Effects of dominant material properties on the stability and transport of TiO2 nanoparticles and carbon nanotubes in aquatic environments: from synthesis to fate
Xuyang
Liu
*a,
Gexin
Chen
a,
Arturo A.
Keller
b and
Chunming
Su
*c
aNational Research Council Resident Research Associate at the U.S. Environmental Protection Agency, 919 Kerr Research Drive, Ada, Oklahoma 74820, USA. E-mail: liu.xuyang@epa.gov; Tel: +1 580 436-8803
bSchool of Environmental Science and Management, University of California, Santa Barbara, CA 93106, USA
cGround Water and Ecosystems Restoration Division, National Risk Management Research Laboratory, Office of Research and Development, U.S. Environmental Protection Agency, 919 Kerr Research Drive, Ada, Oklahoma 74820, USA. E-mail: su.chunming@epa.gov; Fax: +1 580 436-8703; Tel: +1 580 436-8638
First published on 10th December 2012
Abstract
Recently, increasing studies have focused on the environmental stability, transport, and fate of the anthropogenic nanomaterials in the environment, which contributes to the understanding of the potential risks when released. However, applying nanomaterials from different manufacturers and production methods tends to result in inconsistent experimental data and potentially a biased comparison. The aim of this review is to investigate the dominant material properties that determine the aggregation and deposition behavior of nanomaterials. Herein, we focus on two of the most popular anthropogenic nanomaterials, i.e., titanium dioxide (TiO2) and carbon nanotubes (CNTs). We start from the production methods of nanomaterials of different sources, and then examine their influence on the material properties and surface characteristics. The role of the material properties was carefully analyzed and correlated with the stability and transport in aquatic environments. These two case studies may be extended to other nanomaterials with similar surface properties, which will improve our understanding of the impact and risks of anthropogenic nanomaterials in the environment. This study highlights opportunities to design and produce “green” nanomaterials with less environmental risk and no sacrificing of the novel “nano” properties.
 Xuyang Liu | Xuyang Liu (PhD Stevens Institute of Technology, 2009) is a National Research Council resident research associate at the U.S. Environmental Protection Agency. He has been working at the EPA on the National Nanotechnology Initiative sponsored project since he finished his dissertation on the “aggregation and deposition of energetic nanomaterials in Aqueous Environments”. His research interests are environmental nanotechnology, colloidal chemistry, water treatment, and soil and groundwater remediation. |
 Gexin Chen | Gexin Chen (PhD University of California, Riverside, 2009) is a National Research Council resident research associate at the U.S. Environmental Protection Agency. His research interests include environmental implications and applications of nanotechnology, bioremediation, and water treatment. In particular, his work has focused on understanding the factors controlling bacterial and colloidal particle adhesion and transport in natural and engineered aquatic systems. |
 Arturo A. Keller | Arturo A. Keller (PhD Stanford, 1996) is currently the Co-Director of the UC Center for the Environmental Implications of Nanotechnology. He has been a Professor at the Bren School of Environmental Science and Management, University of California, Santa Barbara for 16 years. His research addresses the fate and transport of pollutants in the environment, remediation approaches, and watershed scale management of water quality. Keller's recent work has focused on the nanoscale, both in terms of the implications and the applications of these novel materials. He received his PhD from Stanford University in Civil and Environmental Engineering; he has a previous MS from Stanford and a BS in Chemical Engineering and a BA in Chemistry from Cornell University. He also worked in industry for 11 years. He has over 115 peer-reviewed publications in top journals. Contact information: E-mail: keller@bren.ucsb.edu. |
 Chunming Su | Chunming Su (PhD Washington State University, 1992) is a soil scientist at the U.S. Environmental Protection Agency (EPA), Office of Research and Development (ORD), National Risk Management Research Laboratory (NRMRL), Ground Water and Ecosystems Restoration Division (GWERD). His research has focused on the applications and implications of environmental nanotechnology and in situ groundwater remediation. He has been investigating the fate and transport of engineered nanomaterials and evaluating applications of nanoscale zerovalent iron in treatment of groundwater contaminants. He is a principal author of more than 40 peer-reviewed journal articles and book chapters, and a co-inventor of a U.S. patent. |
Environmental impactIn the current literature, inconsistent experimental data can be found on the fate and transport of anthropogenic nanomaterials (NMs). This study aims to fill the gap of NM production methods and the environmental behavior, and to reconcile the conflicting data from a novel perspective of material properties. Herein, we start the investigation from the synthesis processes of two representative anthropogenic NMs, i.e., titanium dioxide and carbon nanotubes, and focus on the influence of physicochemical properties on the environmental stability and transport. Hence, this study will initiate discussions within the general audiences of Environmental Science: Processes & Impacts on the key material properties determining the environmental safety of anthropogenic NMs, which highlights opportunities to design “green” NMs. |
1 Introduction
The emergence of nanotechnology has the potential to lead to significant advances in many technology sectors, and contribute to substantial economic growth.1 It is estimated that nanotechnology enabled products will achieve a $3 trillion market with six million workers by 2020.2 Besides its economic impact, nanotechnology brings potentially negative effects of nanomaterials (NMs) on the environment and human health. A priority list of anthropogenic NMs for evaluation has been published by the Organization for Economic Co-operation and Development (OECD) based on materials in, or close to, commerce.3 The listed NMs include carbon-based NMs [i.e., C60, single walled carbon nanotubes (SWNTs) and multi walled CNTs (MWNTs)], metal oxides (i.e., TiO2, CeO2, ZnO, SiO2, Al2O3, iron oxide), metals (Ag, Fe, Cu, Au), and quantum dots.Huge gaps exist regarding the environmental implications of anthropogenic NMs, although the quantity of relevant publications has increased in the past few years. However, some of the studies present seemingly conflicting results. For instance, the values of point of zero charge (PZC) of TiO2 varied in a wide range of 2–7 in aqueous solution.4–6 Such conflicting results are probably caused by the large variation of material properties of the same type of NMs due to varying manufacturing processes as well as insufficient characterization.7 Herein, by reviewing the available data in the literature, we investigated two representative anthropogenic NMs, i.e., TiO2 and CNTs, from the perspective of material physicochemical properties, and correlated them with the stability and transport behavior in the environment. Different methods of NM synthesis were analyzed to evaluate the extent to which certain properties determine the stability and transport behavior of the NMs. By doing so, we try to improve our understanding of the life cycle impacts of anthropogenic NMs and to reconcile seemingly contradictory data from different publications. Meanwhile, we provide information that can be used in future studies to design NMs in a greener and safer way without sacrificing their novel functions and applications.8
2 Methods
Published studies considering different methods of NM synthesis and suspension preparation were used in this study (as detailed in the next section), since we assume that the synthesis methods affect the key properties of the NMs, such as primary size and morphology, chemical composition/impurities, crystallinity, specific surface area (SSA), and functional groups. The characteristics of the target NMs under various solution chemistries were summarized, including electrophoretic mobility (EPM) (or ζ potential), aggregate size, PZC, etc. Following the existing characterization data, the aggregation, sedimentation, deposition, and transport of the NMs were compared under similar solution chemistries (Fig. 1). The reported data on nanoparticle (NP)–NP and NP–media interactions were listed in parallel; however, the involved heterogeneities of collector surface and experimental approaches in NP–media interactions are more complex than NP–NP interactions. Compared with other NMs like silver and zinc oxide,9–11 the transformation data on TiO2 NPs and CNTs in the environment are very limited in the literature,12–14 and therefore their transformation processes will not be highlighted in this brief review.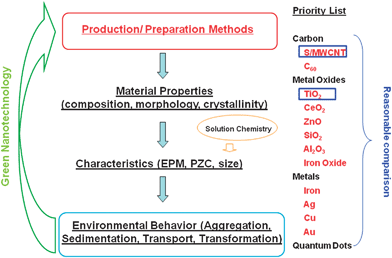 | ||
| Fig. 1 The research strategy steps from synthesis methods to material properties, characteristics, and behavior of NMs in the environment. Feedback from the environmental behavior can serve to guide the production process so that safer NMs are manufactured. | ||
3 Case analysis on TiO2 NPs
3.1 Production process and sources of TiO2 NPs
Commercial TiO2 is generally synthesized by the sulfate process or the increasingly popular chloride process, which can be used to produce both conventional and nano-scale TiO2.15 Ilmenite (FeTiO3) is most economically handled in the sulfate process because it can be easily dissolved in sulfuric acid.16 The chloride process, a gas-phase process, needs considerably less energy than the wet-phase sulfate process.17 Currently, the chloride process accounts for ∼60% of worldwide TiO2 pigment production.18 In addition, many new processes have been developed recently to produce TiO2 NPs, including sol–gel, micelle and inverse micelle, hydrothermal, solvothermal, direct oxidation, chemical vapor deposition (CVD), physical vapor deposition, etc.19 For studies concerning the environmental implication of TiO2 NPs, the most representative synthesis methods are sol–gel and hydrolysis of Ti(IV) salts.20–24 The sol–gel process is also used in industry by Nanogate (Gottelborn, Germany), while spray hydrolysis is used by Altairnano (Reno, Nevada).25,26 Other industrial scale methods include physical vapor synthesis (Nanophase, Romeoville, IL), laser pyrolysis (NanoGram, Milpitas, CA), and mechanical milling (Advanced Nanotech, New York, NY).25 However, some industrial manufacturers still use proprietary synthesis methods, e.g., Degussa (Evonik) and DuPont.25 The TiO2 NPs used in environmental research labs are mostly purchased from a few vendors, including Nano-Structured & Amorphous Materials, Inc. (or NanoAmor, Houston, TX),5,27–35 Evonik Degussa Corp. (NJ or Germany),36–53 and Sigma-Aldrich (St Louis, MO) (Tables 1 and 2).54–57 Although undisclosed synthesis methods in industry pose challenges to the following analysis, an attempt was made to correlate the properties of TiO2 from various sources to the environmental behavior to clarify the current status of understanding of the property–behavior relationship.| Source, production, or preparation methods | Material properties (crystallinity, morphology, composition) | Solution chemistry | Characteristics (EPM, aggregate size, etc.) | Main finding and conclusions | Ref. |
|---|---|---|---|---|---|
| Hydrolysis of titanium ethoxide | Primary particle size: ∼150 nm, crystalline size: on the order of 10 nm, dH: 270–330 nm (>90%) | At pH 6.3, 6.7 and 8.4. | IEP: pH 6.1 | Reasonable agreement between the DLVO theory and experiment could be obtained if an effective interaction radius was used; this corresponds to the size of surface crystallites formed during synthesis. | Snoswell et al. 2005 (ref. 20) |
