DOI:
10.1039/C2FD20153D
(Paper)
Faraday Discuss., 2013,
162, 365-378
Switching-off toluene formation in the solvent-free oxidation of benzyl alcohol using supported trimetallic Au–Pd–Pt nanoparticles
Received
17th December 2012
, Accepted 2nd January 2013
First published on 18th April 2013
Abstract
Trimetallic Au–Pd–Pt nanoparticles have been supported on activated carbon by the sol-immobilisation method. They are found to be highly active and selective catalysts for the solvent-free aerobic oxidation of benzyl alcohol. The addition of Pt promotes the selectivity to the desired product benzaldehyde at the expense of toluene formation. Detailed aberration corrected STEM-XEDS analysis confirmed that the supported particles are indeed Au–Pd–Pt ternary alloys, but also identified composition fluctuations from particle-to-particle which vary systematically with nanoparticle size.
Introduction
The oxidation of primary alcohols to aldehydes is an industrially important process, as aldehydes are valuable as intermediates and as high value components for the perfume industry. Conventionally this reaction is carried out using expensive and toxic oxidants such as chromate or permanganate, causing serious environmental and economical concerns.1–3 Hence, catalytic oxidation of primary alcohols to aldehydes using molecular oxygen/air, or other greener oxidants such as hydrogen peroxide, becomes an attractive alternative. We have previously demonstrated that Au–Pd bimetallic nanoparticles can be very active for the alcohol oxidation reaction, and provide high selectivity toward aldehydes even under solvent-free reaction conditions.4–8 Au–Pd catalysts are generally prepared via co-impregnation4 or sol-immobilization5–8 methods, the latter of which provides much better control over the particle size and allows the possibility of tuning the particle morphology at will, to generate core–shell structures or homogeneous random Au–Pd alloys.9 However previously, in order to achieve high selectivity towards the aldehyde, either a low reaction temperature approach was chosen resulting in a low turnover frequency (TOF), or the conversion was kept at a low level. Herein, using benzyl alcohol oxidation as an example, we demonstrate that when a small amount of Pt metal is alloyed into the Au–Pd sols, a high selectivity toward benzaldehyde can be achieved in the solvent-free oxidation of benzyl alcohol reaction while still preserving high conversion levels.
Experimental details
Catalyst preparation – Sol-immobilisation method
Au-only, Pd-only, Pt-only monometallic catalysts, as well as Au–Pd, Au–Pt, Pt–Pd bimetallic catalysts and Au–Pd–Pt trimetallic catalysts supported on activated carbon were prepared using a colloidal method. For the synthesis of the trimetallic Au–Pd–Pt supported catalysts, an aqueous solution of PdCl2, HAuCl4·3H2O and H2PtCl6 (Johnson Matthey, 99.9% purity) of the desired concentration was prepared. To this solution, polyvinyl alcohol (PVA) (1 wt % solution, Aldrich, weight average molecular weight Mw = 9000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 80% hydrolysed) was added (PVA/Au (wt/wt) = 1.2). Subsequently, 0.1 M of freshly prepared NaBH4 solution (>96% purity, Aldrich, NaBH4/Au (mol/mol) = 5) was then introduced to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilised by adding the activated carbon (G60 Aldrich) support material (acidified to pH 1 using sulfuric acid) under vigorous stirring conditions. The amount of support material required was calculated so as to give a total final metal loading of 1% wt. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and then dried at 120 °C overnight.
000 g mol−1, 80% hydrolysed) was added (PVA/Au (wt/wt) = 1.2). Subsequently, 0.1 M of freshly prepared NaBH4 solution (>96% purity, Aldrich, NaBH4/Au (mol/mol) = 5) was then introduced to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilised by adding the activated carbon (G60 Aldrich) support material (acidified to pH 1 using sulfuric acid) under vigorous stirring conditions. The amount of support material required was calculated so as to give a total final metal loading of 1% wt. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and then dried at 120 °C overnight.
