Charge generation and transport in efficient organic bulk heterojunction solar cells with a perylene acceptor†
Received
23rd July 2013
, Accepted 28th October 2013
First published on 29th October 2013
Broader context
The advantages of non-fullerene derivatives as electron acceptors in BHJ solar-cells include facile synthesis-scaling and purification procedures, large optical absorption and range of tunable energy levels. However, it has been challenging to obtain uniform heterogeneous films of non-fullerene based BHJs which feature efficient photoinduced charge generation and transport. In this work, we investigate a relatively high power-conversion efficiency BHJ solar-cell employing non-planar twisted perylene as the electron acceptor. A combined absorption of a perylene acceptor along with a low bandgap donor-polymer covers nearly the entire visible spectrum. Intimate mixing of donor and acceptor molecules by disrupting the planarity of perylene exhibits strong photoluminescence quenching of both donor and acceptor. Photo-charge generation from both perylene and polymer excitons results in an external quantum efficiency of ≈45%. It was observed that in these blends, electron and hole transfer processes occur on similar time scales (≈few ps). Efficient charge generation and transport are observed in composition optimized devices resulting in a power conversion efficiency of 3.2%. The molecular, interfacial and morphology aspects of BHJs with non-planar perylene as an acceptor suggest its role as a promising model system and pave the way for efficient, fullerene-free BHJ solar-cells.
|
Introduction
Organic photovoltaic cells are a promising and cost-effective alternative to other solar energy technologies. Light-weight, low-cost and flexibility of these devices show advantages for niche applications. During the past five years, significant advances have been made in terms of increasing power conversion efficiency of bulk heterojunction (BHJ) organic solar cells (OSCs) reaching over 9.2%.1 The active layer of high efficiency devices consists of low bandgap donor–acceptor co-polymers as electron donors and a fullerene based derivative as the electron acceptor. Most of these donor–acceptor polymers have extended absorption λ > 800 nm with an absorption gap in the high energy visible region (λ ≈ 400–600 nm). Due to increased absorption in the visible region, [6,6]-phenyl C71-butyric acid methyl ester (C71-PCBM)2 is preferred over [6,6]-phenyl C61-butyric acid methyl ester (C61-PCBM)3 to fill the absorption gap in BHJs. The monopoly of fullerene based materials as successful electron acceptors is due to a variety of factors, with particular emphasis on a favorable nanoscale morphology.4 However, there are important limiting factors for fullerenes beside practical constraints and elaborate purification procedures. This includes relatively low absorption of fullerene acceptors in the visible region. Additionally, the limited tunability of fullerene energy levels limits strategies to minimize energetic losses within the device.5,6 In this regard, perylenes with good electron mobility are potential alternatives to fullerenes as electron acceptors in BHJ OSC's, due to the following aspects: (i) the flexibility of functionalizing the molecule allows control of the electronic structure and tuning of its energy levels;7 this strategy can be implemented to optimize the device Voc and Jsc; (ii) the overall usage of light across the solar spectrum can be enhanced due to the significantly increased acceptor optical absorption in the visible region which can compensate for a donor spectral-gap8,9 and (iii) high photostability.10
Power conversion efficiencies of perylene diimide (PDI) (which until recently have been typically planar) based BHJ OSC's have so far been below those of the corresponding fullerene devices.11–17 We have recently reported on the synthesis and device performance of a twisted perylene (TP), whereby disrupting the planarity of the perylene helped to increase the short circuit current density and device efficiency (>2.75%) (Fig. 1a).18 The relatively high performance of the twisted perylene based BHJ device was attributed to a combination of factors including improved morphology, optimal electronic donor–acceptor (D–A) interface which can reduce early-recombination losses and an increase in PDI dimensionality which can lead to a lower barrier for charge separation. In this paper, we address the impact of these factors upon the enhanced device performance and suggest strategies to overcome the loss processes in these blend systems. Here, we use steady-state absorption and photoluminescence spectroscopy, transient absorption measurements, and nano-morphological probes, and correlate these with device data as a function of blend composition to determine differences in morphology, exciton migration, charge-transfer and charge-recombination dynamics of these polymer–TP blend films at different composition ratios. These data are compared against control data collected with PCBM blend films. The role of morphology in these polymer–TP systems and its correlation with the device performance is highlighted, suggesting routes to higher efficiency device performance for the non-fullerene BHJ solar cells.
 |
| | Fig. 1 (a) Structure of twisted perylene (TP) and PBDTTT-CT. (b) Absorption spectra of TP, PDBTTT-CT and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of PBDTTT-CT:TP films; films were spun on glass substrates at 1500 rpm for 60 s from a 12 mg ml−1 concentrated chlorobenzene solution. :1 ratio of PBDTTT-CT:TP films; films were spun on glass substrates at 1500 rpm for 60 s from a 12 mg ml−1 concentrated chlorobenzene solution. | |
Results
Blend films of electron-donor polymer poly[4,8-bis-(2-ethylhexyloxy)-benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl-alt-4-(2-ethylhexyloxy-1-one)thieno[3,4-b]thiophene-2-yl-2-ethylhexan-1-one] (PBDTTT-CT)19 and electron-acceptor TP (Fig. 1a) were chosen for the present study. Ground state UV-visible absorption spectra of PBDTTT-CT and TP films exhibit maximum absorbance at 720 nm and 540 nm respectively (Fig. 1b). The low-energy absorption corresponds to excitation of the PBDTTT-CT singlet exciton. The blends of different ratios exhibit the additive nature of absorption of the PBDTTT-CT:TP components. The steady state photoluminescence (PL) spectra of neat PBDTTT-CT and TP films indicate emission maxima, λemission, at 820 nm and 650 nm respectively. Comparison of the PL of 1:1 blend films with that of neat films indicates complete quenching (>99%) of both the PBDTTT-CT and TP emission, and suggests efficient charge transfer between PBDTTT-CT and TP driven by both PBDTTT-CT and TP excitons (Fig. 2). PL quenching is an indicator of the efficiency of exciton quenching at the D–A interface; however it is not a reliable measure of the yield of fully dissociated charges. In particular it is insensitive to the non-radiative geminate recombination of the initially generated polaron pairs (or ‘charge-transfer (CT)’ states) prior to their dissociation into separated charges.20 As such PL quenching can only provide an indication of an upper limit to the yield of dissociated charges. In order to quantify this yield further, transient absorption spectroscopy (TAS) is employed to monitor the yield of dissociated polarons in such donor:acceptor blend films.21–23
 |
| | Fig. 2 Steady state PL spectra of films (a) excited at 543 nm showing emission maxima at 650 nm and 820 nm for pristine TP (blue) and PBDTTT-CT (red) respectively and quench to >99% (black) by blending PBDTTT-CT and TP in 1:1 weight ratio [inset: schematic of hole transfer mechanism], and (b) excited at 632 nm showing an emission maximum at 820 nm for pristine PBDTTT-CT (red) which quenches to 99% (black) by blending PBDTTT-CT and TP in 1:1 weight ratio [inset: schematic of electron transfer]. | |
μs-Transient absorption spectroscopy
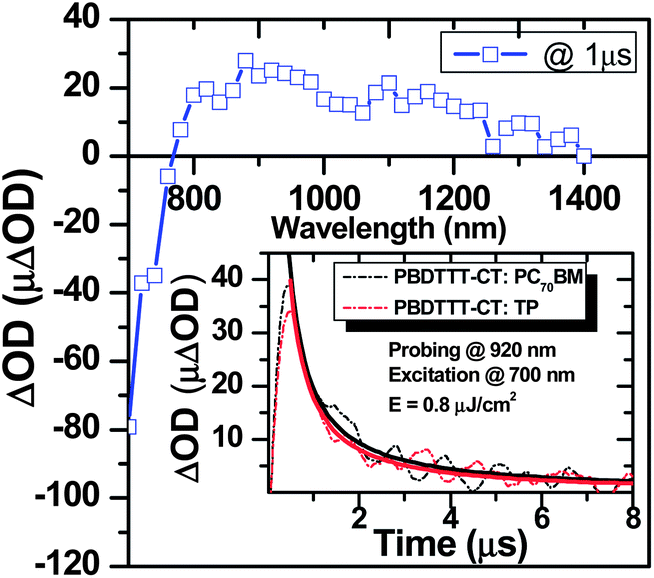
Microsecond transient absorption studies of blend films were carried out under low-intensity (typically 0.8 μJ cm−2) excitation conditions. Control data on neat TP and PCBM gave negligible transient signals on the time scales studied. The transient absorption spectrum ΔOD(t) of a PBDTTT-CT:TP blend film at t ≈ 1 μs also indicates enhanced broad near infrared absorption in the range 800–1400 nm assigned to formation of PBDTTT-CT positive polarons (Fig. 3). The negative magnitude of the ΔOD(t) signal for λ < 700 nm arises from the photobleaching of the PBDTTT-CT ground state (which appears to dominate over any PDI anion absorption expected in this spectral region). The transient signals exhibited oxygen-independent decay dynamics, indicative of their assignment to polarons rather than triplet absorption. These signals are an order of magnitude in excess of those of a neat PBDTTT-CT film measured under same conditions. The amplitudes of the transient signals were observed to vary approximately linearly with excitation density up to 4 μJ cm−2 (ESI, Fig. S1†), indicating that neither saturation effects nor non-geminate recombination losses prior to 1 μs distorted this comparison significantly. It should be noted that for other perylene systems, this saturation occurs at much higher intensities.26 The decay of these absorption transients exhibited power law decays (ΔOD(t) = At−α), consistent with the non-geminate recombination of dissociated charge carriers, as we have discussed previously for other polymer:acceptor blends.21,24,25 This transient absorption signal is therefore assigned to the transient absorption of dissociated PBDTTT-CT polarons.
 |
| | Fig. 3 μs-Transient absorption spectra of a PBDTTT-CT:TP (1:1) film, pumped at 640 nm and probed at 1 μs showing a broad absorption from 740 nm to 1400 nm having a maximum at 900 nm. The inset compares decay dynamics of positive polaron absorption of PBDTTT-CT at 920 nm by blending with TP and PC70BM acceptors, exhibiting power law decay (ΔOD(t) = At−α) (solid lines) with similar amplitudes and decay kinetics. (Excitation at 700 nm, 0.8 μJ cm−2.) | |
The inset to Fig. 3 shows a comparison of this PBDTTT-CT polaron signal between PBDTTT-CT:TP and PBDTTT-CT:PCBM films, employing matched excitation densities (0.8 μJ cm−2). It is apparent that the two transients are very similar in amplitude and decay dynamics, indicating similar charge generation and recombination in these two blend films.
In order to quantify effects of charge generation and charge collection from both PBDTTT-CT and TP excitons in the blend devices, we measured the incident photon to current conversion efficiency (IPCE) of the system at different acceptor composition ratios. In the λ range of 400–600 nm, the larger oscillator strength of TP compared to PBDTTT can result in a higher photoinduced hole transfer from TP excitons. Similarly, in the 600–800 nm range the higher absorption of PBDTTT-CT should predominantly lead to electron transfer from PBDTTT-CT excitons. The IPCE spectrum follows the trend of the blend absorption and indicates charge generation from both TP and PBDTTT-CT excitons (Fig. 4a). A maximum of 46% IPCE is observed for the 1:1 ratio blend at λ ∼ 500 nm, showing higher contribution to photocurrent from TP absorption. Increasing the TP ratio in the blend, while maintaining constant film thickness, decreases the overall IPCE of the device. Also, as the blend composition is increased to TP contents of 66% and 75%, the transient absorption amplitude of blend films decreases (ESI, Fig. S2†). Internal quantum efficiency (IQE) spectra estimated from optical transmission and IPCE data (without correction for optical interference effects) confirm charge generation from both PBDTTT-CT and TP excitons, with slightly higher quantum efficiencies observed following PBDTTT-CT excitation (Fig. 4b). At higher TP contents, the IQE is reduced for both PBDTTT-CT and TP excitation. Overall these trends suggest that 50% by wt is near the optimum TP concentration in the blend films for efficient charge generation, which is also evident from the J–V characteristics (Table 2).
 |
| | Fig. 4 (a) IPCE spectra of PBDTTT-CT:TP at different weight ratios of 1:1 (black), 1:2 (red) and 1:3 (blue), and (b) the corresponding internal quantum efficiencies of these blends. | |
fs-Transient absorption spectroscopy
We employed ultrafast transient absorption spectroscopy to interrogate the dynamics of the charge separation following generation of both TP and PBDTTT-CT excitons.27–32 Transient absorption spectra of both pristine films and the blend film were measured from 200 fs to 6 ns in visible and near IR regions (Fig. 5a–c). The blend films were excited at an excitation intensity of 2 μJ cm−2 at 470 and 700 nm. Data as a function of excitation density are presented in the ESI,† showing the absence of non-linear processes (e.g. exciton–exciton annihilation) at this excitation density. We note that at an excitation wavelength of 470 nm, we are exciting primarily the TP with minor polymer excitation, while at 700 nm excitation, we only excite the polymer.
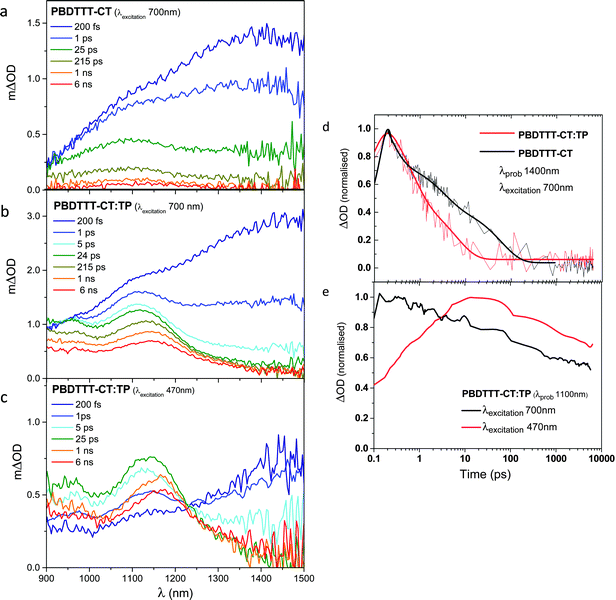 |
| | Fig. 5 fs-Transient absorption spectra of (a) neat PBDTTT-CT excited at 700 nm, (b) blend of PBDTTT-CT:TP excited at 700 nm and (c) at 470 nm. (d) Transient absorption decay dynamics for neat PBDTTT-CT (black) and PBDTTT-CT:TP blend (red) excited at 700 nm and probed at 1400 nm (black). (e) Transient absorption decay dynamics of a PBDTTT-CT:TP blend probed at 1100 nm and excited at 700 nm (black) and 470 nm (red). | |
Fig. 5a shows the transient absorption spectra (ΔOD) of a neat PBDTTT-CT polymer film (excited at 700 nm) as a function of time delay. These ΔOD spectra exhibit a photoinduced (PA1) band peaking at around 1400 nm, which is assigned to PBDTTT-CT exciton absorption. The decay of this photoinduced absorption exhibits rather dispersive, biphasic dynamics, with a half-time of 10 ± 2 ps, assigned to the decay of photogenerated singlet excitons. A small residual, long lived signal is also observed and assigned to polaron/triplet states generated in the neat film. Analogous data for neat TP films showed no measurable signals over this spectral range, indicating that TP excitons do not absorb significantly over this spectral range (TP exciton absorption was observed between 580 and 820 nm, decaying with a 400 ps half-time – see the ESI†).
We now turn to the blend film spectra (Fig. 5b), initially focusing on data collected following PBDTTT-CT excitation at 700 nm. ΔOD spectra of PBDTTT-CT:TP blends at an early time scale (200 fs) have spectra similar to that of a neat polymer film (Fig. 5a), indicating that in the blend, polymer excitons are also initially photogenerated. However, this initial spectrum rapidly evolves to new spectra with absorption maxima at ∼1100 nm and <900 nm, assigned, by comparison with the microsecond data, to PBDTTT-CT positive polarons. This evolution can be monitored most easily from the decay dynamics of the 1350–1400 nm PA1 singlet exciton absorption band (Fig. 5d, black line). It is apparent that in the blend film, this exciton absorption band decays with a 1.6 ± 0.4 ps half-time, assigned to photoinduced electron transfer from the PBDTTT-CT exciton into the TP acceptor. This electron transfer is approximately an order of magnitude faster than the singlet exciton lifetime in the neat film (10 ± 2 ps), consistent with the high efficiency of this photoinduced electron transfer process.
We consider now the transient data collected following 470 nm pumping of the blend film, corresponding to primarily TP excitation. Again at early times, the transient spectrum resembles that of the neat PBDTTT-CT film, although with a relatively small amplitude, consistent with the low PBDTTT-CT absorption at 470 nm (the low amplitude and instrument response limited rise of this PBDTTT-CT singlet exciton absorption indicates that this signal does not originate from energy transfer, i.e., from TP excitons to PBDTTT-CT). As for 700 nm excitation, the long time delay spectrum is indicative of PBDTTT-CT polarons. However it is striking to observe that when probing at 1100 nm, the peak of PBDTTT-CT polaron absorption, a pronounced rise time is observed with a half-time of 1.8 ± 0.4 ps (Fig. 5e). This rise cannot be assigned to spectral evolution from PBDTTT-CT excitons (which absorb more strongly than polarons at this wavelength) and is therefore assigned to hole transfer from TP excitons to PBDTTT-CT. We thus conclude that photoinduced charge separation from TP excitons proceeds with a half-time of 1.8 ± 0.4 ps, on a similar time scale to charge separation from PBDTTT-CT excitons. It is possible that the half-times for these photoinduced charge separation processes may be limited, at least in part, by exciton migration to the polymer–TP interface. Nevertheless, in terms of charge separation efficiency, both charge separation half-times are fast relative to excited state decay to ground, consistent with relatively intimate mixing of the blend on a length scale much less than the material exciton diffusion lengths, consistent with our PL quenching and structural analyses (see below).
Charge transport studies
Ideally, the efficient photoinduced charged generation should be followed by efficient charge extraction processes. The comparable charge carrier yield accompanied by lower efficiency compared to fullerene based BHJs indicates the limiting factors posed by the transport processes. SCLC measurements on the TP blends highlight the difference in carrier mobility brought out by compositional variations.33–36 The electron and hole mobilities were measured separately in electron only and hole only devices (ESI, Fig. S6†). Dark J–V curves of devices are fitted to the SCLC model at low voltages using the Mott–Gurney equation,35J = 9εoεrμV2/8L3where J is space charge limited current, εoεr is the permittivity of the polymer, μ is the carrier mobility, and L is the device thickness. The mobilities are summarized in Table 1. A key aspect of the results is that the 1:1 ratio blend reveals a more balanced charge transport similar to fullerene based BHJs.35 The interesting point to be noted is that the hole mobility is higher than the electron mobility for an optimized 1:1 ratio.
Table 1 Charge mobilities in the blend films of different compositional ratios in electron only and hole only devices
| PBDTTT-CT:TP (wt/wt) |
μ
e (cm2 V−1 s−1) |
μ
h (cm2 V−1 s−1) |
| 1:1 |
1.47 × 10−4 |
2.74 × 10−4 |
| 1:2 |
3.08 × 10−4 |
0.98 × 10−4 |
| 1:3 |
4.47 × 10−4 |
0.72 × 10−4 |
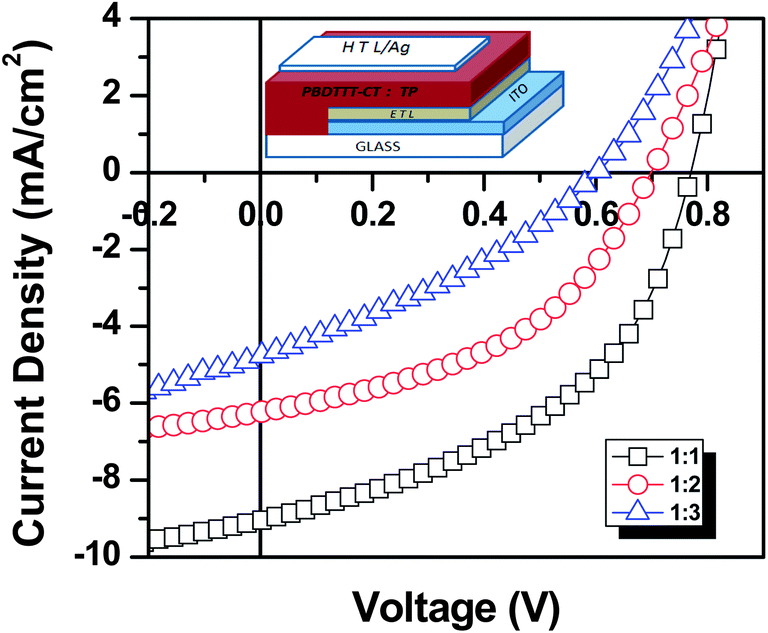
The device characterization of blends of different compositional ratios was carried out in inverted BHJ device geometry (ITO/ZnO/PBDTTT-CT:TP/MoOx/Ag, ESI†). Current density–voltage (J–V) characteristics of the devices measured under AM 1.5G irradiation are shown in Fig. 6. The resulting Jsc, Voc, FF, and PCE values, as determined from the J–V curves, are summarized in Table 2. Increasing the composition of the acceptor in the blend from 50% to 75% decreased all the device parameters and their efficiency. A maximum device efficiency of 3.2% was achieved for 50% composition of TP. It is also apparent from the table that Jsc, Voc and FF decrease drastically for 75% acceptor composition, indicative of a threshold composition for efficient charge transport and collection.
 |
| | Fig. 6
J–V characteristics of PBDTTT-CT:TP devices with different compositional ratios of 1:1 (black), 1:2 (red) and 1:3 (blue) under illumination of an AM 1.5G solar simulator (100 mW cm−2). The inset shows the schematic of the inverted device structure fabricated for testing BHJ-OSC's. | |
Table 2 Device performance of a PBDTTT-CT:TP BHJ solar cell at different acceptor ratios under AM 1.5, 1 sun unit illumination. The final column represents corresponding ΔOD at 1 μs of blends when pumped at 700 nm with E = 0.8 μJ cm−2 and probed at 920 nm
| PBDTTT-CT:TP (wt/wt) |
J
sc (mA cm−2) |
V
oc (V) |
Fill factor (%) |
η (%) |
ΔOD (μΔOD) |
| 1:1 |
9.0 |
0.77 |
0.46 |
3.20 |
25.0 |
| 1:2 |
6.2 |
0.70 |
0.44 |
1.93 |
20.1 |
| 1:3 |
4.8 |
0.60 |
0.32 |
0.94 |
18.6 |
Morphological studies
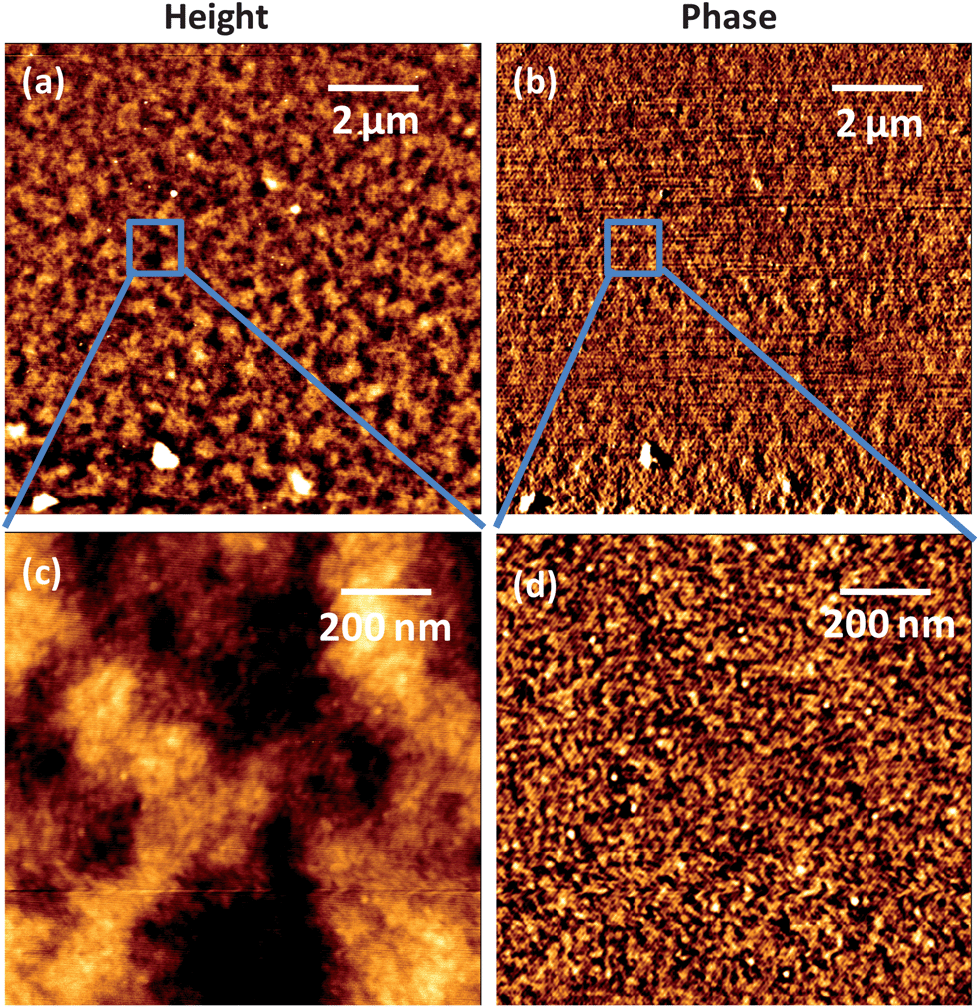
Atomic force microscopy (AFM) scans were carried out for 1:1 ratio blend films spun coated on the ITO substrate under similar fabrication conditions as those of devices (Fig. 7). The large area scans of 10 × 10 μm2 show a smooth surface morphology with an average root mean square roughness of 8 nm, and the corresponding phase image exhibits features smaller than instrument resolution. On scanning the smaller area of 1 × 1 μm2, the phase image displays domains of an average size of about 10 nm. This implies that the system achieves mixing of both TP and PBDTTT-CT in the blend on the 10 nm or less length scale, consistent with high PL quenching. An optimum D–A interfacial area with optimum domain sizes is required not just for efficient charge generation but also charge transport.37,38 We have previously observed that planar perylene blend films typically exhibit larger domains, sometimes extending upto micron size.18 From this study we conclude that, though the intermittent mixing of a blend is efficient in charge generation, and has improved device performance, however, the device efficiencies are still limited by optimised transport through the donor–acceptor percolation network. Recent observations of further fine tuning the morphology during fabrication processes have indicated that the performance of the device can be further enhanced.
 |
| | Fig. 7 AFM images of PBDTTT-CT:TP of a 1:1 weight ratio blend film in intermittent contact mode. (a) Topography and (b) phase image of a 10 × 10 μm2 area scan and (c) topography and (d) phase image of a 1 × 1 μm2 area scan. | |
Conclusions
In summary, TP based BHJs exhibit one of the highest reported power conversion efficiencies for non-fullerene acceptor based organic solar cells. The prerequisite condition of sizable charge generation yields in TP based BHJs was verified by a combination of transient and steady-state photophysical measurements. Strong PL quenching of both the donor and acceptor emissions indicates efficient electron and hole transfer processes at the PBDTTT-CT:TP interface. The near-IR pump–probe experiments and IPCE measurements demonstrate that polarons are generated from both the polymer and TP excitons. It was observed that in these blends, electron and hole transfer processes occured on a similar time scale of a few picoseconds. In the microsecond time scale, the magnitude of polaron yield of PBDTTT-CT:TP blends is comparable to that of fullerene blends and exhibits similar decay dynamics.
Efficient charge generation and transport are observed in devices with 50% by weight composition of the TP acceptor. Based on the charge generation yield and decay dynamics it is reasonable to expect efficiencies comparable to those of fullerene systems. In summary, the molecular, interfacial and morphology aspects of BHJ's with TP as an acceptor suggest their role as a promising model system and pave the way for efficient, fullerene-free BHJ solar cells.
Acknowledgements
All authors acknowledge INDO-UK APEX Project funded by EPSRC (EP/H040281), UK and DST, Government of India.
References
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS PubMed.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- S. Mukhopadhyay, S. Ramachandra and K. S. Narayan, J. Phys. Chem. C, 2011, 115, 17184–17189 CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954–985 CrossRef CAS PubMed.
- A. P. H. J. Schenning, J. v. Herrikhuyzen, P. Jonkheijm, Z. Chen, F. Würthner and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 10252–10253 CrossRef CAS PubMed.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CAS.
- A. J. Das and K. S. Narayan, Adv. Mater., 2013, 25, 2192 CrossRef CAS.
- G. R. J. Muller, C. Meiners, V. Enkelmann, Y. Geerts and K. Mullen, J. Mater. Chem., 1998, 8, 61–64 RSC.
- J. J. Dittmer, E. A. Marseglia and R. H. Friend, Adv. Mater., 2000, 12, 1270–1274 CrossRef CAS.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Lee and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384–390 RSC.
- J. A. Mikroyannidis, M. M. Stylianakis, P. Suresh and G. D. Sharma, Sol. Energy Mater. Sol. Cells, 2009, 93, 1792–1800 CrossRef CAS PubMed.
- G. D. Sharma, P. Suresh, J. A. Mikroyannidis and M. M. Stylianakis, J. Mater. Chem., 2010, 20, 561–567 RSC.
- X. Zhan, Z. A. Tan, E. Zhou, Y. Li, R. Misra, A. Grant, B. Domercq, X.-H. Zhang, Z. An, X. Zhang, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 5794–5803 RSC.
- M. Sommer, S. M. Lindner and M. Thelakkat, Adv. Funct. Mater., 2007, 17, 1493–1500 CrossRef CAS.
- S. Rajaram, P. B. Armstrong, B. J. Kim and J. M. J. Fréchet, Chem. Mater., 2009, 21, 1775–1777 CrossRef CAS.
- S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405–2408 CrossRef CAS.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeney, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030–3042 CrossRef CAS PubMed.
- J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155209 CrossRef.
- T. M. Clarke, A. Ballantyne, S. Shoaee, Y. W. Soon, W. Duffy, M. Heeney, I. McCulloch, J. Nelson and J. R. Durrant, Adv. Mater., 2010, 22, 5287–5291 CrossRef CAS PubMed.
- A. F. Nogueira, I. Montanari, J. Nelson, J. R. Durrant, C. Winder, N. S. Sariciftci and C. Brabec, J. Phys. Chem. B, 2003, 107, 1567–1573 CrossRef CAS.
- S. Shoaee, M. P. Eng, E. Espíldora, J. L. Delgado, B. Campo, N. Martín, D. Vanderzande and J. R. Durrant, Energy Environ. Sci., 2010, 3, 971–976 CAS.
- S. Shoaee, T. M. Clarke, C. Huang, S. Barlow, S. R. Marder, M. Heeney, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2010, 132, 12919–12926 CrossRef CAS PubMed.
- S. Westenhoff, I. A. Howard, J. M. Hodgkiss, K. R. Kirov, H. A. Bronstein, C. K. Williams, N. C. Greenham and R. H. Friend, J. Am. Chem. Soc., 2008, 130, 13653–13658 CrossRef CAS PubMed.
- I. A. Howard, F. d. r. Laquai, P. E. Keivanidis, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2009, 113, 21225–21232 CAS.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS PubMed.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2010, 132, 6154–6164 CrossRef CAS PubMed.
- F. Etzold, I. A. Howard, N. Forler, D. M. Cho, M. Meister, H. Mangold, J. Shu, M. R. Hansen, K. Müllen and F. Laquai, J. Am. Chem. Soc., 2012, 134, 10569–10583 CrossRef CAS PubMed.
- S. M. Mickley Conron, L. E. Shoer, A. L. Smeigh, A. B. Ricks and M. R. Wasielewski, J. Phys. Chem. B, 2013, 117, 2195–2204 CrossRef CAS PubMed.
- C. Melzer, E. J. Koop, V. D. Mihailetchi and P. W. M. Blom, Adv. Funct. Mater., 2004, 14, 865–870 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster, P. W. M. Blom, C. Melzer, B. deBoer, J. K. J. vanDuren and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 795–801 CrossRef CAS.
- V. D. Mihailetchi, H. X. Xie, B. deBoer, L. J. A. Koster and P. W. M. Blom, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS.
- L. M. Andersson and O. Inganas, Appl. Phys. Lett., 2006, 88, 082103 CrossRef.
- X. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS PubMed.
- S. Mukhopadhyay, A. J. Das and K. S. Narayan, J. Phys. Chem. Lett., 2012, 4, 161–169 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Device fabrication procedure, excitation density dependent μs-TAS, blend composition dependent μs-TAS, power law decay fit of different acceptor blends, fs-TA spectra at different excitation wavelengths and intensities, SCLC measurements on e-only and h-only devices. See DOI: 10.1039/c3ee42484g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.