| Controlled hydrolysis | Primarily anatase (70–100%) with some brookite and rutile, crystalline diameter of 5–12 nm for anatase | At pH 1, 3, 7, 10, and 12 | PZC3.6 nm particles = 4.8, PZC8.1 nm particles = 6.2 | PZC of TiO2 changes with nanocrystalline size, with smaller sizes exhibiting lower PZC. The pH, and therefore, surface potential and aggregate size, dominate NP interactions with each other. | Guzman et al. 2006 (ref. 21) |
| Sol–Gel method | Anatase (63.3%)/brookite (36.7%), crystalline size: 5.5 nm for anatase and 4.4 nm for brookite. | At pH 4.5, in NaCl | PZC = 6.8. | Divalent cations may enhance aggregation of nano-TiO2 in soils and surface waters. Branching aggregate sizes ranged 1–10 μm in diameter. | French et al. 2009 (ref. 22) |
| Gaosida Nanomaterial (China) | Anatase, purity: 99.9%, SSA: 32.5 m2 g−1, average size: 35 nm | Twelve surface soils from China, at circumneutral pH | PZC: 4.5, ζ potential: −20.7 to −25.8 mV at pH 7–9. | The stability of TiO2 in soil suspensions was positively relevant to DOC and the clay content of soils, but was negatively correlated with IS, pH, and ζ potential. | Fang et al. 2009 (ref. 59) |
| Sigma-Aldrich | Nominal size: 15 nm | In 0.01 M KCl, 110 mg L−1 NOM, pH 7.8 | D H: 530 nm, ζ potential: −37 mV | 0.01 M KCl induced the aggregation; within 2 h dH increased to more than 1500 nm. In the presence of 1 mg L−1 NOM, TiO2 remained stable within 2 h, because NOM imparted negative charge to TiO2 surfaces. | Zhang et al. 2009 (ref. 54) |
| Aldrich | A mixture of rutile and anatase, size: <100 nm (BET), purity: 99.9% (metal basis) | In 20 mg L−1 SRNOM, TOC: 10 mg C L−1, IS: 7.5 mM NaCl | IEP: pH 4.5 | Depending on pH and TiO2 loading, the adsorption of NOM is controlled by the availability of divalent cations or pre-ozonation of NOM. Non-XDLVO intermolecular bridging by Ca2+ was probably influential. | Kim et al. 2009 (ref. 55) |
| Sigma-Aldrich, #634662 (St Louis, USA) | A mixture of anatase and rutile, purity: 99.9%, spherical and polydisperse, TEM diameter: 15–350 nm, SSA: 19.6 m2 g−1 | In cell culture media (RPMI), containing low amounts of amino acids, vitamins, and glucose | ζ potential: −16 mV at pH 7.4, DH: 793 nm (5 mg L−1) in RPMI media, IEP: pH 2.9 | The aggregation and sedimentation rates for TiO2 increased with NP concentration. The presence of fetal bovine serum (FBS) and human serum albumin (HAS) notably enhanced the stability of TiO2 NPs in suspensions. | Allouni et al. 2009 (ref. 56) |
| Nano-Structured & Amorphous Materials, Inc., (Houston, TX) | Anatase with a nominal size of 5 nm | In 1.0 mg L−1 SRFA, IS = 0.01 M NaNO3, pH 4.0 | EPM: 2.2 μm cm V−1 s−1, PZC: 4.5–5.2 | Aggregation of bare TiO2 increased at pH values near PZC or at high IS. Adsorption of SRFA resulted in less aggregation of TiO2 NPs, presumably due to increased steric repulsion. | Domingos et al. 2009 (ref. 27) |
| Nano-Structured & Amorphous Materials, Inc., (Houston, TX) | Anatase with a nominal size of 5 nm | At pH 2–8, IS = 0.01 M NaCl, SRFA: 1–5 mg L−1 | PZC: 4.5–5.2 | Ca2+ resulted in aggregation of TiO2 NPs, even in the presence of SRFA. Phosphate adsorption resulted in destabilization of TiO2, but for low SRFA concentrations only. | Domingos et al. 2010 (ref. 28) |
| Nano-Structured & Amorphous Materials, Inc., (Houston, TX) | Anatase with a primary size of 5 nm, SSA: 219 m2 g−1 (BET) | In 100 mM citric acid, at pH 2.0, 4.0, 6.0, and 7.5, IS = 0.03 M NaCl | PZC: 4.2, 9.7–3.1 × 1013 citric acid molecules cm−2 at pH 2.0–7.5 | Surface coverage of citric acid is a function of pH and decreases with increasing pH. After equilibration, the fully deprotonated citrate ion is present on the surface regardless of the highly acidic solution pH. | Mudunkotuwa et al. 2010 (ref. 29) |
| Nano-Structured & Amorphous Materials, Inc., (Houston, TX) | 5, 10, and 50 nm anatase; 10 × 40 nm, and 30 × 40 nm rutile | In NaCl or CaCl2 solutions, pH 7 | IEP: 6.0 (5, and 10 nm anatase), 4.4 (10 × 40 nm rutile), 4.7 (30 × 40 nm rutile), and 2.7 (50 nm anatase) | Crystallinity and morphology are not the dominant factors for the stability of TiO2. The chemical composition, or the impurity content of Si and P, is the dominant material property that determined the aggregation and sedimentation of TiO2 in an aquatic environment. | Liu et al. 2011 (ref. 5) |
| (1) NanoAmor, (Houston, TX); (2) Alfa Aesar (Ward Hill, MA) | (1) 5 nm anatase (100%), SSA: 219 m2 g−1; (2) 32 nm anatase (99.9%), SSA: 41 m2 g−1 | At pH 2 (0.01 N HCl), pH 5.5 and 6.5 (25 mM MES and HEPES buffer) | Minimal differences in the amount of adsorbed organic acid on the basis of SSA loading | The 5 nm TiO2 can aggregate to a greater extent than the 32 nm anatase particles, and the presence of organic acids diminished TiO2 suspension stability. | Pettibone et al. 2008 (ref. 30) |
| Degussa P25 and Hombikat UV-100 | Primary diameter: ∼21/<10 nm, SSA BET: 50/>250 m2 g−1, for P25 Evonik/UV-100, respectively. | In 1–10 mg L−1 NOM, 1–5 mM Na4P2O7, 0.1–100 mM NaCl, at 0.01–10 mM CaCl2 at pH 3.5–6.5 | D H: 293/302 nm, ζ potential: 20/19.1 mV, PZC: 5.6/4.8, for P25 Evonik/UV-100, respectively. | Regardless of a 5-fold difference in BET SSA, two tested TiO2 NPs behave in an overall comparable manner. | Kammer et al. 2010 (ref. 36) |
| Evonik Degussa Corp. (#4168063098, US) | Anatase (82%) and rutile (18%), primary size: 27 nm, morphology: semisphere, purity: 98.03, surface area: 51.5 m2 g−1 | Different ambient waters applied | IEP: 6.2, EPM in 1 mM KCl: 2.37 μm cm V−1 s−1), particle size in DI: 194 ± 7 nm | The EPM of the metal oxide NPs was dominated by the presence of NOM and ionic strength. NOM adsorbed onto these NPs significantly reduced the aggregation under many conditions. The transition from reaction to diffusion limited aggregation occurs at an EPM from −2 to −0.8 μm cm V−1 s−1. | Keller et al. 2010 (ref. 37) |
| Evonik Degussa Corp. (#4168063098, US) | Anatase (82%) and rutile (18%), primary size: 30 nm, morphology: semisphere, purity: 98.03, surface area: 51.5 m2 g−1 | In NaCl or CaCl2 solutions, IS = 1, 10, and 100 mM, at pH 5, 6, 7, 8 and 9; 10 mg L−1 SRHA | ζ potential: 20, and −50 mV at pH 4, and 9 in 1 mM NaCl | The CCC[Ca2+]/CCC[Na+] ratios were a proportionality factor of z−7.2 and z−5.6 in the absence and presence of SRHA, respectively. SRHA drastically increased the stability of TiO2 NPs under most conditions. | Thio et al. 2011 (ref. 38) |
| Evonik P25 (Evonik Industries, Germany) | Anatase (88%) and rutile (12%), purity: 99.9%, primary size: 19.8 nm, SSA (BET): 57 m2 g−1, density: 3.8 g cm−3, pH value (40 g L−1): 3.6 | NaCl, CaCl2, or NOM were added, titrated with HCl or NaHCO3, seven natural water samples applied | IEP shifted from pH 5.0 to pH 6.5 with increasing NaCl from 0.1 to 500 M. | The concentration of divalent ions, the pH values and the DOC concentrations would be the parameters with the strongest influence on aggregation behavior. | Ottofuelling et al. 2011 (ref. 39) |
| Evonik P25 (Germany) | Anatase (81%) and rutile (19%), primary size: 20–30 nm (TEM), SSA: 51.5 m2 g−1 (BET), purity: 98.03% | Cell culture media, at IS of 50–270 mM, with Na+, K+, Ca2+, Mg2+, PO43+, SO42+, and Cl− | In DI, ζ potential: 30.2 mV, DH: 209 nm; in cell culture media, DH: 770–1052 nm, ζ potential: ∼10 mV. | Bovine Serum albumin (BSA) improved the stability of TiO2 in cell culture media, and phosphate was one of the key factors. Fetal bovine serum (FBS) was an effective agent due to the synergistic effects of its multiple proteins. | Ji et al. 2010 (ref. 40) |
| P25, Evonik Degussa GmbH (Germany) | Nominal primary size: 21 nm | In fresh (pH 5.0, and 6.1) and brackish (pH ∼8) natural waters, TOC: 4.1–6.1 mg L−1; 1, 10 and 100 mg L−1 NP | D H: 200 nm in brackish water | The aggregation rate in brackish waters was clearly higher at higher TiO2 initial conc. (100 mg L−1). The aggregation is influenced by NP concentration, ionic content, organic substance and pH. | Sillanpää et al. 2010 (ref. 41) |
| P25, Degussa, Germany | Non-porous spheres, D: 30 nm, SSA: 35–45 m2 g−1 | In SRFA, IS = 5 mM NaCl, at pH 4, 6, and 8. | At 0/1.5 mg L−1 SRFA, ζ potential: 19.2/−35.0, −28.8/−42.7, and −31.4/−45.6 at pH 4, 6, and 8, respectively. | Fe(III) stabilized TiO2 NPs at pH 4 due to increased positive charge, but destabilized aggregation at pH 6 and 8 due to bridging. SRFA enhanced aggregation at pH 4, while it stabilized the NPs at higher pH values in the presence of both SRFA and Fe(III). | Li et al. 2011 (ref. 42) |
| P25, Evonik Degussa (USA) | Anatase (82%) and rutile (18%), primary size: 27 nm, SSA: 51.5 m2 g−1, purity: 98.03 | At pH 6.3, C: 100 mg L−1 in NanoPure water | D H: 250–300 nm, EPM: 2.37 × 10−8 m2 V−1 s−1, IEP: pH 6.2 | Sunlight and sonication could partially disagglomerate TiO2 clusters. A cycle of temperature change disagglomerated the compact clusters in the heating phase and reagglomerated NPs as more open fractal structures during the cooling phase. | Zhou et al. 2012 (ref. 43) |
| P25, Evonik Degussa (USA) | Anatase (82%) and rutile (18%), primary size: 27 nm, SSA: 51.5 m2 g−1, purity: 98.04 | In NaNO3, pH 4, and 8 | D H: slightly >200 nm, IEP: pH 6, CCC: 25 mM (pH 4), 10 mM (pH 8) | At pH 4, montmorillonite reduced the stability of both positively charged TiO2 NPs. Such enhanced coagulation only occurs within an intermediate IS range. | Zhou et al. 2012 (ref. 44) |
| Degussa P25, Evonik | Anatase (86.9%) and rutile (13.1%), primary size: 20–30 nm (XRD or SEM) | Salicylic acid concentration: 5–10 ppm, pH: 11 | D H: 100–130 nm (pH: 11), IEP: pH 5.7 and 5 (without/with salicylic acid) | Salicylic acid was shown to inhibit the aggregation of TiO2 NPs. The size of aggregates on the mica surface was smaller than hydrodynamic size measured by DLS. | Almusallam et al. 2012 (ref. 45) |
| Source, production, or preparation methods | Material properties (crystallinity, morphology, composition) | Solution chemistry and media properties | Characteristics (EPM, aggregate size, etc.) | Main finding and conclusions | Ref. |
|---|---|---|---|---|---|
| Controlled hydrolysis | Primarily anatase (70–100%) with some brookite and rutile, crystalline diameter of 5–12 nm for anatase | pH 1, 3, 7, 10, and 12 | PZC3.6 nm particles = 4.8, PZC8.1 nm particles = 6.2 | pH, and therefore, surface potential and aggregate size, dominate NP interactions with surface. | Guzman et al. 2006 (ref. 21) |
| Altair Nanomaterials Inc. (Reno NV) | Anatase, primary size: 40 nm | Spherical silicate glass beads from 300–425 μm (Particle Technology Ltd., Hatton, UK), IS: 0.01 M, pH 7.0 | PCS size: 198 nm, EPM: −0.27 × 10−8 m2 V−1 s−1, PZC: 6.1 | The transport of TiO2 NPs/aggregates was observed to be a function of velocity, which is consistent with predictions of particle deposition based on theory for particle transport and a constant attachment efficiency factor. | Lecoanet and Wiesner 2004 (ref. 65) |
| Anatase, Gaosida Nanomaterial (China) | Purity: 99.9%, SSA: 32.5 m2 g−1, average size: 35 nm | Twelve surface soils from China, at circumneutral pH | PZC: 4.5, ζ potential: −20.7 to −25.8 mV at pH 7–9. | A significant portion of TO2 (18.8–83.0%) passed through soil columns that contain large-size soil particles at low IS. TiO2 was significantly retained by soils with higher clay contents and salinity. | Fang et al. 2009 (ref. 59) |
| Alfa-Aeser 39953, acidified at pH 2.5 with HCl, and supernatant sampled. | Largely anatase with a trace quantity of rutile, primary size: 32 nm, surface area: 47.6 m2 g−1 | At pH 4, 6 or 8; IS = 1, 10, 40, and 100 mM NaCl, silica sand (natural sand of Mios, France) | IEP: pH 5.5, ζ potential: −40 to −12 mV (at pH 8, IS: 10−4 to 1 M) | When surface chemistry favors NP–NP aggregation over NP-collector attachment, the rate of NP aggregation within pores may be comparable to that of deposition at ratios of collector to NP surface area as high as 40. | Solovitch et al. 2010 (ref. 68) |
| NanoAmor (Nanostructured & Amorphous Materials, Inc.) | Nominal diameter: 5 nm | IS = 1–100 mM NaNO3 | D H: 350–750 nm, (depending on IS and pH), DFCS: 7–189 nm, DAFM/TEM: 30 nm, PZC: ∼5 | The deposition behavior of TiO2 onto silica, determined by measurement of frequency and slopes in QCM-D, was in good qualitative agreement with classical DLVO theory at pH 3 and 9. | Fatisson et al. 2009 (ref. 31) |
| Nanostructured & Amorphous Materials, Inc. (Houston, TX) | Anatase, nominal size: 10 nm | Sodium carboxymethyl cellulose (CMC) (weight ratio of CMC/TiO2: 100), quartz sand (290 μm, U.S. Silica, IL), porosity: 0.32. | IEP: pH 5.6 to 2 upon addition of CMC, DH: 162/239 nm in 1/10 mg L−1 TiO2. | The mobility of CMC–TiO2 was retarded by the presence of amorphous Fe and Al hydroxide, Na+ and Ca2+. Chemical-colloidal interaction such as chemi-complexation and ligand exchange was the most important factor. | Joo et al. 2009 (ref. 32) |
| Nanostructured & Amorphous Materials, Inc. (Houston, TX) | Rutile, with nominal size of 10 × 40 nm | At pH 6.0, in NaCl or CaCl2. Ottawa sand (U.S. silica) with 250–300 μm size, soaked in 12 N HCl and baked at 800 °C. | ζ potential: −47.5 to −28.5 mV at 0.003–1.00 mM IS (in NaCl), or −29.9 to −20.3 mV at 0.015–0.06 mM IS (in CaCl2). | The breakthrough curves displayed a transition from blocking to ripening shapes, and the NP retention profiles exhibited a shift of the maximum NP retention segment from the end toward the entrance of the column gradually with increasing IS. | Chen et al. 2011 (ref. 33) |
| Nanostructured & Amorphous Materials, Inc. (Houston, TX) | Rutile, with nominal size of 10 × 40 nm, SSA: 155.11 m2 g−1 | IS: 3–200 mM NaCl, SRHA: 0–10 mg L−1, at pH 5.7 and 9.0 | Uptake of SRHA: 0.042–0.575 mg m−2 at 3–100 mM IS, 1–10 mg L−1 SRHA at pH 5.7. At pH 9.0, very small adsorption of HA even at IS > 60 mM. | The transport of TiO2 was dramatically enhanced at pH 5.7 even at 1 mg L−1 HA. In contrast, this enhancement of transportability was limited at pH 9.0 due to negligible adsorption of HA. NOM and pH are likely key factors to govern the stability and mobility of TiO2 in the natural environment. | Chen et al. 2012 (ref. 34) |
| (1) Nanostructured & Amorphous Materials, Inc. (2) Vive Nano™ | (1) Anatase, 5 nm (TEM), purity: 99%; (2) polyacrylic acid coated, 3–4 nm (TEM), purity: 84% | In NaNO3 with IS of 0.1–1000 mM, or in 1–10 mM CaCl2, at pH 7 (by 1 mM MOPS). Quartz sand with a d50 of 256 μm (Sigma-Aldrich). | (1) DH: 128–1001 nm, DNTA: 155–316 nm, EPM: −1.60 to −0.954 × 10−8 m2 V−1 s−1 at IS of 0.1–100 mM; (2) DH: 18–214 nm, DNTA: 100–108 nm, EPM: −2.38 to −1.48 × 10−8 m2 V−1 s−1 at IS of 0.1–1000 mM, pH 7. | Bare TiO2 NPs exhibited high retention within a saturated matrix at low IS of 0.1 mM NaNO3 and displayed dynamic behavior. In contrast, the polymer coated TiO2 NPs barely aggregated and exhibited significant transport potential at IS as high as 100 mM NaNO3 or 3 mM CaCl2. | Petosa et al. 2012 (ref. 35) |
| (1) Synthesized by an acidic peptization process; (2) Aeroxide P 25 (Evonik Degussa Corp., Parsippany, NJ) | Synthesized TiO2: amorphous, P25: anatase (86.5%) and rutile (13.5%), with spherical shape, primary size: 20 nm (for both). | In 0.9 mM NaCl at pH 6.0 and 10.0. Ottawa #12 Flint silica sand, d50: 529 μm (U.S. Silica), porosity: 0.36 | D H, synthesized: 60 nm; DH, P25: 446 nm in DI, at pH 6.0. IEP: pH 6.6 and 6.7 for synthesized and P25 TiO2, respectively. | Synthesized TiO2 was more stable compared to the P25 TiO2, and P25 TiO2 was deposited at a faster rate than the synthesized TiO2 in the streambed. | Boncagni et al. 2009 (ref. 46) |
| Aeroxide P 25 from Evonik Degussa Corp. (Parsippany, NJ) | Anatase (80%)/rutile (20%), purity: >99.5%, density: 3.8 g cm−3, SSA (BET): 50 m2 g−1, particle size 21 nm (vendor data) | At pH 7 and 9, in Triton X-100 and/or DSBS | At pH 7/9, ζ potential: 20.0/−16.8, 21.2/17.6, and −11.6/−19.2 mV for no surfactant, in Triton X-100, and in SDBS, respectively. | The electrostatic and steric repulsions in connection with the size of TiO2 aggregates and flow velocity affected the transport distance of TiO2 at pH 7 and 9. | Godinez et al. 2011 (ref. 47) |
| Aeroxide P 25 from Evonik Degussa Corp. (NJ) | Primary size: 21 nm (vendor), TEM size: 5–45 nm, with majority in 10–25 nm range, sphere shape. | IS: 1–20 mM KCl, pH 5, and 7. NP conc.: 100–800 mg L−1. Ultra-pure Iota quartz of 275 μm (Unimin Corp., NC), flow rate: 1.9, and 9.5 × 10−4 m s−1 | IEP: pH 6.2 | A combination of mechanisms including straining, blocking, and DLVO-type forces were involved over the range of solution chemistry and NP concentration tested. | Chowdhury et al. 2011 (ref. 48) |
| Aeroxide P 25 from Evonik Degussa Corp. (NJ) | NPs labeled with fluorescein isothiocyanate isomer I, primary size: 17.7 nm | NP: 10 mg L−1, injection: 0.47 m h−1, in 10 mM KCl and CaCl2 at pH 5, and 7, SRHA: 1 mg L−1, Iota quartz | EPM: 1.18, and −1.37 × 10−8 m2 V−1 s−1 at pH 5 and 7 in 10 mM KCl. EPM: −2.78, and −2.54 × 10−8 m2 V−1 s−1 at pH 5 and 7 in 10 mM KCl + SRHA. | The extent of TiO2 NP deposition follows: without NOM or bacteria > with bacteria > with NOM > combined bacteria and NOM. Ca2+ played a significant role in the interactions, promoting the formation of large clusters. | Chowdhury et al. 2012 (ref. 49) |
| TiO2 labeled with fluorescein isothiocyanate (FITC) | Primary size: 17.7 nm, with 70% NP within 10–20 nm. | IS: 1–100 mM KCl, at pH 5, and 7, in a parallel plate (PP) system | IEP: pH 5.6. EPM: 2.19 × 10−8 m2 V−1 s−1 in 1 mM KCl at pH 5 | Deposition of TiO2 NPs on a glass surface in the PP system was controlled by a combination of DLVO and non-DLVO forces, shear rate, aggregation state, and gravitational force. | Chowdhury et al. 2012 (ref. 50) |
| Aeroxide P-25 (Degussa Corp., Germany) | Nominal size of 21 nm | IS: 0.2 mM NaCl, pH 10 | PZC: 6.2, ζ potential: −26.4 mV, DH: 123.2 nm, porosity: 0.37 | Retention of TiO2 increased as saturation decreased, with slower drainage rates corresponding to greater retention at a given saturation. | Chen et al. 2008 (ref. 51) |
| Aeroxide P-25 (Degussa Corp., Germany) | Nominal size of 21 nm | IS: 0.2 mM NaCl, pH 5, 7, and 10, in 0.5 mm spherical glass beads (Scientific Industries Inc., NY), porosity: 0.37 | ζ potential: 29.9, −6.1, and −36.5 at pH 5, 7, and 10 | During saturated transport, retention varied from very strong at pH 5 to no measurable retention at pH 10. Regardless of the mechanisms, release of TiO2 by saturated or unsaturated media was difficult to achieve. | Chen et al. 2010 (ref. 52) |
| Evonik Degussa Corp. (#4168063098, US) | Anatase (82%) and rutile (18%), primary size: 30 nm, morphology: semisphere, purity: 98.03, surface area: 51.5 m2 g−1 | In NaCl or CaCl2 solutions, IS = 1, 10, and 100 mM, at pH 5, 6, 7, 8 and 9; 10 mg L−1 SRHA | ζ potential: 20/−50 mV at pH 4, and 9 in 1 mM NaCl | ζ potential can be used to predict the interaction of NPs with NPs or surfaces in QCM-D experiment. The presence of SRHA significantly hinders the deposition of TiO2 NPs onto silica sensors at high IS in QCM-D. | Thio et al. 2011 (ref. 38) |
| (1) Evonik Degussa Aeroxide P25; (2) Sachtleben Hombikat UV100 | (1) P25: 80% anatase and 20% rutile, (2) UV-100: 10 nm anatase, SSA: 250 m2 g−1 | Natural surface water:, IS: 3.47 mM (Ca2+, Mg2+, Na+, K+, NO3−, Cl−, SO42−, HCO3−) | (1) P25: DH: 1283 nm, ζ potential: −18.9 mV (2) UV-100: DH: 1085 nm, ζ potential: −20.2 mV | The removal rate exceeds the exponential decay (first-order), which was attributed to gravitational settling of large aggregates. Biofilm reduced the travel length. | Battin et al. 2009 (ref. 53) |
| Hombikat TiO2, Sigma-Aldrich | D H: 40 nm | Effluent and sludge from wastewater reclamation facility in the U.S. In batch experiment, 1 mM NaHCO3, at pH 7.2 | IEP: pH 5.2 | Only 12% of Ti passed through the SBR in supernatant, while 88% was associated with biosolid fraction. The removal rate of 69% of Ti in full-scale units was attributed to the contained surfactant and NOM. | Kiser et al. 2009 (ref. 57) |
3.2 Material properties and PZC of TiO2 NPs
The PZC of most TiO2 falls in the weak acidic to neutral pH range (pH 4.2–6.8),5,20,22,27–29,36,37,39,43,45,58,59 and therefore, the NPs are likely to aggregate and settle out of the liquid phase at circumneutral pH. Interestingly, inconsistent data were also observed for some specific TiO2 NPs in reports from different research groups. For instance, a PZC of 2.7 was reported for the 50 nm anatase NPs (NanoAmor),60 2.9 for a mixture of nanocrystalline anatase and rutile (Sigma-Aldrich) in DI,56 and even <2 for one type of anatase (Sigma T8141).6 Some researchers attributed the variation of PZC to nanocrystalline size, with smaller sizes exhibiting lower PZC.58 However, for one group of TiO2 from the same source (NanoAmor), the smallest TiO2 NPs, i.e., 5 and 10 nm anatase, have the largest value of PZC (6.0), while for the largest size of TiO2, the 50 nm anatase has the lowest PZC of 2.7.5 The deviation of PZC towards lower values was most likely due to the chemical impurities of the pristine TiO2, rather than morphology or crystallinity.5 Liu et al. (2011) reported that the PZC decreased with an increase in the extractable silicon and phosphorous contents (Fig. 2),5 which could be ingredients introduced during the synthesis process to promote nanocrystal formation and to control morphology.61,62 This argument is supported by the finding that the PZC shifted from 4.7 for pure TiO2 to 1.8 for SiO2-coated TiO2 NPs.63 Kim and Choi also reported that phosphate loading markedly changed the surface charge of TiO2 towards the negative side, shifting PZC from ca. 6.5 for pure TiO2 to ca. 5.0.64 Besides the presence of phosphorous in the pristine NPs, Ji et al. (2010) reported that the adsorption of phosphate in solution probably changed the surface potential of TiO2 NPs, which enhanced the stabilization.40 In the presence of SRFA, however, Domingos et al. (2010) reported an increased aggregate size of anatase (NanoAmor) covered by Suwannee River fulvic acid (SRFA) with an increase in solution phosphate concentrations, and attributed it to the possible conformational modifications of the FA in the presence of phosphate.28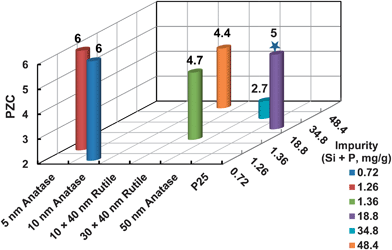 | ||
| Fig. 2 Correlation of impurity (i.e., Si and P) contents with the PZC of TiO2 NPs with various crystallinity, morphology, and compositions. The figure was re-drawn from data in the literature.5,14,64 The star symbol indicates that the surface composition of the P 25 TiO2 was estimated by X-ray photoelectron spectroscopy (XPS) (mass percentage converted from original atom percentage),64 while the rest of the TiO2 NPs were dissolved by alkaline fusion and analyzed by inductively coupled plasma-optical emission spectrometry (ICP-OES).5,14 | ||
In aqueous phase, NPs with lower PZC tend to be more negatively charged in the neutral pH range, and the NPs could be stabilized due to the high electrostatic force and pose higher risks to the environment. Hence to decrease the environmental risks of TiO2 NPs, alternative methods or chemicals that do not contain Si and P are recommended to use during the synthesis. However, in the natural environment, the adsorption of natural organic matter (NOM) can greatly change the surface properties and behavior of TiO2 NPs.27,29,34,38,42 As a result, further study is warranted on the interaction of NOM with TiO2 of varying properties, and to determine whether it is the property of adsorbed NOM or TiO2 that can dominantly influence the stability and transport in the natural environment.
3.3 Material properties and critical coagulation concentration (CCC) of TiO2 NPs
Ionic strength (IS) is another significant factor influencing the stability of NPs. At high IS, the thickness of the electric double layer is compressed and the repulsion is screened. As a result, aggregation and subsequent sedimentation are initiated when NPs collide with each other. Although the nature and valence of the dissolved ions need to be considered, the CCC is a valuable measure to evaluate NP stability in electrolyte solutions. For instance, Zhang et al. (2009) reported that 10 mM KCl was enough to induce aggregation of the 15 nm TiO2 (from Sigma-Aldrich) at pH 5.6 (Fig. 3).54 In contrast, French et al. (2009) reported the micron-sized aggregates of a 5.5 nm anatase/4.4 nm brookite mixture (synthesized using sol–gel method) formed within 15 min in ca. 80 mg L−1 dispersion at 16.5 mM NaCl and pH 4.5.22 At pH 5.8, 6.8, 7.5 and 8.2, the formation of micro-sized aggregates was observed within 5 min in 8.5, 8.4, 9.3 and 9.9 mM NaCl, respectively.22 The CCC was the lowest (8.4 mM) for the anatase/brookite nanocrystalline NPs at pH 6.8, which is closest to the PZC, while the CCC increased with the pH values deviating from PZC towards acid or base (Fig. 3). The observed change in the CCC of TiO2 with pH is consistent with the predictions of the classical Derjaguin–Landau–Verwey–Overbeek (DLVO) theory.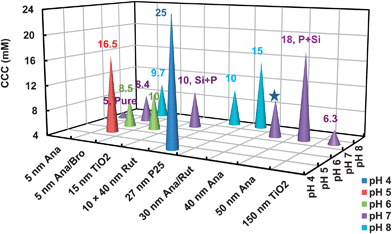 | ||
| Fig. 3 Comparison of CCC for the TiO2 NPs with different crystallinity, morphology, and composition at various pH values as reported in the literature.5,20,22,38,44,54,65 The CCC values at acidic pH (e.g., pH 5) were generally higher than that at circumneutral pH (e.g., pH 7) which is close to the PZC. The CCC for 10 × 40 nm rutile (10 mM) and 50 nm anatase (18 mM) were higher compared with the 5 nm anatase (5 mM), due to the detected impurities of Si and P and consequently more negative surface charge at pH 7.5 Further analysis on the material properties of the other TiO2 is not feasible, due to the insufficient information on the composition of the pristine TiO2. The star symbol stands for the estimated value of CCC, due to the unavailability of the aggregation kinetics data.65 Ana = anatase, Rut = rutile, Bro = brookite. | ||
For the aggregate size, Lecoanet and Wiesner (2004) reported the stable aggregates with a mean diameter of 198 nm anatase (Altair Nanomaterials, Inc.) in 10 mM NaCl at pH 7.65 In contrast, French et al. attributed the observed larger aggregates (sol–gel TiO2) of micron level under similar solution chemistry to the primary particle size, NP concentration, and probable crystalline composition.22 Kammer et al. reported the overall comparable environmental behavior of 21 nm Evonik P25 and Hombikat UV-100 with a primary diameter of <10 nm, which are composed of the same core material, however with a 5-fold difference in BET specific surface area (SSA).36 A recent comprehensive study reported CCC values of 5, 10, and 18 mM NaCl for 5 nm anatase, 10 × 40 nm rutile, and 50 nm anatase (NanoAmor), respectively, in dispersions of 5–20 mg L−1 TiO2 at pH 7 (Fig. 3).5 The variation in CCC was attributed to the impurities of Si and P, which led to more negative surface charge for the 10 × 40 nm rutile and the 50 nm anatase at neutral pH.5 Hence, a characterization of chemical compositions of TiO2 seems to be able to help reconcile the variation in the aggregation of different TiO2 NPs, based on the available data in the literature.
3.4 Material properties and transport of TiO2 NPs
Table 2 presents the deposition and transport results of various TiO2 NPs as affected by material properties. Using 5 nm anatase (NanoAmor, TX), Petosa et al. (2012) reported its high retention (with the C/C0 value of ca. 0.5) at 0.1 mM NaNO3 and pH 7 in saturated quartz sand (d50 = 256 μm).35 In contrast, Chen et al. (2011) found complete breakthrough of 10 × 40 nm rutile (NanoAmor, TX) under similar conditions, i.e., in 0.1 mM NaCl at pH 6 in quartz sand media of 250–300 μm.33 Interestingly, Liu et al. characterized the same NMs of anatase and rutile (NanoAmor), and found that the 10 × 40 nm rutile was more negatively charged than the 5 nm anatase under the same conditions due to the existence of Si impurity in the 10 × 40 nm rutile.5 It is noted that the Darcy velocities of the two studies were 4.2 × 10−3 cm s−1 (or 3.6 m per day) and 5.1 × 10−3 cm s−1 for 5 nm anatase and 10 × 40 nm rutile, respectively, and the effect of velocity could be negligible. In a study by Chowdhury et al., partial transport of Aeroxide P 25 TiO2 (with the C/C0 value of ca. 0.65) was observed in ultrapure quartz sand (Iota quartz, NC) of 275 μm at 1 mM KCl and pH 7.66 In comparison, the C/C0 value of the 5 nm anatase was ≤0.1 at pH 7 in 1 mM NaNO3 as observed by Petosa et al.35 The higher effluent concentration of Aeroxide P25 could again be mainly attributed to higher surface potential of those NPs. For instance, the values of EPM were −1.37 and −1.06 × 10−8 m2 V−1 s−1 for Aeroxide P25 and 5 nm anatase, respectively, at the same IS of 10 mM (the EPM value for Aeroxide P25 at 1 mM IS was not presented). In addition, a larger hydrodynamic force could also possibly contribute to the higher breakthrough,65,67 since the Darcy velocity of 1.9 × 10−2 cm s−1 for P25 was higher than that for the 5 nm anatase, i.e., 4.2 × 10−3 cm s−1 (or 3.6 m per day). Therefore, the TiO2 NPs with higher absolute value of ζ potential (i.e., the 10 × 40 nm rutile, and Aeroxide P 25) show higher breakthrough concentrations than the TiO2 with lower ζ potential (i.e., the 5 nm anatase) at similar solution chemistries in cleaned quartz sand media of similar size with close pore velocities.In another study, Solovitch et al. (2010) applied the 32 nm anatase (Alfa-Aeser 39953) in transport experiment using natural silica sand (France) with an average diameter of 650 μm.68 Complete breakthrough of the Alfa-Aeser TiO2 was observed at 1–20 mM NaCl, and gradually increased deposition was observed at 40–100 mM NaCl (Fig. 4). In contrast, the 10 × 40 nm rutile was retained at much smaller IS, i.e., 0.75–1.0 mM NaCl, onto silica with similar values of ζ potential.33 Careful examination of the characteristics of TiO2 revealed that the transition of breakthrough to deposition happened at the ζ potential range of c.a. −28 to −32 mV for both TiO2 (although at different IS).33,68 It could be concluded that the transport of TiO2 in saturated sand media is sensitive to the ζ potential within the narrow range. Boncagni et al. (2009) applied two types of TiO2, i.e., synthesized amorphous TiO2 and commercial P25 (Evonik Degussa Corp., Parsippany, NJ) NPs in their study.46 The synthesized TiO2 was more negatively charged at neutral to basic pH than the P25 TiO2 NPs, and thus higher electrostatic repulsive force for the synthesized TiO2 is favorable for NP disaggregation and detachment from silica sand at pH 11.3.46 Therefore, the transport of TiO2 is greatly influenced by the surface ζ potential of the NMs, with more negative ζ potential causing larger electrostatic repulsion between NPs and media surfaces and higher breakthrough. We must note here that the conclusion is made based on the ideal experimental conditions, e.g., in the absence of organic matter (OM) and in sand media that were cleaned by concentrated acid before tests.
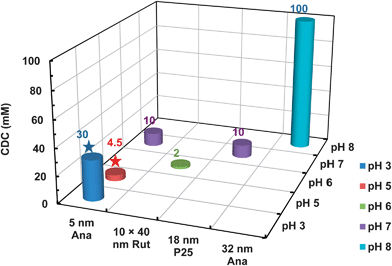 | ||
| Fig. 4 Critical deposition concentration (CDC) for TiO2 NPs with various characteristics.31,33,35,49,68 The star symbols indicate that the data were obtained from QCM-D experiments under favorable conditions (where attractive electrostatic and van der Waals interactions were dominant), while the remaining data were obtained from column experiments. | ||
A unique technique called quartz crystal microbalance with dissipation (QCM-D) has been applied to the study of transport of anthropogenic NPs. Fatisson et al. (2009) reported significant deposition of 5 nm anatase (NanoAmor) on a silica quartz sensor in a QCM-D chamber at pH 3 and 5 but little deposition at pH 9 at IS of 2–30 mM NaNO3.31 Similarly, Thio et al. (2011) reported the deposition of 30 nm TiO2 (Evonik Degussa) on silica sensors at pH 5–7 in both NaCl and CaCl2, while no deposition in NaCl or CaCl2 at pH ≥ 8 on silica in the QCM-D test.38 However, in column experiments, the deposition was observed at similar pH and lower IS. Such deviation of the QCM-D test result could be attributed to surface heterogeneity of sand, NP aggregation, and flow geometries, as detailed in a recent study.69 In addition, under the comparatively ideal condition of QCM-D chambers, non-DLVO interactions, such as straining, were less influential compared with in column experiments, and hence the reported QCM-D test data were in qualitative accordance with the predictions of DLVO theory.31,38
The presence of OM can greatly change the surface charges and structures of both NP and media. The material property of NPs and solution chemistry control the adsorption quantity of the OM, while functional groups of the adsorbed OM also greatly change the surface characteristics of the NPs. In this scenario, the ζ potential alone is not effective to estimate the transport of NPs, since the additional steric effect of adsorbed OM is not accounted for in the classical DLVO theory. In recent studies, extended DLVO (X-DLVO) equations have been applied to fit the experimental data with appropriate assumptions.34 However, the surface heterogeneities of collector media and NPs play a significant role in NP deposition, which is challenging to quantify. For instance, the mobility of carboxymethyl cellulose (CMC) adsorbed 10 nm anatase (NanoAmor, TX) was retarded in porous media composed of Fe and Al hydroxide coated sand.32 Moreover, in the presence of divalent ions such as Ca2+, the possible generation of chelating/bridging of NPs/NPs or NPs/media through OM makes the deposition process even more complicated. The complex interaction of NPs with OM and the effect on the transport is not fully understood, and deserves further investigation.