Catalyst preparation – Impregnation method
For comparative purposes Au-only, Pd-only and Au–Pd, Pt–Pd and Au–Pd–Pt catalysts were also prepared supported on titania (P25 Degussa) or carbon (G60 Aldrich), using a conventional wet impregnation technique. The trimetallic Au–Pd–Pt material was prepared by impregnation of the support using aqueous solutions of PdCl2, HAuCl4·3H2O and PtCl2 (Johnson Matthey). For instance, the detailed procedure for the preparation of 2 g of the 5 wt% (2.27Au:2.27Pd:0.45Pt)/TiO2 catalyst is as follows: PdCl2 (75.7 mg) was dissolved in a stirred and heated aqueous solution (4.55 ml) of HAuCl4·3H2O (5 g in 250 ml water) and an aqueous solution (0.09 ml) of PtCl2 (1 g in 10 ml water). The resultant solution was added to TiO2 (1.9 g) and the slurry formed was dried at 110 °C for 16 h. The recovered powder was ground and calcined (1 g, 6 inch quartz boat) in static air at 400 °C for 3 h at a ramp rate of 20 °C min−1.
Benzyl alcohol oxidation was carried out in a stirred reactor (100 ml, Parr reactor). The vessel was charged with alcohol (40 ml) and catalyst (0.05 g). The autoclave was then purged 5 times with oxygen leaving the vessel at 10 bar gauge. The stirrer was set at 1500 rpm and the reaction mixture was raised to the required temperature. Samples from the reactor were taken periodically via a sampling system. For the analysis of the products, a GC-MS and GC (a Varian star 3400 cx with a 30 m CP-Wax 52 CB column) were employed. The products were identified by comparison with known standards. For the quantification of the amounts of reactants consumed and products generated, the external calibration method was used.
Catalyst characterisation
The aqueous sols, contained in a quartz cuvette, were optically characterised using a UV-vis spectrometer (V-570, JASCO) operating over the 200 to 900 nm wavelength range. Samples for examination by scanning transmission electron microscopy (STEM) were prepared by dispersing the dry catalyst powder onto a holey carbon film supported by a 300 mesh copper TEM grid. STEM high angle annular dark field (HAADF) images of the metallic particles were obtained using an aberration corrected JEOL 2200FS (S)TEM operating at 200 kV. X-ray energy dispersive (XEDS) spectra were acquired from individual nanoparticles larger than 2 nm in size while rastering the beam over the entire particle, using a Thermo Noran Si(Li) XEDS detector. STEM-XEDS spectrum imaging experiments were carried out at 200kV using an aberration corrected JEM-ARM 200F microscope equipped with a JEOL Centurio SDD XEDS spectrometer.
Results and discussion
Supported Au–Pd–Pt trimetallic catalysts were synthesized by the sol-immobilization method as described above. This process involved the co-reduction of three metal precursors (HAuCl4, PdCl2 and H2PtCl6) at the desired concentration in aqueous solutions, using sodium borohydride as chemical reductant and polyvinyl alcohol (PVA) as the protective ligand. The synthesised Au–Pd–Pt colloids were analysed by UV-vis spectrometry (Fig. 1) and the spectrum obtained did not show a characteristic plasmon resonance band for gold, suggesting the formation of trimetallic alloy particles. The colloids were then immobilized onto an activated carbon support, followed by a washing and 120 °C drying process to yield the final catalysts with 1 wt% total metal loading. For comparative purposes Au-only, Pd-only, Pt-only catalysts, as well as Au–Pd, Au–Pt and Pt–Pd bimetallic catalysts were also prepared using a similar sol-immobilization procedure.
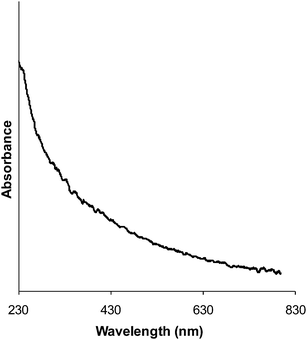 |
| | Fig. 1 Typical UV-vis spectrum from a Au–Pd–Pt aqueous sol preparation (0.45:0.45:0.1 by weight in this case). | |
Oxidations were carried out in a stirred autoclave reactor using solvent-free conditions (40 ml of benzyl alcohol), at 120–140 °C, and pO2 = 10 bar. The catalytic results are given in Table 1, and Table 2 presents the catalytic data at iso-conversion levels. The oxidation of benzyl alcohol with O2 under solvent-free conditions proceeds efficiently with very high catalytic activity using C supported Au–Pd nanoparticles, with TOF values of 50000 to 60000 h−1 being reached. The main product is benzaldehyde and the major by-product is toluene, which primarily comes from the disproportionation of benzyl alchohol10–12 as well as via the hydrogenolysis pathway.13 It is considered that Pd sites are responsible for the disproportionation reaction.10–12 However, we have recently reported that this disproportionation reaction can be controlled to some extent by the choice of catalyst support when using Au–Pd catalysts, as the disproportionation of benzyl alcohol can be switched-off simply by using basic support materials (e.g. ZnO, MgO).8 While carbon supported Au–Pd nanoparticles were always found to catalyse the disproportionation reaction, we observed a very low selectivity towards toluene for carbon supported monometallic Pt and bimetallic Au–Pt (1.1%) and Pd–Pt (0.7%) catalysts. However these desirable selectivities were accompanied by low benzyl alcohol conversion levels of 2.8%, 9.7% and 14.8% respectively. Taking into account the fact that the highest conversion level was observed with the Au–Pd/C catalysts, it was natural to consider the possibility of maintaining high activity while simultaneously keeping the selectivity to toluene at a very low level, simply by incorporating Pt into the Au–Pd supported catalyst. Consequently, we synthesised a series of trimetallic Au–Pd–Pt nanoparticles supported on carbon in which the amount of Pt metal added was systematically varied. By comparing three trimetallic catalyst formulations, namely (0.3Au + 0.4Pd + 0.3Pt)/C, (0.4Au + 0.4Pd + 0.2Pt)/C, (0.45Au + 0.45Pd + 0.1Pt)/C, it is clear from Table 1 that, by decreasing the amount of Pt incorporated, an increase in benzyl alcohol conversion was observed, whereas the selectivity to toluene remained below 1% in all cases. The highest selectivity toward benzaldehyde, measured at iso-conversion levels, was found for the 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C catalyst where the formation of toluene was almost completely switched-off. These results clearly demonstrate that only a small amount of Pt is needed for the effective termination of toluene formation. The best of our knowledge, this is the first report of the effective blocking of toluene formation under solvent-free conditions using Au–Pd nanoparticles modified with the optimum amount of a third alloying component.
Table 1 Benzyl alcohol oxidation results after 2 h reaction with Pd-only, Pt-only, Au–Pd, Au–Pt, Pt–Pd and various Au–Pd–Pt catalysts supported on carbon prepared by the sol-immobilisation methoda
| Catalyst |
Conversion (%) |
Selectivity (%) |
TOF/h−1b |
| Toluene |
Benzaldehyde |
Benzoic acid |
Benzyl benzoate |
|
Reaction conditions: benzyl alcohol, 0.05g of catalyst, T = 120 °C, pO2 = 150 psi, stirring rate 1500 rpm.
Calculation of TOF (h−1) value was done after 0.5 h of reaction, and was based on the total metal loading.
|
| 1 wt% (0.65Au + 0.35Pd)/C |
71.1 |
4.0 |
69.8 |
19.9 |
6.3 |
52700 |
| 1 wt% (0.5Au + 0.5Pd)/C |
80.7 |
3.4 |
67.0 |
23.1 |
6.5 |
63800 |
| 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C |
35.4 |
0.9 |
83.3 |
7.9 |
7.9 |
16000 |
| 1 wt% (0.4Au + 0.4Pd + 0.2Pt)/C |
36.7 |
0.6 |
81.7 |
8.0 |
9.7 |
22100 |
| 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C |
53.9 |
0.1 |
80.2 |
13.1 |
6.6 |
31900 |
| 2 wt% (1Au + 1Pt)/C |
11.8 |
2.0 |
79.9 |
8.4 |
9.7 |
4770 |
| 1 wt% (0.5Au + 0.5Pt)/C |
9.7 |
1.1 |
84.9 |
3.1 |
10.9 |
8450 |
| 1 wt% (0.5Pd + 0.5Pt)/C |
14.8 |
0.7 |
84.7 |
5.7 |
8.9 |
9580 |
| 1 wt% Pt/C |
2.8 |
0.0 |
90.7 |
0.7 |
8.6 |
2910 |
| 1 wt% Pd/C |
59.3 |
6.8 |
74.7 |
10.4 |
8.1 |
12200 |
Table 2 Comparison of benzyl alcohol oxidation at iso-conversion of 30% with Pd-only, Au–Pd and various Au–Pd–Pt catalysts supported on carbon prepared by the sol-immobilisation methoda
| Catalyst |
Selectivity (%) |
| Toluene |
Benzaldehyde |
Benzoic acid |
Benzyl benzoate |
|
Reaction conditions: benzyl alcohol, 0.05g of catalyst, T = 120 °C, pO2 = 150 psi, stirring rate 1500 rpm.
|
| 1% (0.65Au + 0.35Pd)/C |
8.1 |
80.5 |
2.6 |
8.8 |
| 1% (0.5Au + 0.5Pd)/C |
6.9 |
81.5 |
3.6 |
8.0 |
| 1% (0.3Au + 0.4Pd + 0.3Pt)/C |
0.8 |
85.7 |
6.5 |
7.0 |
| 1% (0.4Au + 0.4Pd + 0.2Pt)/C |
0.6 |
83.3 |
6.8 |
9.3 |
| 1% (0.45Au + 0.45Pd + 0.1Pt)/C |
0.4 |
86.7 |
5.9 |
7.0 |
| 1% Pd/C |
5.6 |
78.7 |
5.8 |
9.9 |
Selected catalysts were characterized by aberration corrected scanning transmission electron microscopy (AC-STEM) in order to evaluate their microstructure. Fig. 2 shows representative low magnification STEM-HAADF images and the corresponding measured particle size distribution of various trimetallic (Au–Pd–Pt/C), bimetallic (Au–Pd/C, Au–Pt/C and Pt–Pd/C) and monometallic (Pt/C) catalyst samples analysed in this study. To produce each histogram 500–1000 particles were analysed in each survey. It was found that the trimetallic (Fig. 2a and b) and bimetallic (Fig. 2c–e) samples analyzed all have quite similar particle size distributions with mean diameters in the 2–3 nm range. Although some minor coarsening effects were noted as a result of the 120 °C drying process, especially for the 1% (0.5Au + 0.5Pt)/C (Fig. 2e) and 1% Pt/C samples (Fig. 2f), the vast majority of the particles in each sample resided in the 1–3 nm size range. For these reasons, particle size variation issues from sample-to-sample should not be too significant when comparing the catalytic activities of these samples.
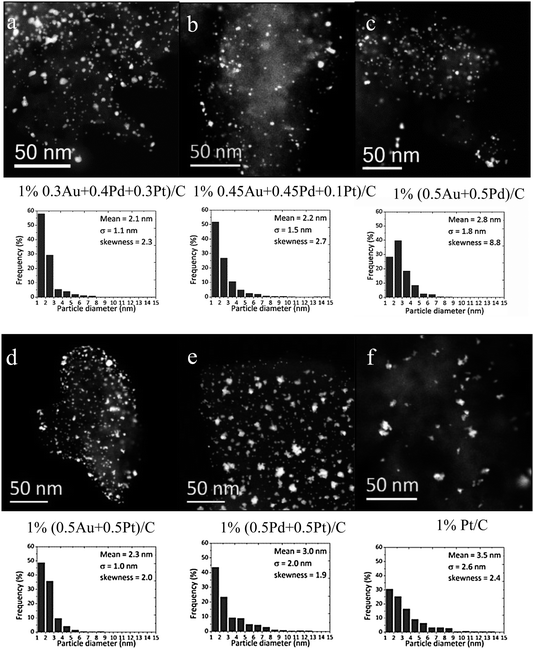 |
| | Fig. 2 Representative low magnification STEM-HAADF images and corresponding particle size distributions of selected trimetallic, bimetallic and monometallic catalysts characterized in this study; namely; (a) 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C; (b) 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C; (c) 1 wt% (0.5Au + 0.5Pd)/C; (d) 1 wt% (0.5Au + 0.5Pt)/C; (e) 1 wt% (0.5Pd + 0.5Pt)/C and (f) 1 wt% Pt/C. | |
It proved possible to collect reasonable quality X-ray energy dispersive point spectra (XEDS) from individual nanoparticles larger than about 2 nm in size in our JEM 2200FS microscope which was equipped with a standard Si(Li) XEDS detector.14 Representative STEM-HAADF and XEDS data from small (∼2 nm), intermediate size (∼5 nm), and larger (∼10 nm) metal particles are shown in Fig. 3a–c respectively for the most active 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C catalyst. From the relative intensity ratios between the overlapping Au and Pt M series and Pd L series, a clear size-dependent composition variation can be seen. The larger (∼10 nm) particles tend to be Pd-rich, while the smaller (∼2 nm) particles were consistently found to be Pd-deficient. This same general size/composition trend was also noted for all the Pd-containing alloys analyzed in the current study; (i.e. the 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C trimetallic sample (Fig. 4); the 1 wt% (0.5Pd + 0.5Pt)/C bimetallic sample (Fig. 5); and the 1 wt% (0.5Au + 0.5Pd)/C bimetallic sample (Fig. 6). It is interesting to note that no comparable composition/size variation was found for the particles in the Au–Pt binary system (Fig. 7). It has been reported previously by Braidy et al.15 that similar size-dependent composition variations in bimetallic Au–Ag nanoparticles can be generated as an