 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic oxygen evolution using BaNbO2N modified with cobalt oxide under photoexcitation up to 740 nm†
Takashi
Hisatomi
a,
Chisato
Katayama
bc,
Yosuke
Moriya
a,
Tsutomu
Minegishi
a,
Masao
Katayama
a,
Hiroshi
Nishiyama
d,
Taro
Yamada
d and
Kazunari
Domen
*a
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, 113-8656 Tokyo, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp; Fax: +81 3 5841 8838; Tel: +81 3 5841 1148
bJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan
cFUJIFILM Corporation, 577, Ushijima, Kaisei-Machi, Ashigarakami-gun, 258-8577 Kanagawa, Japan
dDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan
First published on 30th September 2013
Abstract
BaNbO2N was activated for photocatalytic sacrificial water oxidation and reduction by modifying the starting material for nitridation and loading appropriate cocatalysts. Addition of BaCO3 to the Ba5Nb4O15 precursor improved the crystallinity and uniformity of BaNbO2N as a nitridation product, leading to higher oxygen evolution activity. BaNbO2N generated oxygen from an aqueous AgNO3 solution under illumination up to 740 nm.
Broader contextWater splitting on semiconductor photocatalysts has received much attention for the large-scale production of renewable hydrogen from solar energy and water. Photocatalysts should be active for water splitting under irradiation up to 600 nm or even longer wavelengths in order to achieve a high solar energy conversion efficiency at a reasonable quantum efficiency. In this regard, BaNbO2N is a promising candidate because of its small band gap energy of 1.7 eV, allowing light absorption up to 740 nm. The present study demonstrates that BaNbO2N can be activated for photocatalytic water oxidation and reduction in the presence of sacrificial reagents by modifying the starting material for nitridation and loading appropriate cocatalysts. BaNbO2N generates oxygen from an aqueous AgNO3 solution under illumination up to 740 nm, the longest wavelength ever reported for (oxy)nitride photocatalysts. In addition, BaNbO2N has a conduction band edge that is more negative than the hydrogen evolution potential. This means that BaNbO2N electrodes could drive photoelectrochemical water splitting with a smaller applied voltage than other visible-light-driven oxide photoanodes. The promising semiconducting properties of BaNbO2N in solar water splitting and the critical roles of the cocatalysts in boosting the photocatalytic activity will be key to achieving practical solar fuel production. |
Water splitting on semiconductor photocatalysts has received much attention for large-scale production of renewable hydrogen from solar energy and water.1–8 The hydrogen thus produced is storable and can be efficiently converted into electricity or heat at any desired time using fuel cells. Moreover, hydrogen can be employed to produce feedstock in the chemical industry.2 Since visible and infrared rays account for the greater portion of solar energy, it is necessary to extend the wavelength region available for photocatalytic water splitting by developing narrow band gap semiconductors. In fact, photocatalytic materials active for water splitting under irradiation up to wavelengths of 600 nm or even longer are needed to achieve a high solar energy conversion efficiency at a reasonable quantum efficiency.2 However, photocatalysis is triggered by photoexcited electrons and holes, which relax to the bottom of the conduction band and the top of the valence band, respectively. Thus, it is more challenging to drive photocatalytic water splitting as the band gap energy decreases.
Perovskite-type and relevant semiconductors modified with appropriate cocatalysts have produced a variety of benchmarks in the field of photocatalytic water splitting. La-doped NaTaO3 shows excellent quantum efficiencies for overall water splitting under UV light (56% at 270 nm)9 and Rh-doped SrTiO3 is used as a hydrogen evolution photocatalyst in Z-scheme water splitting under visible light.10 Non-oxide perovskite-type materials also exhibit promising activity in water splitting. Porous single-crystalline LaTiO2N modified with CoOx as an oxygen evolution cocatalyst exhibited a quantum efficiency of 27% at 440 nm for an oxygen evolution reaction in the presence of Ag+ cations as sacrificial electron acceptors.11 BaTaO2N and its solid solutions with BaZrO3 have attracted growing interest owing to its response to long-wavelength photons up to 660 nm and the ability to generate both hydrogen and oxygen in the presence of sacrificial reagents.12 In photoelectrochemical applications, in which necessary electrodes can be readily fabricated via electrophoretic deposition13 and particle transfer14 using photocatalyst powder, photoanodes of SrNbO2N decorated with IrO2 can utilize photons up to 700 nm for water oxidation.13 The various physical and semiconducting properties of perovskite-type materials, a consequence of their adjustable compositions and structural stability, allow flexible synthetic routes and establish the potential of perovskite-type photocatalysts for use in versatile approaches to solar energy conversion. Spectroscopic and computational studies are also active to understand band structures of perovskite-type photocatalysts.15–17
Nb-based perovskite-type oxynitrides have smaller band gap energies than the corresponding Ta-based oxynitrides because of the higher electronegativity of Nb.18 BaNbO2N is of particular interest for solar energy conversion because of its narrow band gap of 1.7 eV although it was found to be inactive for both photocatalytic oxygen and hydrogen evolution reactions.19 In the present study, however, we successfully activated BaNbO2N for both of these reactions by modifying the preparation conditions of the starting material for nitridation and loading suitable cocatalysts such as CoOx (ref. 11 and 20) and Pt. Here, we report the effect of the preparation conditions for BaNbO2N on its physical properties and photocatalytic activity for O2 evolution.
BaNbO2N was prepared by nitriding Ba5Nb4O15 under an NH3 flow. Ba5Nb4O15 was prepared by calcining a mixture of BaCO3 and Nb2O5, with NaCl added as a flux at 1173 K unless otherwise noted. After a slow cool-down to room temperature, the oxide sample was rinsed with copious amounts of distilled water to eliminate the NaCl flux. BaCO3 was added to Ba5Nb4O15 before nitridation to suppress the possible generation of niobium oxynitride.19 The precursor was nitrided at 1202 K for 15 h under an NH3 flow of 200 mL min−1. Excessive Ba species generated during the nitridation were dissolved by aqua regia. Photocatalytic reactions were carried out using a closed-circulation system. BaNbO2N was modified with nanoparticles of CoOx and Pt, which served as cocatalysts for oxygen and hydrogen evolution, respectively. Aqueous solutions of AgNO3 and CH3OH were used for sacrificial oxygen and hydrogen evolution reactions, respectively. The reaction vessel was evacuated to remove air before irradiation. The light source used was a 300 W Xe lamp equipped with cutoff filters. Evolved gases were analysed by gas chromatography. Other experimental details are described in the ESI.†
Fig. 1A shows X-ray diffraction (XRD) patterns of samples at different preparation stages. Ba5Nb4O15 was obtained by calcining the starting materials at 1173 K with a NaCl flux while unreacted BaCO3 remained as a by-product in the absence of the flux. A mixture of Ba5Nb4O15 and BaCO3 at a Ba/Nb ratio of 2.00 was converted into BaNbO2N and by-products originating from excessive Ba-species. XRD-pure BaNbO2N was obtained by dissolving the by-products by treatment with aqua regia. On the other hand, Ba5Nb4O15 calcined at 1423 K remained partly unreacted when nitrided under the same conditions (data not shown), presumably because of the larger particle size of the oxide. Scanning electron microscopy (SEM) observation revealed that particles of the nitridation product were huge aggregates, several micrometres in size, and consisted of partly fused grains (Fig. S1†). After the aqua regia treatment, most of the particles became porous. This is presumably because the excessive Ba species between BaNbO2N domains, which were compressed during the crystallographic conversion from Ba5Nb4O15 to BaNbO2N, were dissolved. XRD-pure BaNbO2N was also obtained at different Ba/Nb ratios between 1.25 and 2.00 after treatment with aqua regia (Fig. S2†). Note that NbOxNy was easily produced as a by-product in our previous work particularly when the Ba/Nb ratio was lower than 2.00.19 The full width at half maximum of the (110) peak at 30.5–30.6° decreased from 0.246 to 0.213, 0.210, and 0.207 as the Ba/Nb ratio increased from 1.25 to 1.50, 1.75, and 2.00, respectively. This suggests that higher Ba/Nb ratios in the precursor oxide could improve the crystallinity and uniformity of the nitridation products.
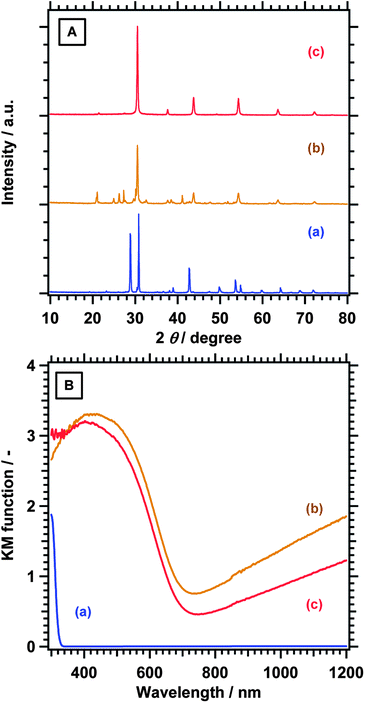 | ||
| Fig. 1 (A) XRD patterns and (B) UV-Vis DRS for (a) Ba5Nb4O15 prepared by the flux method, (b) BaNbO2N nitrided from a mixture of Ba5Nb4O15 and BaCO3 with a Ba/Nb ratio of 2, and (c) BaNbO2N after treatment with aqua regia. | ||
In the diffuse reflectance spectra (DRS) of the nitridation products, the onset of light absorption characteristics of the band gap excitation of BaNbO2N was observed at around 740 nm (Fig. 1B). Additional light absorption was observed in the longer wavelength region, which could be attributed to defects such as reduced B-site cations (Nb) and anion vacancies.18,19,21 The background absorption was weakened by the aqua regia treatment, suggesting that defect species on the surface were eliminated to some extent. However, considerable background absorption remained even after the acid treatment. This indicated that the bulk of BaNbO2N also held reduced Nb species and anion vacancies. Since no diffraction peaks other than those for the perovskite-type material were observed, such defect phases could be expressed nominally as solid solutions of BaNbO2N with BaNbO3 containing Nb4+ states and/or BaNbO2−δ1N1−δ2 with anion deficiencies if crystalline. In addition, amorphous defect by-products could contribute to the background absorption although their impact could not be determined unambiguously. The addition of BaCO3 to Ba5Nb4O15 was expected to weaken the light absorption due to defect species, similar to the case of LaTiO2N nitrided from La2Ti2O7 with La deficiency.22 In reality, however, the intensity of the background absorption increased by the addition of BaCO3 to Ba5Nb4O15 (Fig. S3†). Moreover, the nitridation of Ba5Nb4O15 obtained at higher temperatures than 1173 K tended to result in stronger background light absorption. The generation of defect species was not controllable solely by the Ba/Nb ratio during nitridation, presumably because it was too sensitive to the degree of nitridation from Ba5Nb4O15 to BaNbO2N, which was in turn governed by both the nitridation conditions and the nature of the precursor oxides.
Fig. 2 shows reaction time courses for photocatalytic oxygen evolution using BaNbO2N loaded with the CoOx cocatalyst. Oxygen evolution was observed under visible light illumination (λ > 410 nm) regardless of the Ba/Nb ratio during nitridation. The gradual decrease in the oxygen evolution rates was due to photodeposition of Ag, which blocked the surface of the photocatalyst and shielded the incident light. A small amount of nitrogen (5 μmol at most) was also generated at the initial stage of the reaction because of oxidation of the outmost surface of BaNbO2N. Seemingly, the loading of CoOx as an oxygen evolution cocatalyst suppressed self-oxidation of nitride ions in the oxynitride. Similar behaviours are commonly observed for the oxygen evolution reaction on (oxy)nitride photocatalysts.12,19,20 By contrast, unmodified BaNbO2N did not generate any oxygen under illumination. BaNbO2N loaded with Pt also showed activity for hydrogen evolution although the activity was very low (0.04 μmol h−1, Fig. 3). In our previous work, Pt-loaded BaNbO2N did not show activity for hydrogen evolution.19 The hydrogen evolution observed in the present study may be due to the modifications to the preparation and nitridation conditions for the oxide precursor, which yielded BaNbO2N free from NbOxNy by-products. The above results indicate that BaNbO2N has a band gap straddling the oxygen and hydrogen evolution potentials and that modification with an oxygen evolution cocatalyst is essential for activating BaNbO2N for the reaction. In addition, BaNbO2N was applicable to photoelectrochemical water oxidation under simulated solar irradiation when processed into a photoanode by the particle transfer method14 (Fig. S4†) while having a more negative conduction band edge than the hydrogen evolution potential. It is thus expected that the external bias voltage to drive hydrogen evolution on a counter electrode could be minimized.
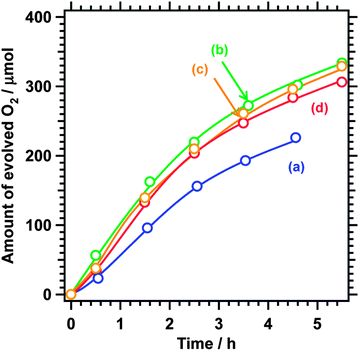 | ||
| Fig. 2 Reaction time courses for oxygen evolution under visible light (λ > 410 nm) using 2 wt% CoOx-loaded BaNbO2N nitrided at a Ba/Nb ratio of (a) 1.25, (b) 1.50, (c) 1.75, and (d) 2.00. Photocatalyst, 0.20 g; solution, 200 mL of 50 mM AgNO3 aqueous solution containing 0.20 g of La2O3; and light source, 300 W Xe lamp. | ||
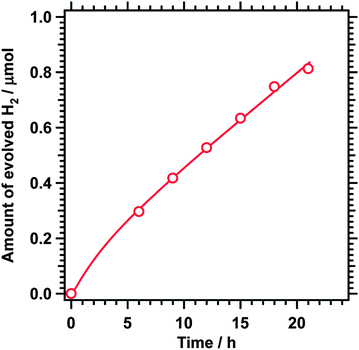 | ||
| Fig. 3 Reaction time course for hydrogen evolution under visible light (λ > 410 nm) using 1 wt% Pt-loaded BaNbO2N nitrided at a Ba/Nb ratio of 1.75. Photocatalyst, 0.20 g; solution, 200 mL of 80 vol% aqueous CH3OH solution; and light source, 300 W Xe lamp. | ||
The oxygen evolution rates were increased by adding BaCO3 to Ba5Nb4O15 as a starting material for nitridation. The XRD patterns of the samples indicated that the crystallinity and uniformity of the nitridation products were improved by the addition of BaCO3 to the precursor. This would improve oxygen evolution rates, because higher crystallinity and uniformity are thought to be favourable for higher photocatalytic activity owing to the lower probability of carrier trapping. Similar results were reported for LaTiO2N (ref. 23) and Ta3N5,24 where the photocatalyst samples with sharper XRD peaks exhibited higher oxygen evolution activity. On the other hand, the background absorption due to defect species was progressively enhanced. The influence of defect species on photocatalytic oxygen evolution in the presence of sacrificial reagents is still controversial. In the present study, they likely did not contribute to charge trapping and recombination significantly. The present results demonstrate that adding an appropriate amount of BaCO3 to the precursor oxide could modify the structural properties and improve the photocatalytic activity of BaNbO2N. More comprehensive synthesis and analysis of BaNbO2N samples are currently underway.
Fig. 4 shows the dependence of the initial oxygen evolution rate on the cutoff wavelength of the incident light, where BaNbO2N nitrided at a Ba/Nb ratio of 1.5 and modified with 2 wt% cobalt oxide was employed. The DRS trace of the sample is also shown for comparison. Photocatalytic oxygen evolution from the AgNO3 solution was observed up to a cutoff wavelength of 740 nm (0.2 μmol h−1) although the oxygen evolution rate decreased as the cutoff wavelength increased. The apparent quantum efficiency of the oxygen evolution reaction was measured to be 0.04% at 640 ± 30 nm. The onset of light absorption reasonably matched the longest wavelength available for photocatalytic oxygen evolution. On the other hand, continuous light absorption by defect species did not contribute to photocatalysis. Thus, it was concluded that water oxidation proceeded via band gap transitions for the BaNbO2N photocatalyst.
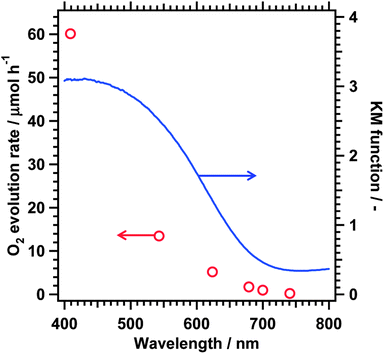 | ||
| Fig. 4 Dependence of the initial oxygen evolution rate of 2 wt% CoOx-loaded BaNbO2N on the cutoff wavelength of the incident light. The DRS trace of the BaNbO2N used is also shown. Photocatalyst, 0.10 g; solution, 200 mL of 50 mM aqueous AgNO3 solution containing 0.20 g of La2O3; and light source, 300 W Xe lamp. | ||
Conclusion
We demonstrated activation of BaNbO2N by loading a CoOx cocatalyst for photocatalytic O2 evolution under visible light. The apparent quantum efficiency of the reaction using CoOx-loaded BaNbO2N was lower than the value for the porous single-crystalline LaTiO2N,11 likely because of the smaller band gap energy of BaNbO2N (smaller by 0.4 eV) in addition to insufficient optimization of the preparation conditions for the material. Nevertheless, BaNbO2N can utilize photons up to 740 nm for photocatalytic O2 evolution. This wavelength is the longest ever reported for an (oxy)nitride photocatalyst. Managing longer wavelength photons could help overcome the barrier against effective production of solar fuel via water splitting.2 Moreover, BaNbO2N had a band gap straddling the hydrogen and oxygen evolution potentials. This means that overall water splitting and unassisted photoelectrochemical water splitting could be achieved under illumination of red light by improving the semiconducting quality of the BaNbO2N photocatalyst. These findings motivate further studies on refinement of the nitridation and cocatalyst loading conditions,11,20 modifications of oxide precursors,23,24 and doping25 to improve the photocatalytic and photoelectrochemical activity of BaNbO2N.Acknowledgements
This work was supported by the Artificial Photosynthesis Project of the Ministry of Economy, Trade and Industry (METI) of Japan, a Grant-in-Aid for Specially Promoted Research (no. 23000009) and for Young Scientists (B) (no. 25810112) of the Japan Society for the Promotion of Science (JSPS). JSPS also contributed through the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST), initiated by the Council for Science and Technology Policy (CSTP).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS PubMed.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS PubMed.
- T. Hisatomi, T. Minegishi and K. Domen, Bull. Chem. Soc. Jpn., 2012, 85, 647–655 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133–137 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- K. Maeda and K. Domen, Angew. Chem., Int. Ed., 2012, 51, 9865–9869 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, B. Siritanaratkul, R. Abe and K. Domen, J. Am. Chem. Soc., 2011, 133, 12334–12337 CrossRef CAS PubMed.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120–1124 RSC.
- I. E. Castelli, D. D. Landis, K. S. Thygesen, S. Dahl, I. Chorkendorff, T. F. Jaramillo and K. W. Jacobsen, Energy Environ. Sci., 2012, 5, 9034–9043 CAS.
- S. Balaz, S. H. Porter, P. M. Woodward and L. J. Brillson, Chem. Mater., 2013, 25, 3337–3343 CrossRef CAS.
- P. Reunchan, S. Ouyang, N. Umezawa, H. Xu, Y. Zhang and J. Ye, J. Mater. Chem. A, 2013, 1, 4221–4227 CAS.
- Y.-I. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 74–78 CrossRef CAS PubMed.
- S. S. K. Ma, K. Maeda, T. Hisatomi, M. Tabata, A. Kudo and K. Domen, Chem.–Eur. J., 2013, 19, 7480–7486 CrossRef CAS PubMed.
- M. Miyauchi, M. Takashio and H. Tobimatsu, Langmuir, 2003, 20, 232–236 CrossRef.
- T. Moriga, K. Ikeuchi, R. Mashima, D. Aoki and K. Murai, J. Ceram. Soc. Jpn., 2007, 115, 637–639 CrossRef CAS.
- A. E. Maegli, T. Hisatomi, E. H. Otal, S. Yoon, S. Pokrant, M. Grätzel and A. Weidenkaff, J. Mater. Chem., 2012, 22, 17906–17913 RSC.
- S. S. K. Ma, T. Hisatomi, K. Maeda, Y. Moriya and K. Domen, J. Am. Chem. Soc., 2012, 134, 19993–19996 CrossRef CAS PubMed.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386–19388 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental, SEM images, XRD patterns, DRS, hydrogen evolution reaction, and J–E curves of the samples. See DOI: 10.1039/c3ee42951b |
| This journal is © The Royal Society of Chemistry 2013 |