4 Case analysis on carbon nanotubes (CNTs)
4.1 Production process and sources of CNTs
Typical synthesis methods for CNTs are carbon electric-arc (arc discharge),70,71 laser ablation (for SWNTs),72 and CVD.73 CNTs synthesized using arc discharge or laser ablation historically have the advantage of better quality; however, the inherent design of these systems poses limitations to their large-scale production due to, e.g., vacuum conditions and replacement of graphite electrodes.74 In contrast, CVD and enhanced CVD techniques show the greatest promise for economical large-scale synthesis.74 Catalytic CVD (CCVD), either thermal or plasma enhanced, is now the standard method for CNT production. Most commercially available CNTs are produced using arc-discharge or CVD techniques as a compromise of cost and quality.74In industry, many vendors have reported to produce CNTs via CVD (Tables 3 and 4), including MER Corporation (Tucson, AZ),75 Shenzhen Nanotech Port Co. (Shenzhen, China),76,77 Cheap Tubes Inc. (Brattleboro, Vermont),78 Nano TechLabs Inc. (Yadkinville, NC),79 NanoLab Inc. (Waltham, MA),80 Chengdu Organic Chemistry Co. (Chinese Academy of Sciences, China),81etc. As another industrially scalable type of CVD, high pressure CO conversion (HiPCO) relies on thermal cracking of the precursor CO gas in the presence of a metal catalyst to produce SWNTs.82 HiPCO has been used by several manufacturers, including Carbon Nanotechnologies, Inc.,83 Tubes@Rice,84 Richard Smalley Institute (at Rice University, Houston, TX),85etc. The commercial raw-CNTs often contain significant amounts of impurities such as amorphous carbon, nanographitic structures, and catalytic metals.82 Consequently, the CNTs are purified following the production process, by use of concentrated acid, such as HCl,86 or a combination of HF/HCl.87
| Type | Source, production, or preparation methods | Material properties | Solution chemistry | Characteristics (EPM, aggregate size, etc.) | Main finding and conclusions | Ref. |
|---|---|---|---|---|---|---|
| SWNTs | Carbon Nanotechnologies, Inc., via HiPCO, refluxed in 70% HNO3 for 1 h, sonicated in HNO3–H2SO4 for 3 h | Purity: 93% (as reported by the vendor) | In Triton X-100 (0.2 T) | Bundle thickness of 15–30 nm | Acid treatment generated hydrophilic carboxyl segments at the ends of shortened tubes and made aggregation more favorable. The adsorption of Triton X-100 resulted in repulsion and increased dispersion. | Chen et al. 2004 (ref. 83) |
| SWNTs | Cheap Tubes Inc. | Diameter: 0.8–1.6 nm, length: 5–30 μm | TOC: 2.5 mg L−1 of LB, SRHA, BSA, and alginate, at pH 6.0 in NaCl or CaCl2 | EPM: −1.24 to −0.46 μm cm V−1 s−1 at 1–55 mM NaCl; G/D (Raman) ratio decreased after sonication (0.90 → 0.83); relatively pure (by TGA) | The presence of biomacromolecules significantly retarded the aggregation rate of SWNTs. BSA protein molecules were most effective, followed by SRHA, LB, and alginate. | Saleh et al. 2010 (ref. 103) |
| SWNTs | Cheap Tubes Inc. | Average outside diameter: 1.1 nm, purity: 90% (reported by vendors) | At pH 7.3 ± 0.2 in 0.001–0.1% SDS | D h: 164–197 nm, EPM: −3.49 to −4.10 μm cm V−1 s−1 | CCC increased with SDS for Na+, but was less sensitive for Ca2+. SDS-SWNTs remained suspended over 6 weeks in surface water. | Bouchard et al. 2012 (ref. 102) |
| MWNTs | Cheap Tubes Inc. (VT), some MWNTs heavily oxidized by 30% HNO3 at 80 °C for 6 h. | Purity: >95%, length: 10–30 μm, outer diameter: 10–30 nm. | In Milli-Q filtered water, moderately hard reconstituted freshwater (MHRW), or MHRW containing HA and 20‰ seawater. | Effective diameter and ζ potential: 208–223 nm and −22.7 to −24.6 mV (MWNTs), 181–187 nm and −23.0 to −23.6 mV (MWNT–OH), 181–185 nm and −19.7 to −23.2 mV (MWNT–COOH), 187–196 nm and −21.7 to −25.1 mV (oxidized MWNTs). | Raw MWNTs settled more rapidly than carbon black and activated carbon particles. The presence of functional groups slowed the settling of MWNTs with the stability order: OH > COOH > raw, especially in combination with NOM. | Kennedy, et al. 2008 (ref. 118) |
| MWNTs | MER Corporation (Tucson, AZ), by CVD method | Diameter: 140 ± 30 nm, length: 7 ± 2 μm, purity: >90% | In 1% SDS, 100 mg C per L SRNOM | N/A | Dispersal of carbon-based NMs might occur to an unexpected extent. | Hyung, et al., 2007 (ref. 75) |
| MWNTs | Nano TechLabs, Inc. | Diameter: 20–40 nm, length: 50 μm | At pH 6.0 in NaCl, CaCl2 or MgCl2, and 5 mg L−1 SRHA | EPM from −0.28 to −3.75 μm cm V−1 s−1 at pH 3–11 in 1 mM NaCl | CCC as 25 mM NaCl, 2.6 mM CaCl2, and 1.5 mM MgCl2. SRHA enhanced stability of MWNTs due to steric repulsion. | Saleh et al. 2008 (ref. 97) |
| O-MWNTs | NanoLab Inc., refluxed by 50% HNO3 at 140 °C for 1.5 h | Outer diameter: 15 ± 5 nm, length: 5–20 μm, impurities: <5% (Fe or Ni) | In various electrolytes at pH 6 | EPM: −1.7 to −3.2 μm cm V−1 s−1 at pH 3–5, with little change above pH 6 | Colloidal properties observed, and CCCs were 93 mM NaCl, 1.8 mM MgCl2, and 1.2 mM CaCl2 at pH 6. | Smith et al. 2009 (ref. 87) |
| O-MWNTs | NanoLab Inc., acid-washing treatment by 0–70% HNO3 at 140 °C, KMnO4 at 150 °C, or H2SO4–HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 70 °C. :1) at 70 °C. |
Purity: >95% carbon (treated with HF/HCl to remove most of the residual catalysts by the vendor), outer diameter: 15 ± 5 nm, length: 5–20 μm | At pH 6 and 8 in NaCl | Surface O concentration of 6.1%, 10.8%, and 9.0% by refluxing with H2SO4–HNO3, the mixed acid in microwave, and oxidized with KMnO4, respectively. | Linear correlation exists between CCC, total O concentration, and surface charge of O-MWCTs. EPM did not prove to be a useful metric of stability. The distribution of O-containing functional groups influences the stability, with COOH groups playing the most important role. | Smith et al. 2009 (ref. 91) |
| MWNTs | NanoLab (PD30L5-20) | Diameter: 30 ± 15 nm, length: 5–20 μm | At circumneutral pH, OM or surfactant stabilized CNTs in various coagulant | N/A | Higher influent concentrations of kaolin and alginate improved MWNT removal, while OM hindered the coagulation. | Holbrook et al. 2010 (ref. 104) |
| MWNTs | NanoLab (PD15L5-20), refluxed in 98% H2SO4 and 69% HNO3 (3:1) at 70 °C for 8 h |
Outer diameter: 15 ± 5 nm, length: 5–20 μm, total O: 10.6% and 6.3% (HO-MWNTs and LO-MWNTs) | Oxygen groups comprised of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH, and OH groups O, COOH, and OH groups |
At pH 7.1 ± 0.2 (buffered with 0.15 mM NaHCO3) | CCC (210 mM) and CDC (330 mM) of HO-MWNTs are higher than the CCC (53 mM) and CDC (150 mM) of LO-MWNTs in NaCl. However, similar CCC and CDC for HO- and LO-MWNTs obtained in CaCl2. | Yi and Chen 2011 (ref. 98) |
| MWNTs | NanoLab Inc. (PD15L5-20), refluxed in 98% H2SO4 and 69% HNO3 (3:1) at 70 °C for 8 h |
Mean length: 376 nm, surface oxygen content: 10.3% (carboxyl, carbonyl and hydroxyl groups made up 70%, 15%, and 10%) | At pH 5.2 ± 0.2 in Hematite NP suspensions or/and HA solutions | D h: 109.8 nm, EPM: −2.09 × 10−8 m2 V−1 s−1 (for CNTs), Dh: 80.7 nm, EPM: 1.79 × 10−8 m2 V−1 s−1 (for HemNPs) | An Increase in the CNT/HemNP ratio resulted in a corresponding increase in the heteroaggregation rate through bridging. Further increase of the ratio (when >0.0316) led to a decrease in the heteroaggregation rate, likely through blocking. | Huynh and Chen 2012 (ref. 111) |
| MWNTs | Shenzhen Nanotech Port Co. (by CVD from CH4/H2 at 700 °C using Ni catalyst, and purified by mixed HNO3 and H2SO4) | Five MWNTs with outer diameter of <10, 10–20, 20–40, 40–60, and 60–100 nm, with length of 1–2 μm | In 20 mg L−1 tannic acid | PZC: 4.7–6.4 | CCC of 6.9, 0.33, 0.29, and 0.014 mM for Na+, Ca2+, Mg2+, and La3+, respectively. | Lin et al. 2009 (ref. 76) |
| MWNTs | Shenzhen Nanotech Port Co., (by CVD from CH4/H2 at 700 °C using Ni catalyst, and purified by mixed HNO3 and H2SO4) | Outer diameter: 28 nm, length: 1–2 μm | At pH 6 in CTAB, TX100, or SDBS | PZC of 4.8 (pristine), 5.3 (sonicated), 9.9 (CTAB-MWNTs), and <2 (TX1000- or SDBS-MWNTs) | For CTAB-MWNTs, montmorillonite and kaolinite greatly deposited the suspension. Montmorillonite partially deposited the TX-100 suspended MWNTs; either clay mineral could change the stability of SDBS-MWNTs | Han et al. 2008 (ref. 77) |
| O-SWNTs | Shenzhen Nanotech Port, treated by O3/ultrasound for 24 h | TOC of 92 ppm for O-SWNT suspension (100 ppm) | At pH 3.2, in NaCl, CaCl2, or AlCl3 | The adsorption of HA did not change the ζ potential of O-SWNTs greatly. | CCC of 160, 4.2, and 0.054 mM for NaCl, CaCl2, and AlCl3, respectively. The aggregation was not sensitive to pH (3–8). HA enhanced the stability in NaCl, rather than in CaCl2, or AlCl3. | Li and Huang, 2010 (ref. 95) |
| MWNTs | Nanostructured & Amorphous Materials Inc., USA | (1) 10–30 nm diameter, 5–20 μm length; (2) 20–200 nm; (3) 240–500 nm, length unknown | In 1% SDS, 21 mg L−1 DOC of SR NOM, or BR NOM, at pH 6.8 | N/A | In 1% SDS, 5, 21 and 29% of CNTs suspended after 5 h for 10–30, 20–200, and 240–500 nm CNTs, respectively. But a reverse trend was observed in SR or BR NOM solutions. | O'Driscoll et al. 2010 (ref. 114) |
| MWNTs | CVD at 750 °C for 1 h using acetylene and ferrocene; carboxylated by H2SO4 and HNO3 at 25, 60 and 80 °C | Diameter: 20–30 nm, length: 1 μm | At pH 1–13 in HCl, HCOOH, or CH3COOH | Acid treatments generated COOH and OH groups, and caused damage to the structure (TEM, TGA). | Acid treatment exhibited more negative ζ potential and improved dispersion. | Shieh et al. 2010 (ref. 101) |
| MWNTs | Chengdu Organic Chemistry Co. Ltd, Chinese Academy of Sciences | Purity: >95%, length: 0.5–2 μm, diameter: 5–10 nm. | In 10 mg L−1 HA, FeCl3: 0–0.3 mM, pH: 7.0 | N/A | The intervention of HA-MWNTs increased the degree of polymerization and the particle size of the produced hydrous ferric oxide. Effective sequestration of MWNTs by coagulation–flocculation–sedimentation was demonstrated. | He et al. 2012 (ref. 119) |
| MWNTs | Chengdu Organic Materials Co. Ltd., Chinese Academy of Sciences, purified by HNO3–H2SO4. | Purity: >95%, pristine (P) and COOH–MWCTs (C) with outer diameters of <8, 20–30, and >50 nm | At pH 6.6–7.0, in 10 mg L−1 HA | ζ potential: 32.1/ −13.7 mV for P30 (pristine, with OD of 30 nm) and 33.6/ −11.8 mV for C30, before/ after sonication. More negatively charged in HA. | Suspended surface area concentration followed an order of OD8 > OD30 > OD50 for both pristine and COOH–MWNTs. | Zhou et al. 2012 (ref. 120) |
| SWNTs and MWNTs | Chengdu Organic Chemistry Co. Ltd., Chinese Academy of Sciences, by CVD from CH4/H2 at 700 °C using Ni catalyst | Outer diameters: 2 and 5 nm, lengths: 1–3 and 0.5–2 μm, tap densities: 0.14 and 0.27 g cm−3 for SWNTs and MWNTs, respectively. | In 40 mg L−1 SDBS, at pH 6.3; 1, 5, and 20 mg L−1 HA (Aldrich) added. | IEP: <pH 3 for SDBS prepared CNTs | CCC values of NaCl were 90 and 120 mM for SDBS prepared SWNTs and MWNTs, and CCC of CaCl2 were 0.82 and 2.6 mM, respectively. In 1, 5, and 20 mg L−1 HA, CCC of NaCl for MWNTs increased to 130, 160, and 240 mM, due to steric effect, while the effect was negligible in CaCl2 solutions. | Ju et al. 2012 (ref. 81) |
| Type | Source, production, or preparation methods | Material properties | Solution chemistry and media properties | Characteristics (EPM, aggregate size, etc.) | Main finding and conclusions | Ref. |
|---|---|---|---|---|---|---|
| SW- and MWNTs | SWNTs by HiPCO from Tubes@Rice, MWNTs by CVD | SWNTs' diameter: 1.4 nm; MWNTs' diameter: 35 nm | In HA of <25 mg L−1, at IS of 0.10 mM | Negative ζ potential of HA coated NTs | CNTs showed considerably higher breakthrough and lower deposition rates than the natural soil colloids, indicating higher mobility within porous media. | Wang et al. 2008 (ref. 84) |
| SWNTs | HiPCO synthesized at the Richard Smalley Institute at Rice University | Diameter: 0.8 nm, length: 400 nm (AFM) | In 1% SDBS, fine to coarse sand media | ζ potential: −42.84 mV in 1% SDBS | SDBS-dispersed SWNTs were highly mobile. Film straining only occurred at very low moisture content; pore straining was not observed. | Tian et al. 2011 (ref. 85) |
| COOH–SWNTs | Cheap Tubes Inc. | Diameter: 0.9–1.6 nm | At pH 7.0 (by 0.1 mM KHCO3), IS: 0.1–55 mM KCl, in quartz sand (from Fisher, sonicated in conc. HCl) | Relative defect level (Raman), EPM of −2.68 to −2.08 μm cm V−1 s−1 at IS 0.1–55 mM | Both physicochemical filtration and straining play a role at low IS, while filtration dominated at higher IS. CDC was 10 mM KCl. | Jaisi et al. 2008 (ref. 115) |
| COOH–SWNTs | Cheap Tubes Inc. | Diameter: 0.9–1.6 nm, length: 0.6–8 μm | At pH 5.5–5.8 in natural soils (Hamden, CT) | G/D (Raman) ratio decreased after sonication; BET: 407 m2 g−1 | Physical straining governs the transport of SWNTs in soil columns, due to its large aspect ratio, highly bundled aggregated state and heterogeneity of soil particles. | Jaisi and Elimelech 2009 (ref. 116) |
| SWNTs | Cheap Tubes Inc., via CCVD and acid purification | Purity: 90%, with trace amounts of Al (0.08%), Cl (0.41%), Co (2.91%), and S (0.29%). D: 1.2 nm, L: 5–30 μm, SSA: 407 m2 g−1. | Leachate composed of 20–800 mg C L−1 HA, IS: 50–400 mM NaCl, pH: 6, 7, and 8, buffered with 25–50 mM P buffer. Ottawa sand with D of 0.32 mm. | EPM: ∼−4 × 10−8 m2 V−1 s−1 in 20–800 mg L−1 HA at pH 6–8 | High molecular weight organics, e.g. HA, and IS acts to stabilize CNTs present in leachate. CNTs may be mobile through solid waste. | Lozano and Berge 2012 (ref. 78) |
| COOH–SWNTs and MWNTs | COOH–SWNTs from Cheap Tubes Inc. MWNTs synthesized via CVD with Ni and Mg catalysis and oxidized by H2SO4–HNO3 (3:1). |
N/A | At pH 5.6, 8.0 and 10; silica sand of 0.5–0.6 mm, porosity of 0.40. Part of sand was baked at 550 °C. | PZC: 2.9 and 2.4 for SW- and MWNTs. EPM: −2.71, −2.69, and −2.75 × 10−8 m2 V−1 s−1 for SWNTs, and −2.99, −2.82 and −2.89 × 10−8 m2 V−1 s−1 for MWNTs at pH 5.6, 8, and 10, respectively. | CNTs were highly mobile in the acid-cleaned sand but showed little transport in both natural and baked sand. Retention reduced when pH increased, which suggested the electrostatic and/or hydrogen-bonding attractions between function groups of CNTs and metal oxyhydroxide impurities on sand surfaces. | Tian et al. 2012 (ref. 93) |
| MWNTs | Cheap Tubes Inc., functionalized by H2SO4–HNO3 (3:1) |
Outer diameters: 36 ± 11 nm, length: 540 ± 340 nm (by SEM) and 10–20 μm by vendor | At pH 7.5, IS: 0.1 and 7.5 mM, quartz sands of 50, 80, 175 and 476 μm, velocities: 0.42 and 4.2 m d−1 | Catalyst impurities: 0.8% (TGA), O: 6.2 ± 1.4% (XPS) | A dual model coupled with site blocking greatly improves model fits. The contribution of straining is minor. | Mattison et al. 2011 (ref. 117) |
| MWNTs | CVD with Ni and Mg catalysts, purified by HCl and functionalized by H2SO4–HNO3 (3:1) |
Carbon content: 99.6–99.9%, diameter: 7–70 nm, average length of 407 nm | IS: 10 mM | EPM: −3.80 × 10−8 m2 V−1 s−1 at pH 10 | Pore water velocity strongly influenced MWNT transport, with high mobility at pore water velocities greater than 4.0 m d−1. | Liu et al. 2009 (ref. 86) |
| MWNTs | Modified CVD, purified by HCl, and sonicated in conc. H2SO4–HNO3 (3:1) |
SSA: 111–118 m2 g−1 (BET), length: 100–300 nm, O: 7.5–8.6% | Canadian peat with 47.5% of TOC, pH 5.7 | PZC: ∼1.57, ζ potential: −29.7, −20.3, and −12.0 mV in 0, 4, and 40 mM Na+, respectively | MWNTs aggregated at Na+ > 4.0 mM or at pH 4.0. DOM from peat stabilized MWNTs. The interaction became apparent when IS was high. | Zhang et al. 2011 (ref. 94) |
| MWNTs | Modified CVD, purified by HCl, and sonicated in conc. H2SO4–HNO3 (3:1) |
SSA: 111 m2 g−1 (BET), purity: 99.7% (TGA), length: 353 ± 452 nm, O: ∼7.4% (XPS) | Soil minerals of kaolinite (16.2 m2 g−1), smectite (240 m2 g−1) and shale (6.60 m2 g−1); Na+: 0.4–40 mM | N/A | The removal tendency of CNTs correlated with soil mineral's surface potential, IS, and hydrophobicity. The removal can be interpreted by extended DLVO for kaolinite and smectite. | Zhang et al. 2012 (ref. 121) |
Several techniques have been suggested to improve the dispersibility of CNTs in aqueous media,88 including mechanical grinding (such as ultrasonication89 and ball milling90), covalent bonding of functional groups, and adsorption of mediating molecules onto CNT surfaces. A typical treatment is the application of reflux or sonication in concentrated HNO3, or a mixture of H2SO4–HNO3 (3:1), to enhance the stability of CNTs through the addition of hydroxyl and carboxyl functional groups on a CNT surface.83,86,91–94 Other chemical treatment methods include the application of oxidants. For instance, MWNTs treated with (NH4)2S2O8, H2O2, and O3 yield higher concentrations of carbonyl and hydroxyl groups,95 while stronger oxidants, e.g., HNO3 and KMnO4, lead to higher fractional concentrations of carboxyl groups.96 It should be noted that as the dispersion efficiencies of CNTs are improved, CNTs are shortened and wall structures damaged. Their intrinsic properties may also be significantly altered by these functionalizations. Chemical processing, ultrasonication, or a combination of both treatments leads to “worm-eaten” or “ragged” tube walls or walls with cuts, buckles and irreversible bends.89
These surface modifications will affect the stability and fate of the CNTs in the environment. Recent studies on the stability (or aggregation) and transport (deposition) of CNTs are summarized in Tables 3 and 4, respectively. Available data in the literature have revealed the environment behavior of different types of CNTs, or CNTs treated by different methods.
4.2 Colloidal properties of CNTs and challenges
As one significant category of anthropogenic NMs, CNTs generally exhibit colloidal properties as indicated by the data in the literature. For instance, the reaction and diffusion controlled (or unfavorable and favorable) regimes were observed in the aggregation kinetics studies of MWNTs (from NanoLab Inc. and NanoTechLabs Inc., respectively).87,97,98 In addition, the CCCs of oxidized MWNTs (O-MWNTs) in monovalent and divalent electrolytes are consistent with the Schulze-Hardy rule of the classical DLVO theory.87 This rule states that it is the valence of the ion of opposite charge to colloid that has the principal effect on the stability of the colloid.99 The CCC value varies inversely with the sixth power of the valence of the ions in solutions (z−6). In practice, the dependence of CCC on z should be between z−6 and z−2 for a range of NMs.100In spite of recent progress, there are still some issues to predict the stability of CNTs within the framework of the DLVO theory. The electrostatic repulsion and van der Waals attraction are fundamental to NP–NP or CNT–CNT interactions in the classical colloidal theory,21 and surface potential or surface charge is a key parameter to consider. Many CNTs are negatively charged at a wide pH range of 3–10, including MWNTs (NanoTechLabs Inc.),97 O-MWNTs (NanoLab),87,91 and acid treated MWNTs (synthesized by thermal CVD).101 Nevertheless, some other CNTs have positive surface charge at pH 3–5, e.g., MWNTs (Shenzhen Nanotech Port).76,77 Saleh et al. (2008) and Yi and Chen (2011) independently reported that the absolute value of EPM for MWNTs (from NanoTechLabs Inc., and NanoLab Inc., respectively) decreased with an increase in IS due to the compression of the electric double layer and screening of the surface charge in mono- and divalent electrolyte solutions at circumneutral pH (e.g., pH 6–7).97,98 The trend is in accordance with the aggregation kinetics for CNTs and the DLVO theory.97,98 However, Smith et al. (2009) found a lack of change in EPM as the pH values increased from neutral to basic, which was not consistent with the continued increase in colloidal stability of the O-MWNTs (from NanoLab, Inc.).87 In contrast, the surface charge density obtained from acid–base titration increased monotonically with increasing pH, which was consistent with the increased colloidal stability of O-MWNTs above pH 6.87 As a result, Smith et al. argued that EPM did not prove to be a useful metric of colloid stability of CNTs, in contrast with surface charge.91 On the other hand, for other NMs, such as metal oxide, EPM (or ζ potential) could be used to roughly evaluate the stability of the NPs in aqueous media as reported by Keller et al. (2010).37 Hence, the inconsistency of surface potential and surface charge for CNTs, as well as the source of its surface charge, deserves further investigation.
4.3 Stability and aggregation of CNTs in aquatic environments
In natural aquatic media the dispersal of CNTs is relatively stable,97 or it even might occur to an unexpected extent,75 which has significant environmental implications for the CNTs. The suspended CNTs can become bioavailable and may result in toxicity to organisms in the natural environment. For instance, adsorption of Triton X-100, a nonionic surfactant, causes its hydrophilic part to cover a SWNT (from Carbon Nanotechnologies, Inc.) bundle over its axial length, and enhances repulsion and dispersion.83 The concentration of surfactant does not need to be high. Sodium dodecyl sulfate (SDS) is effective in dispersing SWNTs (Cheap Tubes, Inc.) even at 0.001%.102 Saleh et al. (2010) studied the effect of natural and biological OM on the aggregation kinetics of SWNTs (Cheap Tubes, Inc.), and reported the order of their reducing aggregation rate, i.e., bovine serum albumin (BSA) protein > SR-NOM > Luria-Bertani (LB) > alginate.103 The remarkably enhanced stability of SWNTs by BSA was attributed to its globular molecular structure that increased steric repulsion.103 However, in the presence of alginate and high Ca2+ concentrations, the aggregation of MWNTs was enhanced and removal efficiency was improved (Cheap Tubes, and Nanolab, Inc.).103,104 Hence, the interactions of CNTs with various types of NOM are complicated in complex environment conditions. Moreover, almost all of the reported experiments were conducted in the presence of only one type of NOM, and there is uncertainty with regard to the behavior of CNTs in the presence of various NOM with different properties. Two opposite effects, enhanced aggregation or electrosteric effect, are largely dependent on the competitive adsorption of NOM, dominant structural properties of the adsorbed NOM, and the interactions with the surface of CNTs.77,105The heteroaggregation of CNTs with or deposition onto the solid fraction of natural colloids largely determines the fate and transport relative to homoaggregation.106,107 The reason is that natural colloids are ubiquitous and the concentration can be higher than that of released CNTs under natural conditions.44 Han et al. (2008) found that both montmorillonite and kaolinite destabilized the MWNTs (Shenzhen Nanotech Port Co.) suspended by CTAB, while montmorillonite could partially deposit the MWNTs suspended by TX100.77 However, neither montmorillonite nor kaolinite could change the stability of MWNTs suspended by SDBS. The destabilization mechanism was attributed to the competitive adsorption of surfactants by mineral colloids and bridging between minerals and MWNTs by surfactants.77 Holbrook et al. (2010) reported that higher concentrations of kaolin and alginate improved MWNT (Nanolab) coagulation.104 The heteroaggregation between negatively charged O-MWNTs (NanoLab) and positively charged hematite (Hem) NPs was recently reported by Huynh et al. (2012).108 As the ratio of CNT/HemNP concentrations increased to 0.0316, the heteroaggregation rate increased through the bridging of HemNPs by CNT strands.108 However, further increasing the CNT/HemNP ratio, at above the optimal value (0.0316), led to a dramatic decrease in the heteroaggregation rate. Due to the large aspect ratio, the bridging mechanism involved in the heteroaggregation of CNTs with NPs/colloids is different from OM bridging for (quasi-)spherical NPs with a smaller aspect ratio. Divalent cations with appropriate size, such as Ca2+, are indispensable for OM bridging in NP suspensions, which has been reported to occur with nanoscale hematite,109 fullerene C60,110 silver,111 boron,60 silicon,112 silica,113etc.
Systematic studies on the effect of material properties on the stability and transport of CNTs are very limited. O'Driscoll et al. (2010) studied the sedimentation of MWNTs (Nanostructured and Amorphous Materials Inc., or NanoAmor) with three diameter ranges, i.e., 10–30 nm, 20–200 nm, and 240–500 nm, and found that the stability increased with increasing diameter in 1% SDS solutions at pH 6.8.114 In contrast, opposite trends were observed in 21 mg L−1 DOC of SR–NOM or Black River (BR) NOM. It is not clearly understood why the MWNTs behave radically differently in these OM solutions. In addition, significant information on the characteristics, such as the surface charge/potential of CNTs or the adsorption quantity of NOM, is not available. Hence results from a particular study may not be extended to other types of CNTs under different environmental conditions. Further research is needed to focus on the competitive adsorption of NOM and natural colloids, and their synergistic influence on the stability of CNTs in complex environments.
4.4 Transport of CNTs in porous media
Compared with stability and aggregation studies on CNTs, there are fewer studies on the transport of CNTs in porous media (Table 4). Wang et al. (2008) and Jaisi et al. (2008, 2009) reported that physicochemical filtration plays a significant role in the deposition of CNTs (from Tubes@Rice and Cheap Tubes Inc.), which was indicated by the increased deposition with increasing IS.84,115 In addition, due to their specific large aspect ratio, the transport mechanisms are probably different from colloids. For instance, Jaisi et al. suggested the mechanism of physical straining, which was indicated by the incomplete breakthrough curves of COOH–SWNTs (Cheap Tubes Inc.) in DI water and by the relatively stable effluent concentration at low IS.115 Compared with fullerene NPs, the higher deposition rate and its relative insensitivity to IS change further verified the straining mechanism in the transport of COOH–SWNTs in soil media.116 Nevertheless, other researchers proposed different mechanisms. Mattison et al. (2011) argued that straining appears to be a relatively minor mechanism, due to the relatively small change in the effective (or hydrodynamic) diameter of the MWNTs (Cheap Tubes Inc.) from the influent and effluent.117 Instead, site blocking may be an operative mechanism, because retarded MWNT breakthrough happened only for the first pulse in all high IS experiments. Furthermore, Tian et al. (2011) reported that pore straining of SDBS-SWNTs (Richard Smalley Institute at Rice University) was not observed in the porous media (quartz sand), because SWNTs may orient parallel to streamlines to reduce retention.85 Specifically, it is the electrostatic and/or hydrogen bonding attraction between functional groups of CNTs and metal oxyhydroxide on sand surfaces that controlled the retention and transport of CNTs.93 Hence, different conclusions were obtained by researchers who applied different CNTs in different environments.In summary, current research is far from a complete understanding of the effects and mechanisms of key material properties of CNTs on their stability and transport in aquatic environments. Based on the currently available information, MWNTs synthesized by CVD (e.g., Shenzhen Nanotech Port) seem to be safer from a perspective of fate and transport. The reason is that the PZC is close to neutral pH, and hence this type of CNT might easily aggregate and settle out of aqueous phase than other CNTs with more negative charge at neutral pH. In addition, CTAB appears to be a better dispersant for the MWNTs (by CVD), since montmorillonite and kaolinite can more effectively co-precipitate the CTAB suspended MWNTs compared with SDBS and TX100.77 MWNTs produced by CVD (e.g., NanoLab, Inc.) were also reported to heteroaggregate with kaolin or hematite NPs.104,108 However, it is largely unknown whether such heteroaggregation can occur with all the CNTs synthesized by CVD and by other processes as well.
5 Conclusions
This study systematically reviewed the recent progress on the aggregation, deposition, and transport of TiO2 nanoparticles and carbon nanotubes from different manufacturers and synthesis processes, which affect their material properties. TiO2 spherical NPs and nano-rods with low aspect ratio (1:4) show consistent colloidal stability and aggregation kinetics with the predictions of traditional DLVO theory in the absence of organic macromolecules. TiO2 containing high amounts of impurities such as Si and P (e.g., 10 × 40 nm rutile, NanoAmor) has more negative charge, and therefore shows greater stability and has higher breakthrough behavior in porous media compared with the TiO2 with lower amounts of impurities and less negative charge (e.g., 5 nm anatase, NanoAmor) at neutral pH. Hence, ingredients containing Si and P should be prevented during synthesis, and novel materials should be considered to control the crystallinity and morphology. However, the interactions of OM with the TiO2 surface in divalent (such as Ca2+) or trivalent ion solutions make the aggregation and deposition more complicated to predict in complex environmental media. On the other hand, manipulating NMs with novel polymers can be another possible approach to control the environmental behavior of NMs.
For CNTs, the morphology of high aspect ratio makes it challenging to apply traditional DLVO theory and the filtration model to quantify the aggregation and deposition process. Some CVD synthesized CNTs have PZC close to neutral pH, and can heteroaggregate and co-precipitate with natural colloids, which can be regarded as relatively environmentally safer CNTs than the stable CNT suspensions from the perspective of fate and transport. However, studies on comprehensive comparisons of the environmental transport behavior of different CNTs are barely available. For instance, what are the key parameters of CNTs that determine the transport in a defined system? Is it morphology, surface potential (or charge), surface functional groups, impurities, or other factors (such as dispersant or specific interactions with media surface)? Can SWNTs and MWNTs with similar morphology and surface charges behave similarly under the same media conditions? What is the critical ratio of tube length/diameter to pore throat dimension leading to possible straining? Answers to these questions are urgently needed to relate the stability and transport of CNTs in natural environmental media with detailed characterization of their surface properties.
NP toxicity is also dependent on material properties. Those material properties include chemical composition, size, morphology, surface charge, dissolution, and reactive oxygen species.122 Although this topic is not the focus of this study, it is critical to determine those material properties that dominate the NM toxicity towards organisms. In the literature, only limited studies have been conducted to discuss this significant aspect.122–125 In addition, standard protocols are needed for NP dispersion preparation and exposure to address the disparities in the data reported from different labs within the research community on NM environmental implications.
Acknowledgements
This study was funded by the National Nanotechnology Initiative through the U.S. Environmental Protection Agency (US EPA). It has not been subjected to the Agency's peer and administrative review and, therefore, does not necessarily reflect the views of the Agency, and no official endorsement should be inferred. Dr Keller's contribution was in part supported by the National Science Foundation and the Environmental Protection Agency under Cooperative Agreement number DBI-0830117. We thank two anonymous reviewers for their help in improving the quality of this work.References
- NRC, A Research Strategy for Environmental, Health, and Safety Aspects of Engineered Nanomaterials, The National Academies Press, 2012 Search PubMed.
- M. C. Roco, C. A. Mirkin and M. C. Hersam, Nanotechnology Research Directions for Societal Needs in 2020: Retrospective and Outlook, Springer, New York, 2011 Search PubMed.
- Nanosafety at the OECD: The First Five Years 2006–2010, Organization for Economic Cooperation and Development, Paris, France, 2011 Search PubMed.
- M. Kosmulski, J. Colloid Interface Sci., 2009, 337, 439–448 CrossRef CAS.
- X. Liu, G. Chen and C. Su, J. Colloid Interface Sci., 2011, 363, 84–91 CrossRef CAS.
- M. Skocaj, M. Filipic, J. Petkovic and S. Novak, Radiol. Oncol., 2011, 45, 227–247 CrossRef CAS.
- C. Som, P. Wick, H. Krug and B. Nowack, Environ. Int., 2011, 37, 1131–1142 CrossRef CAS.
- P. J. J. Alvarez, V. Colvin, J. Lead and V. Stone, ACS Nano, 2009, 3, 1616–1619 CrossRef CAS.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS.
- I. A. Mudunkotuwa, T. Rupasinghe, C.-M. Wu and V. H. Grassian, Langmuir, 2011, 28, 396–403 CrossRef.
- J. Schmidt and W. Vogelsberger, J. Phys. Chem. B, 2006, 110, 3955–3963 CrossRef CAS.
- J. Labille, J. Feng, C. Botta, D. Borschneck, M. Sammut, M. Cabie, M. Auffan, J. Rose and J.-Y. Bottero, Environ. Pollut., 2010, 158, 3482–3489 CrossRef CAS.
- X. Liu, G. Chen, J. Erwin, N. Adam and C. Su, in preparation.
- J. M. Davis, T. C. Long, J. A. Shatkin, A. Wang, J. A. Graham, M. Gwinn and B. Ranalli, Nanomaterial Case Studies: Nanoscale Titanium Dioxide in Water Treatment and in Topical Sunscreen, U.S. Environmental Protection Agency, Research Triangle Park, NC, 2010 Search PubMed.
- Titanium Dioxide Manufacturing, Emerson Process Management, 2008 Search PubMed.
- N. Osterwalder, C. Capello, K. Hungerbühler and W. Stark, J. Nanopart. Res., 2006, 8, 1–9 CrossRef CAS.
- P. M. Hext, J. A. Tomenson and P. Thompson, Ann. Occup. Hyg., 2005, 49, 461–472 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- D. R. E. Snoswell, J. Duan, D. Fornasiero and J. Ralston, J. Colloid Interface Sci., 2005, 286, 526–535 CrossRef CAS.
- K. A. Dunphy Guzman, M. P. Finnegan and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 7688–7693 CrossRef CAS.
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS.
- E. A. Barringer and H. K. Bowen, Langmuir, 1985, 1, 414–420 CrossRef CAS.
- S. L. Isley and L. R. Penn, J. Phys. Chem. B, 2006, 110, 15134–15139 CrossRef CAS.
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4227–4233 CrossRef CAS.
- D. Verhulst, B. J. Sabacky, T. M. Spitler and J. Prochazka, A New Process for the Production of Nano-Sized TiO2 and Other Ceramic Oxides by Spray Hydrolysis, Altair Nanomaterials Inc., Reno, NV, 2003 Search PubMed.
- R. F. Domingos, N. Tufenkji and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 1282–1286 CrossRef CAS.
- R. F. Domingos, C. Peyrot and K. J. Wilkinson, Environ. Chem., 2010, 7, 61–66 CrossRef CAS.
- I. A. Mudunkotuwa and V. H. Grassian, J. Am. Chem. Soc., 2010, 132, 14986–14994 CrossRef CAS.
- J. M. Pettibone, D. M. Cwiertny, M. Scherer and V. H. Grassian, Langmuir, 2008, 24, 6659–6667 CrossRef CAS.
- J. Fatisson, R. F. Domingos, K. J. Wilkinson and N. Tufenkji, Langmuir, 2009, 25, 6062–6069 CrossRef CAS.
- S. H. Joo, S. R. Al-Abed and T. Luxton, Environ. Sci. Technol., 2009, 43, 4954–4959 CrossRef CAS.
- G. Chen, X. Liu and C. Su, Langmuir, 2011, 27, 5393–5402 CrossRef CAS.
- G. Chen, X. Liu and C. Su, Environ. Sci. Technol., 2012, 46, 7142–7150 CrossRef CAS.
- A. R. Petosa, S. J. Brennan, F. Rajput and N. Tufenkji, Water Res., 2012, 46, 1273–1285 CrossRef CAS.
- F. von der Kammer, S. Ottofuelling and T. Hofmann, Environ. Pollut., 2010, 158, 3472–3481 CrossRef CAS.
- A. A. Keller, H. Wang, D. Zhou, H. S. Lenihan, G. Cherr, B. J. Cardinale, R. Miller and Z. Ji, Environ. Sci. Technol., 2010, 44, 1962–1967 CrossRef CAS.
- B. J. R. Thio, D. Zhou and A. A. Keller, J. Hazard. Mater., 2011, 189, 556–563 CrossRef CAS.
- S. Ottofuelling, F. Von Der Kammer and T. Hofmann, Environ. Sci. Technol., 2011, 45, 10045–10052 CrossRef CAS.
