–853 CrossRef CAS.
- Z. Zhu, F. Tao, F. Zheng, R. Chang, Y. Li, L. Heinke, Z. Liu, M. Salmeron and G. A. Somorjai, Nano Lett., 2012, 12, 1491–1497 CrossRef CAS.
- M. Cabié, S. Giorgio, C. R. Henry, M. R. Axet, K. Philippot and B. Chaudret, J. Phys. Chem. C, 2010, 114, 2160–2163 Search PubMed.
- G. Rupprechter and H.-J. Freund, Top. Catal., 2000, 14, 3–14 CrossRef CAS.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS.
- X. Shao, S. Prada, L. Giordano, G. Pacchioni, N. Nilius and H. J. Freund, Angew. Chem., Int. Ed., 2011, 50, 11525–11527 CrossRef CAS.
- G. Pacchioni, L. Giordano and M. Baistrocchi, Phys. Rev. Lett., 2005, 94, 226104–226107 CrossRef.
- X. Shao, N. Nilius and H. J. Freund, J. Am. Chem. Soc., 2012, 134, 2532–2534 CrossRef CAS.
- X. Shao, P. Myrach, N. Nilius and H. J. Freund, J. Phys. Chem. C, 2011, 115, 8784–8789 CAS.
- X. Shao, N. Nilius, P. Myrach, H. J. Freund, U. Martinez, S. Prada, L. Giordano and G. Pacchioni, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245407–245411 CrossRef; X. Shao, N. Nilius and H. J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115444–115451 CrossRef.
- N. U. Zhanpeisov, V. Staemmler and M. Baerns, J. Mol. Catal. A: Chem., 1995, 101, 51–60 CrossRef CAS.
- L. Wang, Appl. Surf. Sci., 2011, 257, 5499–5502 CrossRef CAS.
- B. C. Stipe, M. A. Rezaei, W. Ho, S. Gao, M. Persson and B. I. Lundqvist, Phys. Rev. Lett., 1997, 78, 4410–4413 CrossRef CAS.
- P. Frondelius, H. Häkkinen and K. Honkala, Angew. Chem., Int. Ed., 2010, 49, 7913–7916 CrossRef CAS.
- F. Stavale, X. Shao, N. Nilius, H. J. Freund, S. Prada, L. Giordano and G. Pacchioni, J. Am. Chem. Soc., 2012, 134, 11380–11383 CrossRef CAS.
- D. R. Lide (Ed), CRC Handbook of Chemistry and Physics, (Boca Raton, CRC Press, 2010) Search PubMed.
- H.-M. Benia, P. Myrach, A. Gonchar, T. Risse, N. Nilius and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 241415–241418 CrossRef.
- R. M. Jaeger, J. Libuda, M. Bäumer, K. Homann, H. Kuhlenbeck and H. J. Freund, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 217–225 CrossRef CAS.
- H.-J. Freund, Th. Klüner, R. Wichtendahl, S. Thiel, M. Adelt, W. Drachsel, M. Bäumer, H. Kuhlenbeck, T. Risse, K. Al-Shamery, M. Kampling, H. Hamann, ‘Metal–Ligand Interactions in Chemistry, Physics and Biology’, NATO ASI Ser., Vol. E, p. 91–128, (Kluwer, Academic Publ, 2000) Search PubMed.
- A. Gonchar, T. Risse, H. J. Freund, L. Giordano, C. Di Valentin and G. Pacchioni, Angew. Chem., Int. Ed., 2011, 50, 2635–2638 CrossRef CAS.
- J. M. Gottfried, K. J. Schmidt, S. L. M. Schroeder and K. Christmann, Surf. Sci., 2002, 511, 65–82 CrossRef CAS.
- G. Wulff, Z. Kryst., 1901, 34, 449–530 CAS.
- R. Ferrando, G. Rossi, F. Nita, G. Barcaro and A. Fortunelli, ACS Nano, 2008, 2, 1849–1856 CrossRef CAS.
- F. Silly and M. R. Castell, Phys. Rev. Lett., 2006, 96, 086104–086107 CrossRef.
- F. Silly and M. R. Castell, Appl. Phys. Lett., 2005, 87, 213107–213103 CrossRef; F. Silly, A. C. Powell, M. G. Martin and M. R. Castell, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165403–165408 CrossRef.
- N. Cabrera, Surf. Sci., 1964, 2, 320–345 CrossRef CAS.
- H. Wolter, K. Meinel, C. Ammer, K. Wandelt and H. Neddermeyer, Phys. Rev. B: Condens. Matter, 1997, 56, 15459–15470 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |