 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of hydrogen peroxide as a sustainable solar fuel from water and dioxygen†
Satoshi
Kato
a,
Jieun
Jung
a,
Tomoyoshi
Suenobu
a and
Shunichi
Fukuzumi
*ab
aDepartment of Material and Life Science, Division of Advanced Science and Biotechnology, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
bDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: wwnam@ewha.ac.kr
First published on 14th October 2013
Abstract
Hydrogen peroxide was produced as a solar fuel from water and dioxygen using solar energy by combination of a water oxidation catalyst and a photocatalyst for two-electron reduction of O2 in acidic aqueous solutions. Photocatalytic production of H2O2 occurred under photoirradiation of [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) used as a photocatalyst with visible light in the presence of Ir(OH)3 acting as a water oxidation catalyst in an O2-saturated H2SO4 aqueous solution. Photoinduced electron transfer from the excited state of [RuII(Me2phen)3]2+ to O2 results in the formation of [RuIII(Me2phen)3]3+ and a superoxide radical anion (O2˙−) which is protonated to produce H2O2via disproportionation of HO2˙ in competition with back electron transfer (BET) from O2˙− to [RuIII(Me2phen)3]3+. [RuIII(Me2phen)3]3+ oxidises water with the aid of catalysis of Ir(OH)3 to produce O2. The photocatalytic reactivity of H2O2 production was improved by replacing Ir(OH)3 nanoparticles by [CoIII(Cp*)(bpy)(H2O)]2+ in the presence of Sc(NO3)3 in water. The optimised quantum yield of the photocatalytic H2O2 production at λ = 450 nm was determined using a ferrioxalate actinometer to be 37%. The value of conversion efficiency from solar energy to chemical energy was also determined to be 0.25%.
Broader contextPhotocatalytic production of hydrogen peroxide from earth-abundant water and dioxygen using solar energy as an ideally sustainable solar fuel has remained a great challenge. We report herein for the first time photocatalytic production of hydrogen peroxide from water and dioxygen under visible light using [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst and Ir(OH)3 nanoparticles or [CoIII(Cp*)(bpy)(H2O)]2+ (Cp* = η5-pentamethylcyclopentadienyl, bpy = 2,2-bipyridine) as a water oxidation catalyst in water containing H2SO4 or Sc(NO3)3. A high turnover number and quantum yield have been attained by combining an efficient water oxidation catalyst with a photosensitiser and a Lewis acid in water. |
1. Introduction
Renewable and clean energy resources are urgently required in order to solve global energy and environmental issues.1,2 Among renewable energy resources, solar energy is by far the largest exploitable resource,1–6 and thereby it is quite important to obtain sustainable solar fuels such as hydrogen (H2) or others. Hydrogen is a clean energy source to reduce the dependence on fossil fuels and the emissions of greenhouse gases in the long term.7–17 However, the storage of hydrogen has been very difficult, because hydrogen is a gas having a low volumetric energy density.18,19 In contrast to hydrogen, hydrogen peroxide (H2O2), soluble in water, can be an ideal energy carrier alternative to oil or hydrogen, because it can be used in a one-compartment fuel cell leading to the generation of electricity.20–27 The output potential of H2O2 fuel cells theoretically achievable is 1.09 V, which is somewhat smaller but comparable to those of a hydrogen fuel cell (1.23 V) and a direct methanol fuel cell (1.21 V).22–27 Thus, a combination of hydrogen peroxide production using solar energy and power generation with a hydrogen peroxide fuel cell provides an ideally sustainable solar fuel.22 However, photocatalytic production of hydrogen peroxide from water (H2O) and dioxygen (O2) using solar energy has remained a great challenge.H2O2 is currently manufactured in industry by the autoxidation of 2-alkyl anthrahydroquinone by O2 to the corresponding 2-alkylanthraquinone (the so-called anthraquinone process) using a noble metal such as palladium to regenerate the anthrahydroquinone with H2.28 H2 used as a reductant is normally produced by steam reforming of natural gases, which emits a significant amount of CO2.28 H2O2 can also be produced by two-electron photoreduction of O2 with use of a semiconductor photocatalyst29 or a homogeneous photocatalyst.30–33 In this case, organic reductants such as 2-propanol, acetaldehyde and oxalate are required as sacrificial electron sources, resulting in unwanted emission of CO2.29–33
We report herein for the first time photocatalytic production of H2O2 from H2O and O2, both of which are earth abundant, without emission of CO2 by two-electron photoreduction of O2 by H2O that is used as an electron source in acidic aqueous solutions. The high turnover number and quantum yield have been attained by combining an efficient water oxidation catalyst (WOC) with a photosensitiser and a scandium ion that acts as a Lewis acid and facilitates two-electron reduction of O2. The present study will pave a way to utilise H2O2 produced from H2O and O2 as a sustainable solar fuel in an H2O2 fuel cell.22
2. Experimental section
2.1 Materials
All chemicals commercially available were used without further purification unless otherwise noted. H2IrCl6·5.5H2O (99.99%) and IrO2 (99%) were purchased from Furuya Metal. RuCl3·3H2O (38.220 wt% Ru) was purchased from Tanaka Kikinzoku Kogyo K.K. 4,7-Dimethyl-1,10-phenanthroline (Me2phen, 98%), tris(2,2′-bipyridyl)ruthenium(II) dichloride hexahydrate ([RuII(bpy)3]Cl2·6H2O), tert-butyllithium (1.7 M in n-pentane), Ag2SO4 (99.9%), (NH4)2SO4 (99.99%), n-pentane, Y(NO3)3·4H2O (99.99%), Lu(NO3)3·nH2O (99.999%) and Mg(NO3)2·6H2O (99.999%) were supplied by Aldrich Chemicals. MnO2 (85%), CoCl2 (95.0%), 2,2′-bipyridine (bpy, 99.0%), tetrahydrofuran and Ca(NO3)2·4H2O (99.9%) were purchased from Wako Pure Chemical Industries Ltd. Pentamethylcyclopentadiene was obtained from Kanto Chemical Co., Inc. Oxo[5,10,15,20-tetra(4-pyridyl)porphinato]titanium(IV) ([TiO(tpyp)]) was supplied by Tokyo Chemical Industry Co., Ltd. (TCI). Sc(NO3)3·4H2O (99.9%) was supplied by Mitsuwa Chemicals Co., Ltd. PbO2 (90%) and Zn(NO3)2·6H2O (99%) were supplied by Nacalai Tesque. 18O18O gas (≥98 at.%) was purchased from TAIYO NIPPON SANSO Corporation as a compressed gas in a cylinder (0.5 L). Carbon monoxide (CO) gas was purchased from Sumitomo Seika Chemicals Co., Ltd. as a compressed gas in a cylinder (3.4 L). Purification of water (18.2 MΩ cm) was performed with a Milli-Q system (Millipore, Direct-Q 3 UV).2.2 Synthesis of iridium hydroxide nanoparticles
Iridium hydroxide nanoparticles were synthesised according to the literature.34 The pH of an aqueous solution of H2IrCl6 was adjusted to ∼10 by adding 5.0 M NaOH solution with vigorous stirring at 100 °C. After 1.0 h stirring, precipitates appeared were collected by centrifugation. Then, the precipitates were washed by water three times, dried in vacuo at room temperature and kept at 65 °C for 10 h.2.3 Synthesis of [RuII(Me2phen)3]SO4
The tris(4,7-dimethyl-1,10-phenanthroline)ruthenium(II) sulphate ([RuII(Me2phen)3]SO4) complex was synthesised according to the literature.35 RuCl3 was refluxed under N2 overnight in ethanol–water (v/v 80/20) with 6 equiv. of ligand, Me2phen, to form the red-orange [RuII(Me2phen)3]Cl2 complex. After evaporation of the solvent, the product was readily precipitated from acetone with ether. The precipitate, [RuII(Me2phen)3]Cl2, was added to water to be completely dissolved and Ag2SO4 (65 mg) solubilised in water was added to the solution. After stirring for 12 h, AgCl was filtered off as a precipitate. An aqueous solution of (NH4)2SO4 was added to the reaction solution to obtain a crystalline product.2.4 Synthesis of [Co(Cp*)(bpy)(OH2)]SO4
The [Co(Cp*)(bpy)(OH2)]SO4 complex was synthesised according to the literature.36 Pentamethylcyclopentadiene (Cp*, 25 mL) and tert-butyllithium (∼1.7 M in n-pentane, 90 mL) were combined in an equimolar amount (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in n-pentane at 203 K. The solution was stirred and slowly allowed to warm up to room temperature. After stirring for further 24 h at room temperature, a white suspension was filtered through an inert gas frit and pentamethylcyclopentadienyllithium (Cp*Li) was filtered off. The anhydrous CoCl2 (1.32 g) was added to the solution of Cp*Li (1.42 g) in 20 mL of tetrahydrofuran. The mixture was stirred for 3 h at room temperature until the brown solution became green-brown. Afterwards the solution was concentrated to a smaller volume under reduced pressure and extracted with 100 mL of n-pentane. The brown extracts were bubbled by CO gas for 30 min through the solution. Di-μ-chloro-bis[chloro(η5-pentamethylcyclopentadienyl)cobalt] ([(μ-Cl)(CoCp*Cl)]2) was obtained as a green powder. The [(μ-Cl)(CoCp*Cl)]2 (100 mg) in 20 mL of water was stirred with 1.5 equimolar amount of 2,2′-bipyridine (88 mg) for 1 h at room temperature under N2. After filtering off free 2,2′-bipyridine, Ag2SO4 (118 mg) was added to the filtrate. After stirring for 12 h, AgCl was filtered off as a precipitate. An aqueous solution of (NH4)2SO4 was added to the reaction solution to obtain a crystalline product. 1H-NMR (300 MHz, D2O): δ (ppm) = 1.34 (s, 15H), 8.10 (t, J = 5.86 Hz, 2H), 8.43 (t, J = 8.06 Hz, 2H), 8.51 (d, J = 8.06 Hz, 2H), 9.85 (d, J = 5.50 Hz, 2H).
:1) in n-pentane at 203 K. The solution was stirred and slowly allowed to warm up to room temperature. After stirring for further 24 h at room temperature, a white suspension was filtered through an inert gas frit and pentamethylcyclopentadienyllithium (Cp*Li) was filtered off. The anhydrous CoCl2 (1.32 g) was added to the solution of Cp*Li (1.42 g) in 20 mL of tetrahydrofuran. The mixture was stirred for 3 h at room temperature until the brown solution became green-brown. Afterwards the solution was concentrated to a smaller volume under reduced pressure and extracted with 100 mL of n-pentane. The brown extracts were bubbled by CO gas for 30 min through the solution. Di-μ-chloro-bis[chloro(η5-pentamethylcyclopentadienyl)cobalt] ([(μ-Cl)(CoCp*Cl)]2) was obtained as a green powder. The [(μ-Cl)(CoCp*Cl)]2 (100 mg) in 20 mL of water was stirred with 1.5 equimolar amount of 2,2′-bipyridine (88 mg) for 1 h at room temperature under N2. After filtering off free 2,2′-bipyridine, Ag2SO4 (118 mg) was added to the filtrate. After stirring for 12 h, AgCl was filtered off as a precipitate. An aqueous solution of (NH4)2SO4 was added to the reaction solution to obtain a crystalline product. 1H-NMR (300 MHz, D2O): δ (ppm) = 1.34 (s, 15H), 8.10 (t, J = 5.86 Hz, 2H), 8.43 (t, J = 8.06 Hz, 2H), 8.51 (d, J = 8.06 Hz, 2H), 9.85 (d, J = 5.50 Hz, 2H).
2.5 Quantum yield and quantum efficiency measurements
The quantum yield (QY) of the photocatalytic production of hydrogen peroxide (Φ) was determined under irradiation of monochromatised light using a Shimadzu spectrofluorophotometer (RF-5300PC) through a band-pass filter transmitting λ = 450 nm, and estimated as| QY (%) = (2 × R/I) × 100 | (1) |
2.6 Measurements of conversion efficiency from solar energy
Typically, [CoIII(Cp*)(bpy)(H2O)]SO4 (10 mM) was added to distilled water (3.0 mL) containing [RuII(Me2phen)3]SO4 (100 μM) and Sc(NO3)3 (100 mM) in a quartz cuvette (light path length = 1 cm). The solution was saturated by bubbling with oxygen gas for ∼30 min. The photocatalyst was irradiated with a solar simulator (HAL-320, Asahi Spectra Co., Ltd.). The light intensity was adjusted to be 10 mJ cm−2 s−1 (Air Mass 1.5 (AM1.5)) at the sample position for the whole irradiation area (1.0 × 3.0 cm2) using a 1 SUN checker (CS-20, Asahi Spectra Co., Ltd.) at room temperature. The amount of produced hydrogen peroxide was determined by the titration with the Ti-TPyP reagent (vide supra).2.7 Spectral measurements
Typically, an H2SO4 aqueous solution (2.0 M, 3.0 mL) containing [RuII(Me2phen)3]SO4 (20 μM) in a quartz cuvette (light path length = 1 cm) was saturated with O2 by bubbling with oxygen gas for ∼30 min. The photocatalysts were irradiated with a Shimadzu spectrofluorophotometer (RF-5300PC) at λ = 450 nm with a slit width of 5.0 mm at room temperature. The generation of [RuIII(Me2phen)3]3+ was monitored by the decrease of the absorption band at λ = 445 nm due to [RuII(Me2phen)3]2+ (ε = 2.5 × 104 M−1 cm−1)39 as well as the increase in the absorption band at λ = 630 nm due to [RuIII(Me2phen)3]3+ (ε = 1.6 × 103 M−1 cm−1) using a Hewlett Packard 8453 diode array spectrophotometer.2.8 Isotope-labelling experiments
Ir(OH)3 (3.0 mg) was dispersed in an H2SO4 aqueous solution (2.0 M, 3.0 mL) containing [RuII(Me2phen)3]SO4 (20 μM) in a quartz cuvette (light path length = 1 cm). The solution was saturated by bubbling with 18O18O gas. The solution was irradiated with a xenon lamp (USHIO Optical Modulex SX-UID 501XAMQ) through a cut-off filter (Asahi Techno Glass L42) transmitting λ > 420 nm at room temperature. After 3 h, the solution was deaerated by bubbling with He gas for ∼30 min to exclude the 18O18O gas. At 10 min after addition of an excess amount of manganese oxide (MnO2) to the solution, 50 mL of the gas in a headspace was sampled using a gas-tight syringe for gas analysis. The ratio of 16O16O, 16O18O and 18O18O was determined based on the intensity of mass spectra (m/z = 32, 34 and 36) obtained using a Shimadzu GC-17A gas chromatograph [He carrier, TC-FFAP column (GL Science, 1010-15242) at 313 K] equipped with a mass spectrometer (Shimadzu, QP-5000).2.9 EPR measurements
The EPR spectrum was taken on a JEOL X-band spectrometer (JES-RE1XE) under nonsaturating microwave power conditions (1.0 mW) operating at 9.2 GHz. Distilled water (1.0 mL) containing [RuII(Me2phen)3]SO4 (20 μM) and Sc(NO3)3 (100 mM) in the EPR tube was saturated by bubbling with pure O2 for ∼30 min. The solution was frozen at 77 K after visible light irradiation (λ > 420 nm). The magnitude of the modulation was chosen to optimise the resolution and the signal to noise ratio (S/N) of the observed spectrum (modulation width, 2.0 G; modulation frequency, 100 kHz). The g values were calibrated using an Mn2+ marker.2.10 Characterisation of Ir(OH)3 nanoparticles
X-ray photoelectron spectra (XPS) were recorded using a Kratos Axis 165 with a 165 mm hemispherical electron energy analyser. The incident radiation was Mg Kα X-ray (1253.6 eV) at 200 W and a charge neutraliser was turned on for acquisition. Each sample was attached on a stainless stage with a double-sided carbon scotch tape. The binding energy of each element was corrected by the C 1s peak (284.6 eV) from residual carbon. TG/DTA data were recorded on an SII TG/DTA 7200 instrument. Each sample (∼3.0 mg) was heated from 298 K to 373 K (held at 373 K for 10 min) and from 373 K to 873 K with a ramp rate of 2 K min−1. A certain amount of γ-Al2O3 was used as a reference for DTA measurements. Nitrogen adsorption–desorption at 77 K was performed with a Belsorp-mini (BEL Japan, Inc.) within a relative pressure range from 0.01 to 101.3 kPa. A sample mass used for adsorption analysis was pretreated at 333 K for 30 min under vacuum conditions and kept in a N2 atmosphere until N2-adsorption measurements. The sample was exposed to a mixed gas of He and N2 with a programmed ratio and the adsorbed amount of N2 was calculated from the change of pressure in a cell after reaching equilibrium (at least 5 min). The transmission electron microscopy (TEM) image of iridium hydroxide, which was mounted on a copper microgrid coated with elastic carbon, was observed using a JEOL JEM-2100 operating at 200 keV.3. Results and discussion
3.1 Characterisation of Ir(OH)3 nanoparticles
The TG curve of Ir(OH)3 nanoparticles is shown in Fig. 1a, which exhibits two consecutive steps. The first step of weight loss with an endothermic peak at 393 K corresponds to the removal of physisorbed water. The weight loss at the second step of Ir(OH)3 starting from 583 K was attributed to dehydration of Ir(OH)3. The TG curve of commercially available IrO2 in Fig. 1b showed no such dehydration step.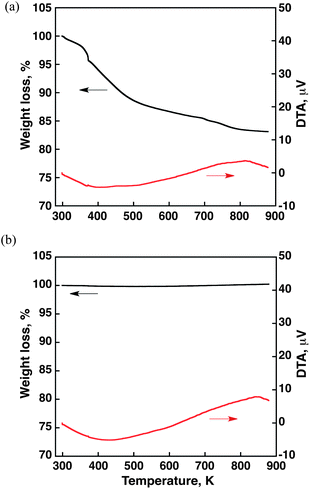 | ||
| Fig. 1 TG/DTA data for (a) Ir(OH)3 and (b) IrO2 (TG curve: black, DTA curve: red). The temperature increased from 298 K to 873 K with a ramp rate of 2 K min−1. | ||
To determine surface conditions of Ir(OH)3, X-ray photoelectron spectroscopy (XPS) measurements were carried out for the energy regions of Ir 4f, O 1s and C 1s with reference to commercially available IrO2. As reported previously, the binding energy of Ir 4f5/2 reflects the valence of Ir ions sensitively where the binding energies of Ir 4f5/2 for Ir0, IrIII and IrIV are reported to be 61.0 eV, 62.0 eV and 63.7 eV, respectively.40–42 The XPS spectra of Ir 4f and O 1s for the iridium hydroxide and IrO2 are shown in Fig. 2. The binding energy of Ir 4f5/2 of both the iridium hydroxide and IrO2 was 62.2 eV, which is close to the reported binding energy of 62.0 eV for Ir(III) species. The O 1s peaks of the iridium hydroxide and IrO2 appeared at 531.5 eV and 530.5 eV, respectively. The higher binding energy of the O 1s peak of the iridium hydroxide results from the formation of hydroxide species as often reported previously.43,44
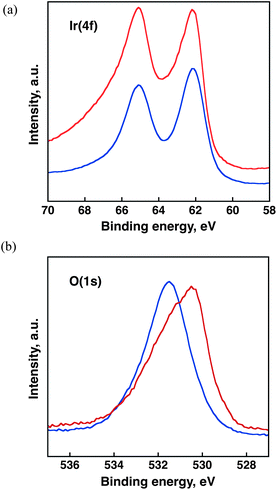 | ||
| Fig. 2 X-ray photoelectron spectra in the binding energy regions of Ir 4f and O 1s for (a) Ir(OH)3 (blue line) and (b) IrO2 (red line). | ||
Iridium hydroxide was investigated by transmission electron microscopy (TEM). The TEM image is displayed in Fig. 3, which indicates that the size is in the range of 50–100 nm with an undefined shape. BET surface areas of Ir(OH)3 and IrO2 (commercially available) are shown in Table S1 in the ESI.† The BET surface area of Ir(OH)3 was 28 times higher than that of IrO2.
 | ||
| Fig. 3 TEM image of Ir(OH)3 nanoparticles. | ||
3.2 Photocatalytic production of H2O2 from H2O and O2 with Ir(OH)3 nanoparticles
Photoexcitation of [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) results in the generation of [RuIII(Me2phen)3]3+ by oxidative quenching of the photoexcited state ([RuII(Me2phen)3]2+*: * denotes the excited state) with O2 in the presence of H2SO4 to produce H2O2 as reported for the case of [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine).45 The time courses of [RuIII(Me2phen)3]3+ generation and H2O2 production are shown in Fig. 4. One-half equiv. of H2O2 was produced, accompanied by the formation of [RuIII(Me2phen)3]3+ under photoirradiation of [RuII(Me2phen)3]2+ in an air-saturated aqueous solution containing 2.0 M H2SO4. Thus, the stoichiometry of the photochemical reaction is given by eqn (2). | (2) |
![Time courses of [RuIII(Me2phen)3]3+ generation (red line) and H2O2 production (blue line) under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ = 450 nm) in an air-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 0.25 mM).](/image/article/2013/EE/c3ee42815j/c3ee42815j-f4.gif) | ||
| Fig. 4 Time courses of [RuIII(Me2phen)3]3+ generation (red line) and H2O2 production (blue line) under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ = 450 nm) in an air-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 0.25 mM). | ||
The quantum yield (Φ) of generation of [RuIII(Me2phen)3]3+ (21%) at the initial stage (0–1 min) was significantly larger than that of [RuIII(bpy)3]3+ (1.6%) in the presence of 2.0 M H2SO4 (Fig. S1 in the ESI†) because the one-electron oxidation potential of [Ru(Me2phen)3]2+* (E0red* = −1.01 V vs. NHE) is more negative than [Ru(bpy)3]2+* (E0red* = −0.84 V vs. NHE).46 The Φ value at the initial stage (0–1 min) increased with increasing the concentration of H2SO4 to reach 72% in the presence of 4.0 M H2SO4 (Table S2 in the ESI†).
With regard to a water oxidation catalyst (WOC), an efficient but acid-stable catalyst must be used for the photocatalytic production of H2O2 from H2O and O2. Ir(OH)3 nanoparticles34 are found to be suitable for this purpose. The photocatalytic production of H2O2 from H2O and O2 [eqn (3)] was examined using [RuII(Me2phen)3]SO4 as a photosensitiser for the two-electron reduction of O2 and Ir(OH)3 nanoparticles as a WOC in an O2-saturated H2SO4 aqueous solution (2.0 M). The sulphate complex was used to avoid possible oxidation of the chloride anion during the photocatalytic water oxidation.
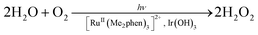 | (3) |
The time courses of H2O2 production in the solution are shown in Fig. 5a. The turnover number (TON) based on [RuII(Me2phen)3]2+ was determined to be 307 after 18 h photoirradiation. No H2O2 production was observed from a reaction solution without [RuII(Me2phen)3]2+. The photocatalytic reactivity increased with increasing the concentration of H2SO4 and exhibits a maximum value at 2.0 M H2SO4 (Fig. S2 in the ESI†). At H2SO4 concentrations higher than 2.0 M, the catalytic reactivity of Ir(OH)3 nanoparticles decreased because Ir(OH)3 nanoparticles became partially dissolved under strongly acidic conditions. Ir(OH)3 nanoparticles were clearly dissolved in water with 3.0 M H2SO4. The photocatalytic reactivity increased with increasing the amount of Ir(OH)3 nanoparticles, but it decreased through a maximum value with further increase in the amount of Ir(OH)3 (Fig. 5b) because of the competition of the visible light absorption of [RuII(Me2phen)3]2+ with that of Ir(OH)3 nanoparticles. The Φ value of the photocatalytic H2O2 production at λ = 450 nm was determined using a ferrioxalate actinometer to be 20% (0–30 min, Fig. S3 in the ESI†), which agrees with the Φ value of generation of [RuIII(Me2phen)3]3+ without WOC (vide supra).
![(a) Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (1.0 μM) in the presence of Ir(OH)3 (3.0 mg) (red circles) and its absence (blue squares) in an O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM). A control experiment without [RuII(Me2phen)3]2+ has been done in the presence of Ir(OH)3 (3.0 mg) (green triangles). (b) Dependence of the amount of H2O2 produced after 1 h on the amounts of Ir(OH)3 under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in an O2-saturated H2SO4 aqueous solution.](/image/article/2013/EE/c3ee42815j/c3ee42815j-f5.gif) | ||
| Fig. 5 (a) Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (1.0 μM) in the presence of Ir(OH)3 (3.0 mg) (red circles) and its absence (blue squares) in an O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM). A control experiment without [RuII(Me2phen)3]2+ has been done in the presence of Ir(OH)3 (3.0 mg) (green triangles). (b) Dependence of the amount of H2O2 produced after 1 h on the amounts of Ir(OH)3 under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in an O2-saturated H2SO4 aqueous solution. | ||
Isotope-labelling experiments using 18O18O instead of 16O16O were conducted to obtain direct evidence for the photocatalytic H2O2 production, in which the produced H2O2 comes from O2 in the gas phase. After the reaction, the solution was carefully deaerated by bubbling with He gas to remove 18O18O and an excess of MnO2, which catalysed the decomposition of H2O2 to O2, was added to the solution. The evolved oxygen in the headspace of a reaction tube was separated using a gas chromatograph equipped with a molecular sieve column and analysed using a mass spectrometer (Fig. 6). Evolved O2 was 18O18O (99%). There was no 16O18O or 16O16O incorporated from H216O. This has confirmed that the produced H2O2 came from O2 in the gas phase (not from water). The amount of evolved O2 (0.76 mmol) in water oxidation reaction was only 0.71% as compared with the amount of introduced 18O18O (106 mmol). This is the reason why the evolution of O2 containing 16O from water was negligible.
![Comparison of relative abundance of 18O-labelled and unlabelled O2, which was evolved by disproportionation of H2O2 produced in the photocatalytic reduction of 18O18O gas (≥98 at.%) containing [RuII(Me2phen)3]2+ (20 μM) and Ir(OH)3 (3.0 mg) in an 18O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM) under photoirradiation (λ > 420 nm) for 1 h.](/image/article/2013/EE/c3ee42815j/c3ee42815j-f6.gif) | ||
| Fig. 6 Comparison of relative abundance of 18O-labelled and unlabelled O2, which was evolved by disproportionation of H2O2 produced in the photocatalytic reduction of 18O18O gas (≥98 at.%) containing [RuII(Me2phen)3]2+ (20 μM) and Ir(OH)3 (3.0 mg) in an 18O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM) under photoirradiation (λ > 420 nm) for 1 h. | ||
3.3 Effect of Lewis acidity of metal ions on photocatalytic production of H2O2 from H2O and O2
The photocatalytic production of H2O2 was also examined using metal nitrates, (M(NO3)n), acting as Lewis acids in water instead of H2SO4 under otherwise the same reaction conditions. The dependence of the photocatalytic reactivity of H2O2 production on the Lewis acidity of metal ions under the same pH conditions (pH 2.8) was examined as shown in Fig. 7, where Sc(NO3)3 exhibited the highest reactivity, which is in agreement with the strongest Lewis acidity of Sc3+.47–51 The Φ value of the photocatalytic H2O2 production in the presence of Sc(NO3)3 under visible light at λ = 450 nm was determined to be 12% (0–3 h, Fig. S4 in the ESI†).![H2O2 production in the absence and presence of metal ions with Lewis acidity under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) for 1 h in the presence of Ir(OH)3 (3.0 mg) and M(NO3)n (Mn+ = Sc3+, Y3+, Yb3+, Lu3+, Zn2+, Mg2+ and Ca2+, 100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM). The pH values of the solutions were adjusted to 2.8 by adding HNO3.](/image/article/2013/EE/c3ee42815j/c3ee42815j-f7.gif) | ||
| Fig. 7 H2O2 production in the absence and presence of metal ions with Lewis acidity under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) for 1 h in the presence of Ir(OH)3 (3.0 mg) and M(NO3)n (Mn+ = Sc3+, Y3+, Yb3+, Lu3+, Zn2+, Mg2+ and Ca2+, 100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM). The pH values of the solutions were adjusted to 2.8 by adding HNO3. | ||
The effect of Sc(NO3)3 on photoinduced electron transfer from [RuII(Me2phen)3]2+* to O2 was examined by nanosecond laser flash photolysis measurements. The rate constants (ket) of photoinduced electron transfer were determined by the emission quenching in the absence and presence of Sc(NO3)3 (Fig. S5 in the ESI†). The ket values in both the absence and presence of Sc(NO3)3 are close to the diffusion-limited value (Table S3†). Thus, Sc(NO3)3 does not affect the oxidative quenching of [RuII(Me2phen)3]2+* by O2.
The effect of Sc(NO3)3 on back electron transfer from O2˙− to [RuIII(Me2phen)3]3+ was examined by nanosecond laser flash photolysis measurements. The bleaching of absorption at λ = 445 nm due to [RuII(Me2phen)3]2+ and the rapid recovery was observed in photoexcitation of [RuII(Me2phen)3]2+ under O2 due to back electron transfer from O2˙− to [RuIII(Me2phen)3]3+ (Fig. 8a). In the presence of Sc3+, however, the recovery of absorption becomes significantly slower because of the binding of Sc3+ to O2˙−, which prohibits the back electron transfer (Fig. 8b). The slower recovery was also observed for other metal ions (Fig. 9).
![Transient absorption spectra of [RuII(Me2phen)3]2+ (20 μM) in O2-saturated H2O (a) in the absence of Sc(NO3)3 and (b) in the presence of Sc(NO3)3 (10 mM) after laser excitation at λ = 430 nm.](/image/article/2013/EE/c3ee42815j/c3ee42815j-f8.gif) | ||
| Fig. 8 Transient absorption spectra of [RuII(Me2phen)3]2+ (20 μM) in O2-saturated H2O (a) in the absence of Sc(NO3)3 and (b) in the presence of Sc(NO3)3 (10 mM) after laser excitation at λ = 430 nm. | ||
![Time profiles at λ = 445 nm for back electron transfer from [RuIII(Me2phen)3]3+ to O2˙− in O2-saturated H2O containing [RuII(Me2phen)3]2+ (20 μM) in the presence of various concentrations of (a) Sc(NO3)3 (0–5.0 μM), (b) Y(NO3)3 (0–10 mM) and (c) Lu(NO3)3 (0–10 mM).](/image/article/2013/EE/c3ee42815j/c3ee42815j-f9.gif) | ||
| Fig. 9 Time profiles at λ = 445 nm for back electron transfer from [RuIII(Me2phen)3]3+ to O2˙− in O2-saturated H2O containing [RuII(Me2phen)3]2+ (20 μM) in the presence of various concentrations of (a) Sc(NO3)3 (0–5.0 μM), (b) Y(NO3)3 (0–10 mM) and (c) Lu(NO3)3 (0–10 mM). | ||
Thus, the acceleration effect of Sc(NO3)3 on the photocatalytic production of H2O2 from H2O and O2 results from the inhibition of back electron transfer by binding of Sc3+ to O2˙−, which leads to more efficient generation of [RuIII(Me2phen)3]3+. In addition, the scandium ion inhibited disproportionation of H2O2 by Ir(OH)3 under the same pH conditions as shown in Fig. 10.
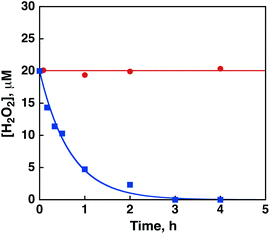 | ||
| Fig. 10 Time courses of the concentration of H2O2 in the presence of Ir(OH)3 (3.0 mg) in H2O (3.0 mL) containing H2O2 (20 μM) and Sc(NO3)3 (100 mM) (red circles) at pH 2.8 and in an HNO3 aqueous solution (pH 2.8, 3.0 mL) containing H2O2 (20 mM) (blue squares). | ||
The formation of the O2˙−–Sc3+ complex during the photooxidation of [RuII(Me2phen)3]2+ with O2 in the presence of Sc(NO3)3 was confirmed by EPR measurements. When O2-saturated H2O of [RuII(Me2phen)3]2+ in the presence of Sc(NO3)3 was irradiated with visible light (λ > 420 nm), the O2˙−–Sc3+ complex was detected by the EPR spectrum in frozen H2O at 77 K as shown in Fig. 11, where the superhyperfine structure due to the binding of Sc3+ (I = 7/2) was observed at gzz. The gzz value of O2˙−–Sc3+ (2.036) is larger than the value reported in frozen acetonitrile at 143 K (2.030),47,52 because Sc3+ is easily hydrolysed in H2O and the Lewis acidity of Sc3+ in H2O is weaker than that in acetonitrile.41
![EPR spectrum of the O2˙−–Sc3+ complex under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) in the presence of Sc(NO3)3 (100 mM) in frozen O2-saturated H2O at 77 K.](/image/article/2013/EE/c3ee42815j/c3ee42815j-f11.gif) | ||
| Fig. 11 EPR spectrum of the O2˙−–Sc3+ complex under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) in the presence of Sc(NO3)3 (100 mM) in frozen O2-saturated H2O at 77 K. | ||
3.4 Improvement of photocatalytic reactivity with a cobalt complex as WOC
The photocatalytic reactivity of H2O2 production was further improved by replacing Ir(OH)3 nanoparticles by [CoIII(Cp*)(bpy)(H2O)]2+.36 The time courses of H2O2 production using [CoIII(Cp*)(bpy)(H2O)]2+ as a WOC are shown in Fig. 12. The reactivity using [CoIII(Cp*)(bpy)(H2O)]2+ is 4.3 times higher than that using Ir(OH)3 at the initial stage (1 h). The TON based on [RuII(Me2phen)3]2+ was determined to be 612 after 9 h photoirradiation.![Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (1.0 μM) in the presence of Ir(OH)3 (3.0 mg) in an O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM) (red circles), in the presence of Ir(OH)3 (3.0 mg) and Sc(NO3)3 (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) (blue squares), and in the presence of [CoIII(Cp*)(bpy)(H2O)]2+ (10 mM) and Sc(NO3)3 (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) (green diamonds).](/image/article/2013/EE/c3ee42815j/c3ee42815j-f12.gif) | ||
| Fig. 12 Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (1.0 μM) in the presence of Ir(OH)3 (3.0 mg) in an O2-saturated H2SO4 aqueous solution (2.0 M, 3.0 mL, [O2] = 1.2 mM) (red circles), in the presence of Ir(OH)3 (3.0 mg) and Sc(NO3)3 (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) (blue squares), and in the presence of [CoIII(Cp*)(bpy)(H2O)]2+ (10 mM) and Sc(NO3)3 (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) (green diamonds). | ||
After photoirradiation for 3 hours, the NMR peaks assignable to bpy and Cp* still remained, indicating that [CoIII(Cp*)(bpy)(H2O)]2+ remains as a homogeneous catalyst during the reaction (Fig. S6 in the ESI†). The photocatalytic reactivity increased with increasing the concentration of [CoIII(Cp*)(bpy)(H2O)]2+ (Fig. S7 in the ESI†). The concentration of [CoIII(Cp*)(bpy)(H2O)]2+ is limited less than ca.10 mM because of the solubility.
The TON based on [CoIII(Cp*)(bpy)(H2O)]2+ was determined to be 61 after 9 h photoirradiation. Thus, [RuII(Me2phen)3]2+ and [CoIII(Cp*)(bpy)(H2O)]2+ act as an efficient homogeneous photocatalyst and water oxidation catalyst, respectively. The Φ value of the photocatalytic H2O2 production under visible light irradiation at λ = 450 nm was determined using a ferrioxalate actinometer to be 37% (0–30 min, Fig. S8a in the ESI†). The value of conversion efficiency from solar energy to chemical energy was also determined to be 0.25% (0–10 min, Fig. S8b in the ESI†). This value has reached the solar energy conversion efficiency of switchgrass (0.2%),53 a promising crop for biomass fuel.
In conclusion, efficient photocatalytic production of H2O2 from H2O and O2 has been achieved using [RuII(Me2phen)3]2+ as a photocatalyst and Ir(OH)3 nanoparticles or [CoIII(Cp*)(bpy)(H2O)]2+ as a WOC in water containing H2SO4 or Sc(NO3)3 as shown in Scheme 1. The photocatalytic production of H2O2 from H2O and O2 using solar energy reported in this paper provides the most convenient and sustainable solar fuel that can be converted to electricity using an H2O2 fuel cell. Further improvement of the catalytic reactivity and the more detailed elucidation of the catalytic mechanism are now in progress.
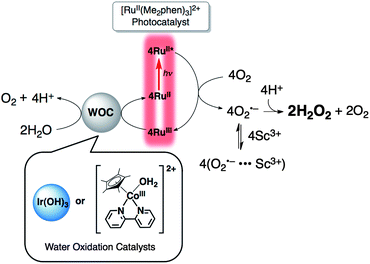 | ||
| Scheme 1 | ||
Acknowledgements
This work was supported by a Grant-in-Aid (no. 20108010), and a Global COE program, “the Global Education and Research Centre for Bio-Environmental Chemistry” from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by NRF/MEST of Korea through CRI, WCU (R31-2008-000-10010-0), GRL (2010-00353) Programs. We sincerely acknowledge Research Centre for Ultra-Precision Science & Technology for TEM measurements.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- D. G. Nocera, Chem. Soc. Rev., 2009, 38, 13–15 RSC.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351–1362 CrossRef CAS.
- T. E. Mallouk, J. Phys. Chem. Lett., 2010, 1, 2738–2739 CrossRef CAS.
- B. Sorensen, Hydrogen and fuel cells, Elsevier Academic Press, London, UK, 2005 Search PubMed.
- Hydrogen as a Future Energy Carrier, ed. A. Zuttel, A. Borgschulte, L. Schlapbach, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Fukuzumi, Y. Yamada, T. Suenobu, K. Ohkubo and H. Kotani, Energy Environ. Sci., 2011, 4, 2754–2766 CAS.
- S. Fukuzumi and Y. Yamada, J. Mater. Chem., 2012, 22, 24284–24296 RSC.
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295–295 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- W. J. Youngblood, S. H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photon., 2012, 6, 511–518 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- S. Fukuzumi and T. Suenobu, Dalton Trans., 2013, 42, 18–28 RSC.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 CAS.
- R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS PubMed.
- A. E. Sanli and A. Aytac, Int. J. Hydrogen Energy, 2011, 36, 869–875 CrossRef CAS PubMed.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334–7336 RSC.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 CAS.
- S. A. M. Shaegh, N. T. Nguyen, S. M. M. Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225–8228 Search PubMed.
- S. Yamazaki, Z. Siroma, H. Senoh, T. Loroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Chem.–Eur. J., 2013, 19, 11733–11741 CrossRef CAS PubMed.
- F. Sandelin, P. Oinas, T. Salmi, J. Paloniemi and H. Haario, Ind. Eng. Chem. Res., 2006, 45, 986–992 CrossRef CAS.
- S. Anandan and M. Miyauchi, Chem. Commun., 2012, 48, 4323–4325 RSC.
- H. Kotani, K. Ohkubo and S. Fukuzumi, Appl. Catal., B, 2008, 77, 317–324 CrossRef CAS PubMed.
- Y. Yamada, A. Nomura, T. Miyahigashi and S. Fukuzumi, Chem. Commun., 2012, 48, 8329–8331 RSC.
- Y. Yamada, A. Nomura, T. Miyahigashi, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2013, 117, 3751–3760 CrossRef CAS PubMed.
- Y. Yamada, A. Nomura, K. Ohkubo, T. Suenobu and S. Fukuzumi, Chem. Commun., 2013, 49, 5132–5134 RSC.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708–5716 CAS.
- C. Turro, J. M. Zaleski, Y. M. Karabatsos and D. G. Nocera, J. Am. Chem. Soc., 1996, 118, 6060 CrossRef CAS.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y. M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS.
- C. Turro, J. M. Zaleski, Y. M. Karabatsos and D. G. Nocera, J. Am. Chem. Soc., 1996, 118, 6060–6067 CrossRef CAS.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781–1784 RSC.
- B. Saha and D. M. Stanbury, Inorg. Chem., 2000, 39, 1294–1300 CrossRef CAS.
- M. Hara, K. Asami, K. Hashimoto and T. Masumoto, Electrochim. Acta, 1983, 28, 1073–1081 CrossRef CAS.
- H. Y. Hall and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 135–152 RSC.
- G. Q. Wei, Y. X. Wang, C. D. Huang, Q. J. Gao, Z. T. Wang and L. Xu, Int. J. Hydrogen Energy, 2010, 35, 3951–3957 CrossRef CAS PubMed.
- J. Yang, H. W. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CAS.
- Y. Yamada, K. Yano, Q. A. Xu and S. Fukuzumi, J. Phys. Chem. C, 2010, 114, 16456–16462 CAS.
- A. Das, V. Joshi, D. Kotkar, V. S. Pathak, V. Swayambunathan, P. V. Kamat and P. K. Ghosh, J. Phys. Chem. A, 2001, 105, 6945–6954 CrossRef CAS.
- C. T. Lin, W. Boettcher, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1976, 98, 6536–6544 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, J. Am. Chem. Soc., 2002, 124, 10270–10271 CrossRef CAS PubMed.
- K. Ohkubo, S. C. Menon, A. Orita, J. Otera and S. Fukuzumi, J. Org. Chem., 2003, 68, 4720–4726 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Nat. Chem., 2010, 2, 756–759 CrossRef CAS PubMed.
- S. Fukuzumi, M. Patz, T. Suenobu, Y. Kuwahara and S. Itoh, J. Am. Chem. Soc., 1999, 121, 1605–1606 CrossRef CAS.
- A. Melis, Plant Sci., 2009, 177, 272–280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c3ee42815j |
| This journal is © The Royal Society of Chemistry 2013 |