artifact in the electron microscope if one of the constituent elements has a tendency to be depleted preferentially from the particle by electron beam bombardment. Since only Pd, but not Au or Pt, has a knock-on damage threshold below 200 keV,16 we examined the possibility of beam damage occurring by taking two consecutive XEDS spectra from a single Au–Pd–Pt particle about 2 nm in size. As shown in Fig. 8, even though significant structural modification can be clearly seen from the consecutive HAADF images, the two sequentially obtained XEDS spectra gave similar composition results within the limits of experimental error. This suggests that the composition variation noted in our study is inherent to the material itself and not an artifact of electron beam damage. This particle size/composition trend with respect to Pd concentration is also consistent with XEDS composition measurements presented in our previous study of the sol-immobilized bimetallic Au–Pd system.14 Interestingly, as shown in Fig. 3 and 4 for the Au–Pt–Pd trimetallic samples, and Fig. 7 for the bimetallic 1 wt% 0.5Au + 0.5Pt/C sample, the Pt L series peak can be detected in all spectra and the relative intensities of the Au Lα (9.71 keV) to Pt Lα (9.44 keV) peaks was found to be relatively constant with particle size, suggesting that no systematic particle size/composition variation develops for this particular pair of elements.
 |
| | Fig. 3 High magnification STEM-HAADF images and the corresponding X-ray energy dispersive spectra acquired from representative (a) 2 nm, (b) 5 nm and (c) 10 nm particles in the sol-immobilized 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C catalyst dried at 120 °C. | |
 |
| | Fig. 4 Representative high magnification STEM-HAADF images and the corresponding X-ray energy dispersive spectra acquired from representative (a) 2 nm, (b) 5 nm and (c) 10 nm particles in the sol-immobilized 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C trimetallic catalyst dried at 120 °C. | |
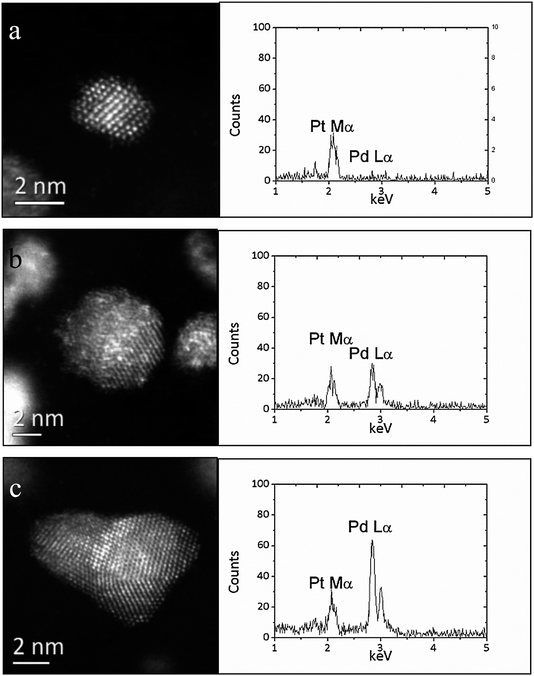 |
| | Fig. 5 Representative high magnification STEM-HAADF images and the corresponding X-ray energy dispersive spectra acquired from representative (a) 2 nm, (b) 5 nm and (c) 10 nm particles in the sol-immobilized 1 wt% (0.5Pd + 0.5Pt)/C bimetallic catalyst dried at 120 °C. | |
 |
| | Fig. 6 Representative high magnification STEM-HAADF images and the corresponding X-ray energy dispersive spectra acquired from representative (a) 2 nm, (b) 5 nm and (c) 10 nm particles in the sol-immobilized 1 wt% (0.5Au + 0.5Pd)/C bimetallic catalyst dried at 120 °C. | |
 |
| | Fig. 7 Representative high magnification STEM-HAADF images and the corresponding X-ray energy dispersive spectra acquired from representative (a) 2 nm and (b) 5 nm and (c) 10 nm particles in the sol-immobilized 1 wt% (0.5Au + 0.5Pt)/C bimetallic catalyst dried at 120 °C. | |
 |
| | Fig. 8 (a) An HAADF image and the corresponding XEDS spectra from a 2 nm Au–Pd–Pt particle in a 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C trimetallic catalyst prepared by the sol-immobilization method; (b) A second consecutive HAADF image and corresponding XEDS spectra of the same particle. A comparison of these two datasets indicates that the composition quantification from XEDS spectra was not significantly affected by electron beam damage under the experimental conditions employed. | |
The elemental distributions within individual particles, which have a profound effect on the resultant catalytic activities, can also be probed using STEM. It has been demonstrated by our group9 and others17–19 that Pd (Z = 46) segregation with respect to Au (Z = 79) or Pt (Z = 78) in multi-metallic nanoparticles can be easily distinguished by STEM-HAADF imaging, which provides mass-thickness contrast approximately proportional to Z2. From the HAADF images presented in Fig. 3, 4, 5, 6 and 7 no Pd segregation or chemical ordering was detected within trimetallic or bimetallic alloy nanoparticles containing Pd, suggesting that the Pd atoms were intimately mixed with the Au and Pt components. Indeed, this homogeneous alloy morphology is to be expected for materials generated by the co-reduction colloidal route and is consistent with our previous studies.9,14 Although the HAADF Z-contrast between Au and Pt is minimal, the relative distribution of Au and Pt within individual alloy nanoparticles can in principle be probed by STEM-XEDS mapping. However, in practice, small nanoparticles are rapidly damaged when subjected to an intense electron beam, whereas the use of low probe currents provides insufficient X-ray generation for XEDS analysis. Furthermore, conventional Si(Li) XEDS detectors only have a very small solid angle of collection (i.e. ∼0.13 sr) which captures less than 1% of the total characteristic X-rays emitted. For these two reasons, STEM-XEDS mapping on nanoparticles smaller than about 6 nm is extremely challenging in our aberration corrected JEOL 2200FS (S)TEM. This situation has dramatically improved recently with the installation of our aberration corrected JEM-ARM 200F (S)TEM which is equipped with a Centurio SDD XEDS spectrometer, having a collection solid angle of 0.9 sr, which represents a 5–10 fold improvement in X-ray collection efficiency.20 Therefore it has now become feasible to routinely acquire STEM-XEDS mapping data on nanoparticles as small as 2–3 nm. Fig. 9 shows representative STEM-XEDS spectrum images of 3 nm, 5 nm and 10 nm diameter alloy particles in the 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C sol-immobilized catalyst taken using this new instrument. This sample was deliberately chosen, instead of the most active 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C catalyst, because it had the highest Pt loading allowing the distribution of all three elements to be more clearly visualized. These results conclusively demonstrate that regardless of size, the particles are all homogenous trimetallic alloys which show no apparent signs of elemental segregation. It is also reasonable to extrapolate that even for the catalyst with the most dilute Pt loading (i.e. 1 wt% (0.45Au + 0.45Pd + 0.1Pt)/C), the Pt atoms get randomly incorporated into the Au–Pd nanoparticles. Even though a wide miscibility gap exists in the ‘bulk’ binary phase diagram for the Au–Pt binary system,21 both experimental22 and theoretical23,24 investigations now suggest that a wide range of homogeneous alloy compositions between Pt, Au and Pd can be prepared and stabilized at low temperature in the nano-regime.
 |
| | Fig. 9 STEM-XEDS spectrum image analysis of the 1 wt% (0.3Au + 0.4Pd + 0.3Pt)/C trimetallic sample. The first column shows HAADF images from representative particles that are (a) ∼3 nm, (b) ∼5 nm, and (c) ∼10 nm in size. The scale bar represents 2 nm. The next three columns show the corresponding XEDS maps derived from the Au L (9.64–9.84 keV), Pd L (2.75–3.05 keV) and Pt L (9.35–9.55 keV) peaks. The final column shows the corresponding RGB overlay maps where Au is red, Pd is green and Pt is blue. | |
The beneficial effects of adding some Pt into Au–Pd nanoparticles, in terms of activity and selectivity for the benzyl alcohol oxidation reaction, are quite remarkable. The incorporated Pt atoms seem to act as either as an electronic or strain modifier to the Au–Pd nanoparticles, which nullifies the Pd sites that are normally active for disproportionation reactions. One issue that hinders a more detailed mechanistic understanding of this process is that the co-reduction colloidal preparation route employed provides only limited composition control as the larger particles are consistently Pd-rich, while the smaller ones are always Pd-deficient. Therefore although the nominal composition of the trimetallic catalysts can be modified by adjusting the molar ratios of the three metal precursors, the resultant materials still contain a complex distribution of particle sizes and compositions. One possible cause of such size dependent composition variations in nanoalloys was proposed by Alloyeau et al.,25 who found that in Pt–Co nanoparticles, the Co has a higher rate of Ostwald ripening than the Pt component on the surface of the carbon support, causing the Co to preferentially segregate onto larger particles after high temperature (c.a. 700 °C) heat treatments. If this type of effect were happening in our situation, we suspect that the selective Ostwald ripening process for Pd is more likely to occur in the solution phase during the sol synthesis process instead of while on the carbon support after immobilization, because the catalyst only underwent a very mild drying treatment at 120 °C. Hence, the ability to synthesize trimetallic Au–Pd–Pt nanoparticles where there is a tighter control over the composition from particle-to-particle, while preserving the mean particle size below 5 nm, still remains as a significant technical challenge.
Finally, we have also extended the scope of using Pt as a third alloying element by synthesizing trimetallic Au–Pd–Pt nanoparticles supported on C and TiO2 using a conventional impregnation method. This synthesis method would be preferred by industry due to its greater simplicity, so we investigated if the same catalytic trends could be obtained with impregnation. The catalytic results for the impregnated catalysts are presented in Table 3, and it is evident that the impregnated trimetallic catalysts also significantly decreased the formation of toluene. However, it should be noted that the catalytic activity of the impregnated supported Au–Pd–Pt catalysts are much lower than the corresponding sol-immobilized catalysts, probably due to (i) a significant increase in mean particle size and (ii) wider composition variations as compared to the colloid derived materials.
Table 3 Benzyl alcohol oxidation results after 3 h reaction with, Au–Pd, Pt–Pd and various Au–Pd–Pt catalysts supported on carbon and titania as prepared by the impregnation method (calcined at 400 °C in air)a
| Entry |
Catalyst |
Conversion (%) |
Selectivity (%) |
TOF/h−1b |
| Toluene |
Benzaldehyde |
Benzoic acid |
Benzyl benzoate |
|
Reaction conditions: benzyl alcohol, 0.025g of catalyst, T = 140 °C, pO2 = 150 psi, stirring rate 1500 rpm.
Calculation of TOF (h−1) value was done after 0.5 h of reaction, and was based on the total metal loading.
|
| 1 |
5% (2.5Au + 2.5Pd)/TiO2 |
45.1 |
13.6 |
74.2 |
9.4 |
2.8 |
6420 |
| 2 |
5% (1.67Au + 1.67Pd + 1.67Pt)/TiO2 |
8.95 |
0.4 |
94.5 |
2.8 |
2.2 |
1410 |
| 3 |
5% (2.5Au + 2.5Pd + 0.5Pt)/TiO2 |
18.6 |
2.7 |
86.9 |
3.3 |
7.1 |
2470 |
| 4 |
5% (2.27Au + 2.27Pd + 0.45Pt)/TiO2 |
10.6 |
1.8 |
86.1 |
5.1 |
7.0 |
1150 |
| 5 |
5% (2.5Pd + 2.5Pt)/TiO2 |
8.6 |
1.1 |
91.3 |
4.3 |
3.3 |
1220 |
| 6 |
5% (2.5Au + 2.5Pd)/C |
55.0 |
16.6 |
79.7 |
1.8 |
1.9 |
7830 |
| 7 |
5% (2.27Au + 2.27Pd + 0.45Pt)/C |
6.2 |
2.7 |
87.9 |
4.8 |
4.6 |
910 |
Conclusions
In summary, Au–Pd–Pt/C trimetallic catalysts were successfully synthesized using the sol-immobilization method. Aberration corrected STEM characterization reveals that the nanoparticles were tightly controlled in size, with mean diameters around 2–3 nm, and they were all random ternary alloys. However, a systematic size-dependant composition variation with respect to Pd was observed, as larger particles were always found to be Pd-rich and smaller particles were consistently Pd-deficient. It was shown that the addition of a small amount of Pt can significantly enhance the selectivity of benzaldehyde in the solvent-free oxidation of benzyl alcohol reaction, while still maintaining a high alcohol conversion.
References
-
W. J. Mijs and C. R. H. I. de Jonge, Organic syntheses by oxidation with metal compounds, New York, Plenum Press, 1986 Search PubMed.
-
R. A. Sheldon and J. K. Kochi, Metal-catalyzed oxidations of organic compounds: mechanistic principles and synthetic methodology including biochemical processes, Academic Press, New York, 1981 Search PubMed.
-
G. Cainelli and G. Cardillo, Chromium oxidations in organic chemistry, Springer-Verlag, Berlin; New York, 1984 Search PubMed.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142–5153 RSC.
- J. C. Pritchard, Q. He, E. N. Ntainjua, M. Piccinini, J. K. Edwards, A. A. Herzing, A. F. Carley, J. A. Moulijn, C. J. Kiely and G. J. Hutchings, Green Chem., 2010, 12, 915–921 RSC.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem.–Eur. J., 2011, 17, 6524–6532 CrossRef CAS.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63–86 RSC.
- S. Meenakshisundaram, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. W. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341–356 RSC.
- S. S. Hladyi, M. K. Starchevsky, Y. A. Pazdersky, M. N. Vargaftik and I. I. Moiseev, Mendeleev Commun., 2002, 12, 45–46 CrossRef.
- A. Villa, D. Wang, N. Dimitratos, D. S. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15 CrossRef CAS.
- J. Chen, Q. H. Zhang, Y. Wang and H. L. Wan, Adv. Synth. Catal., 2008, 350, 453–464 CrossRef CAS.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS.
- N. Braidy, Z. J. Jakubek, B. Simard and G. A. Botton, Microsc. Microanal., 2008, 14, 166–175 CrossRef CAS.
- R. F. Egerton, P. Li and M. Malac, Micron, 2004, 35, 399–409 CrossRef CAS.
- D. Ferrer, D. A. Blom, L. F. Allard, S. Mejia, E. Perez-Tijerina and M. Jose-Yacaman, J. Mater. Chem., 2008, 18, 2442–2446 RSC.
- D. Ferrer, A. Torres-Castro, X. Gao, S. Sepulveda-Guzman, U. Ortiz-Mendez and M. Jose-Yacaman, Nano Lett., 2007, 7, 1701–1705 CrossRef CAS.
- M. R. Ward, T. Hyde, E. D. Boyes and P. L. Gai, ChemCatChem, 2012, 4, 1622–1631 CrossRef CAS.
- M. Watanabe, A. Yasuhara and E. Okunishi, Microsc. Microanal., 2012, 18, 974–975 CrossRef.
- Z. M. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS.
- N. Braidy, G. R. Purdy and G. A. Botton, Acta Mater., 2008, 56, 5972–5983 CrossRef CAS.
- H. B. Liu, U. Pal and J. A. Ascencio, J. Phys. Chem. C, 2008, 112, 19173–19177 CAS.
- K. Yun, Y. H. Cho, P. R. Cha, J. Lee, H. S. Nam, J. S. Oh, J. H. Choi and S. C. Lee, Acta Mater., 2012, 60, 4908–4916 CrossRef CAS.
- D. Alloyeau, G. Prevot, Y. Le Bouar, T. Oikawa, C. Langlois, A. Loiseau and C. Ricolleau, Phys. Rev. Lett., 2010, 105, 255901 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a