- Z. Ji, X. Jin, S. George, T. Xia, H. Meng, X. Wang, E. Suarez, H. Zhang, E. M. V. Hoek, H. Godwin, A. E. Nel and J. I. Zink, Environ. Sci. Technol., 2010, 44, 7309–7314 CrossRef CAS.
- M. Sillanpää, T. M. Paunu and P. Sainio, J. Phys.: Conf. Ser., 2011, 304, 012018 CrossRef.
- S. Li and W. Sun, J. Hazard. Mater., 2011, 197, 70–79 CrossRef CAS.
- D. Zhou, S. W. Bennett and A. A. Keller, PLoS One, 2012, 7, e37363 CAS.
- D. Zhou, A. I. Abdel-Fattah and A. A. Keller, Environ. Sci. Technol., 2012, 46, 7520–7526 CrossRef CAS.
- A. S. Almusallam, Y. M. Abdulraheem, M. Shahat and P. Korah, J. Dispersion Sci. Technol., 2011, 33, 728–738 CrossRef.
- N. T. Boncagni, J. M. Otaegui, E. Warner, T. Curran, J. Ren and M. M. Fidalgo de Cortalezzi, Environ. Sci. Technol., 2009, 43, 7699–7705 CrossRef CAS.
- I. G. Godinez and C. J. G. Darnault, Water Res., 2011, 45, 839–851 CrossRef CAS.
- I. Chowdhury, Y. Hong, R. J. Honda and S. L. Walker, J. Colloid Interface Sci., 2011, 360, 548–555 CrossRef CAS.
- I. Chowdhury, D. M. Cwiertny and S. L. Walker, Environ. Sci. Technol., 2012, 46, 6968–6976 CrossRef CAS.
- I. Chowdhury and S. L. Walker, J. Colloid Interface Sci., 2012, 369, 16–22 CrossRef CAS.
- L. Chen, D. A. Sabatini and T. C. G. Kibbey, Environ. Sci. Technol., 2008, 42, 1916–1921 CrossRef CAS.
- L. Chen, D. A. Sabatini and T. C. G. Kibbey, J. Contam. Hydrol., 2010, 118, 199–207 CrossRef CAS.
- T. J. Battin, F. v. d. Kammer, A. Weilhartner, S. Ottofuelling and T. Hofmann, Environ. Sci. Technol., 2009, 43, 8098–8104 CrossRef CAS.
- Y. Zhang, Y. Chen, P. Westerhoff and J. Crittenden, Water Res., 2009, 43, 4249–4257 CrossRef CAS.
- J. Kim, W. Shan, S. H. R. Davies, M. J. Baumann, S. J. Masten and V. V. Tarabara, Environ. Sci. Technol., 2009, 43, 5488–5494 CrossRef CAS.
- Z. E. Allouni, M. R. Cimpan, P. J. Høl, T. Skodvin and N. R. Gjerdet, Colloids Surf., B, 2009, 68, 83–87 CrossRef CAS.
- M. A. Kiser, P. Westerhoff, T. Benn, Y. Wang, J. Pérez-Rivera and K. Hristovski, Environ. Sci. Technol., 2009, 43, 6757–6763 CrossRef CAS.
- K. A. D. Guzman, M. R. Taylor and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 1401–1407 CrossRef.
- J. Fang, X.-q. Shan, B. Wen, J.-m. Lin and G. Owens, Environ. Pollut., 2009, 157, 1101–1109 CrossRef CAS.
- X. Liu, M. Wazne, Y. Han, C. Christodoulatos and K. L. Jasinkiewicz, J. Colloid Interface Sci., 2010, 348, 101–107 CrossRef CAS.
- A. Teleki, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Langmuir, 2008, 24, 12553–12558 CrossRef CAS.
- D. L. Liao, G. S. Wu and B. Q. Liao, Colloids Surf., A, 2009, 348, 270–275 CrossRef CAS.
- S. Sheen, S. Yang, K. Jun and M. Choi, J. Nanopart. Res., 2009, 11, 1767–1775 CrossRef CAS.
- J. Kim and W. Choi, Appl. Catal., B, 2011, 106, 39–45 CAS.
- H. F. Lecoanet and M. R. Wiesner, Environ. Sci. Technol., 2004, 38, 4377–4382 CrossRef CAS.
- I. Chowdhury, D. M. Cwiertny and S. L. Walker, Environ. Sci. Technol., 2012, 46, 6968–6976 CrossRef CAS.
- X. Liu, M. Wazne, C. Christodoulatos and K. L. Jasinkiewicz, J. Colloid Interface Sci., 2009, 330, 90–96 CrossRef CAS.
- N. Solovitch, J. Labille, J. Rose, P. Chaurand, D. Borschneck, M. R. Wiesner and J. Y. Bottero, Environ. Sci. Technol., 2010, 44, 4897–4902 CrossRef CAS.
- X. Liu, G. Chen and C. Su, Environ. Sci. Technol., 2012, 46, 6681–6688 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS.
- P. M. Ajayan, J. M. Lambert, P. Bernier, L. Barbedette, C. Colliex and J. M. Planeix, Chem. Phys. Lett., 1993, 215, 509–517 CrossRef CAS.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. H. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tománek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483–487 CAS.
- K. Saeed, J. Chem. Soc. Pak., 2010, 32, 559–564 CAS.
- C. H. See and A. T. Harris, Ind. Eng. Chem. Res., 2007, 46, 997–1012 CrossRef CAS.
- H. Hyung, J. D. Fortner, J. B. Hughes and J. H. Kim, Environ. Sci. Technol., 2007, 41, 179–184 CrossRef CAS.
- D. Lin, N. Liu, K. Yang, L. Zhu, Y. Xu and B. Xing, Carbon, 2009, 47, 2875–2882 CrossRef CAS.
- Z. Han, F. Zhang, D. Lin and B. Xing, Environ. Sci. Technol., 2008, 42, 6869–6875 CrossRef CAS.
- P. Lozano and N. D. Berge, Waste Manag., 2012, 1699–1711 Search PubMed.
- NanoTechLabs, http://www.nanotechlabs.com/pages/products/nanotubes.html.
- NanoLab, http://www.nano-lab.com/nanotubes-research-grade.html.
- L. Ju, W. Zhang, X. Wang, J. Hu and Y. Zhang, Colloids Surf., A, 2012, 409, 159–166 CrossRef CAS.
- A. R. Köhler, C. Som, A. Helland and F. Gottschalk, J. Clean. Prod., 2008, 16, 927–937 CrossRef.
- Q. Chen, C. Saltiel, S. Manickavasagam, L. S. Schadler, R. W. Siegel and H. Yang, J. Colloid Interface Sci., 2004, 280, 91–97 CrossRef CAS.
- P. Wang, Q. Shi, H. Liang, D. W. Steuerman, G. D. Stucky and A. A. Keller, Small, 2008, 4, 2166–2170 CrossRef CAS.
- Y. Tian, B. Gao and K. J. Ziegler, J. Hazard. Mater., 2011, 186, 1766–1772 CrossRef CAS.
- X. Liu, D. M. O'Carroll, E. J. Petersen, Q. Huang and C. L. Anderson, Environ. Sci. Technol., 2009, 43, 8153–8158 CrossRef CAS.
- B. Smith, K. Wepasnick, K. E. Schrote, A. R. Bertele, W. P. Ball, C. O'Melia and D. H. Fairbrother, Environ. Sci. Technol., 2009, 43, 819–825 CrossRef CAS.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS.
- J. Hilding, E. A. Grulke, Z. G. Zhang and F. Lockwood, J. Dispersion Sci. Technol., 2003, 24, 1–41 CrossRef CAS.
- Á. Kukovecz, T. Kanyó, Z. Kónya and I. Kiricsi, Carbon, 2005, 43, 994–1000 CrossRef.
- B. Smith, K. Wepasnick, K. E. Schrote, H.-H. Cho, W. P. Ball and D. H. Fairbrother, Langmuir, 2009, 25, 9767–9776 CrossRef CAS.
- E. J. Petersen, J. Akkanen, J. V. K. Kukkonen and W. J. Weber, Environ. Sci. Technol., 2009, 43, 2969–2975 CrossRef CAS.
- Y. Tian, B. Gao, Y. Wang, V. L. Morales, R. M. Carpena, Q. Huang and L. Yang, J. Hazard. Mater., 2012, 213–214, 265–272 CrossRef CAS.
- L. Zhang, E. J. Petersen and Q. Huang, Environ. Sci. Technol., 2011, 45, 1356–1362 CrossRef CAS.
- M. Li and C. P. Huang, Carbon, 2010, 48, 4527–4534 CrossRef CAS.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7963–7969 CrossRef CAS.
- P. Yi and K. L. Chen, Langmuir, 2011, 27, 3588–3599 CrossRef CAS.
- P. C. Hiemenz and R. Rajagopalan, Principles of Colloid and Surface Chemistry, CRC Press, Boca Raton, 3rd edn, 1997 Search PubMed.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS.
- Y.-T. Shieh, H.-M. Wu, Y.-K. Twu and Y.-C. Chung, Colloid Polym. Sci., 2010, 288, 377–385 CAS.
- D. Bouchard, W. Zhang, T. Powell and U. s. Rattanaudompol, Environ. Sci. Technol., 2012, 46, 4458–4465 CrossRef CAS.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2010, 44, 2412–2418 CrossRef CAS.
- R. D. Holbrook, C. N. Kline and J. J. Filliben, Environ. Sci. Technol., 2010, 44, 1386–1391 CrossRef CAS.
- A. Deonarine, B. L. T. Lau, G. R. Aiken, J. N. Ryan and H. Hsu-Kim, Environ. Sci. Technol., 2011, 45, 3217–3223 CrossRef CAS.
- E. J. Petersen, L. Zhang, N. T. Mattison, D. M. O'Carroll, A. J. Whelton, N. Uddin, T. Nguyen, Q. Huang, T. B. Henry, R. D. Holbrook and K. L. Chen, Environ. Sci. Technol., 2011, 45, 9837–9856 CrossRef CAS.
- J. T. K. Quik, M. C. Stuart, M. Wouterse, W. Peijnenburg, A. J. Hendriks and D. van de Meent, Environ. Toxicol. Chem., 2012, 31, 1019–1022 CrossRef CAS.
- K. A. Huynh, J. M. McCaffery and K. L. Chen, Environ. Sci. Technol., 2012, 46, 5912–5920 CrossRef CAS.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS.
- K. L. Chen and M. Elimelech, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS.
- X. Liu, M. Wazne, T. Chou, R. Xiao and S. Xu, Water Res., 2011, 45, 105–112 CrossRef CAS.
- T. Abe, S. Kobayashi and M. Kobayashi, Colloids Surf., A, 2011, 379, 21–26 CrossRef CAS.
- N. O'Driscoll, T. Messier, M. Robertson and J. Murimboh, Water, Air, Soil Pollut., 2010, 208, 235–241 CrossRef CAS.
- D. P. Jaisi, N. B. Saleh, R. E. Blake and M. Elimelech, Environ. Sci. Technol., 2008, 42, 8317–8323 CrossRef CAS.
- D. P. Jaisi and M. Elimelech, Environ. Sci. Technol., 2009, 43, 9161–9166 CrossRef CAS.
- N. T. Mattison, D. M. O'Carroll, R. Kerry Rowe and E. J. Petersen, Environ. Sci. Technol., 2011, 45, 9765–9775 CrossRef CAS.
- A. J. Kennedy, M. S. Hull, J. A. Steevens, K. M. Dontsova, M. A. Chappell, J. C. Gunter and C. A. Weiss, Environ. Toxicol. Chem., 2008, 27, 1932–1941 CrossRef CAS.
- M. He, R. Zhou and X. Guo, J. Colloid Interface Sci., 2012, 366, 173–178 CrossRef CAS.
- X. Zhou, L. Shu, H. Zhao, X. Guo, X. Wang, S. Tao and B. Xing, Environ. Sci. Technol., 2012, 46, 3891–3897 CrossRef CAS.
- L. Zhang, E. J. Petersen, W. Zhang, Y. Chen, M. Cabrera and Q. Huang, Environ. Pollut., 2012, 166, 75–81 CrossRef CAS.
- A. l. Simon-Deckers, S. Loo, M. Mayne-L'hermite, N. Herlin-Boime, N. Menguy, C. c. Reynaud, B. Gouget and M. Carrière, Environ. Sci. Technol., 2009, 43, 8423–8429 CrossRef CAS.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409–6413 CrossRef CAS.
- L. M. Pasquini, S. M. Hashmi, T. J. Sommer, M. Elimelech and J. B. Zimmerman, Environ. Sci. Technol., 2012, 46, 6297–6305 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |