 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green chemistry for organic solar cells
Daniel J.
Burke
and
Darren J.
Lipomi
*
Department of NanoEngineering, University of California, San Diego, 9500 Gilman Drive, Mail Code 0448, La Jolla, CA 92093, USA. E-mail: dlipomi@ucsd.edu
First published on 15th May 2013
Abstract
Essentially all methods of energy production—e.g., fracking, damming, drilling, nuclear fission, and excavation of rare elements for photovoltaics—are associated with some degree of environmental degradation. Organic solar cells (OSCs) are regarded as low-cost and potentially environmentally benign sources of power. π-Conjugated (semiconducting) polymers—the components of OSCs responsible for absorbing light and transporting charge—are not typically synthesized in laboratories in ways that are amenable to manufacturing with low environmental impact. This article discusses strategies for producing conjugated polymers using green chemistry. That is, reaction methodology with low energy intensity, with minimal production of toxic waste, and at low cost. This article briefly reviews the major findings in the literature on the energy intensity and carbon emissions associated with fabricating OSCs on the laboratory scale, and identifies several strategies and materials invented by the community to lower the cost and environmental impact of the components of the devices. The principles of green chemistry, applied to the synthesis of conjugated polymers, are identified as important guidelines for the multi-tonne manufacturing of these materials. A general theme in both green chemistry and process research is that low cost can be correlated to environmental benignity when the costs of disposing wastes are high. This Perspective then highlights five synthetic strategies that satisfy several of the criteria of green chemistry: (1) polymerization using metal-mediated cross-coupling reactions that reduce or eliminate stoichiometric organotin waste; (2) the use of heterogeneously catalyzed polymerizations; (3) polymerization involving activation of C–H bonds; (4) use of biofeedstock-derived starting materials; and (5) polycondensation reactions that evolve water as a byproduct.
 Daniel J. Burke | Daniel J. Burke received his BS in chemistry from the University of Massachusetts, Amherst in 2006, where he conducted research with Prof. Dhandapani Venkataraman of the Chemistry Department and Prof. Todd Emrick of the Polymer Science and Engineering Department. He earned is PhD in 2012 at the University of California, Santa Barbara, under the direction of Prof. Craig J. Hawker, where his research focused on both small-molecule and polymer synthesis for solar cells and other energy capture and storage applications. He is currently a postdoctoral scholar the laboratory of Prof. Darren J. Lipomi in the Department of NanoEngineering at the University of California, San Diego. |
 Darren J. Lipomi | Darren J. Lipomi earned his BA in chemistry with a minor in physics from Boston University in 2005. Under Prof. James S. Panek, his research focused on total synthesis and heterogeneous catalysis for efficient asymmetric synthesis. He earned his PhD in chemistry at Harvard University in 2010, with Prof. George M. Whitesides, where he developed unconventional, green approaches to fabricate nanostructures for electronic and optical applications. From 2010 to 2012, he was a postdoctoral fellow in the laboratory of Prof. Zhenan Bao at Stanford University, where his research was directed toward increasing the mechanical compliance of organic photovoltaic devices. He is now an assistant professor in the Department of NanoEngineering at the University of California, San Diego. |
Broader impactsOrganic solar cells have achieved efficiencies in the research laboratory of ten percent. Great progress in this field has been driven by the development of new organic semiconductors that maximize the absorption of light and the transport of charge, transparent electrodes that do not rely on rare and toxic elements, barrier materials that reduce the rate of photochemical degradation, and substrate materials that improve mechanical robustness. One aspect of organic solar cells that has received far less attention by the research community is the production energy, toxicity, and costs associated with synthesizing organic semiconductors at the scale required to satisfy even a small fraction of the growing worldwide need for energy. For organic solar cells to play a significant role in the worldwide production of carbon-neutral energy, organic solar panels will have to be manufactured to cover land on the scale of perhaps thousands of square kilometers. The merit of high-performance organic semiconductors should thus be judged not only by the highest demonstrated efficiency, but also in light of the costs and environmental impact of these materials when produced at scale. Identifying chemical reactions that can produce semiconducting polymers with minimal production of waste and toxic byproducts—that is, green chemistry—should help the research community ensure that building a global infrastructure for clean energy (which includes a substantial contribution from organic solar cells) does not cause further damage to the environment. |
1 Introduction and background
1.1 Photovoltaics at large scale
Researchers in the area of organic photovoltaic materials and devices are motivated by the potential of the technology to generate power renewably and at low cost.1,2 The numerous attractive features of organic solar cells (OSCs) cells easily justify the resources committed to this research, which one can judge by the roughly exponential increase in the number of articles published per year on “organic solar cells” since 1995, according to ISI Web of Knowledge. The ultimate goals of this research are (1) to reduce the environmental degradation brought about by the combustion of fossil fuels and (2) to do so at low economic and environmental cost, and with high profitability. Global photovoltaic capacity was 70 GW at the end of 2011; installed capacity has grown an average of 58 percent per year over the last five years.3 In 2011, photovoltaic systems accounted for more new capacity for electricity in the European Union than any other source.3 The market potential for solar energy is immense, and could exceed USD 1 trillion over the next several years. If organic systems eventually comprise even a small fraction of the total photovoltaic market, substantial public investments in research will be justified.1.2 Organic solar cells
An organic solar cell is a photovoltaic device whose active layer comprises π-conjugated polymers and small molecules (Fig. 1).4 Among the arguments for pursuing research on OSCs are that most—if not all—of the components can be deposited from solution in a roll-to-roll manner,5,6 that the materials are in principal earth-abundant,7 that devices can be semitransparent8 or aesthetically pleasing,9 that the devices are ultra-flexible10 and even stretchable,10–13 and that the materials and whole devices can be extraordinarily lightweight.10 Kaltenbrunner et al., for example, have shown that ultrathin OSCs (2 μm total thickness, including the 1.4 μm plastic substrate) have a specific power of ∼10 W g−1—which is about an order of magnitude greater than that of any other photovoltaic technology.10 Organic photovoltaic devices are thus unique not only in that they could have low costs per module, but that their thinness and extremely small mass could also reduce the costs associated with transportation and installation of modules (part of the balance-of-system costs, which are generally independent of the particular photovoltaic technology of the module).14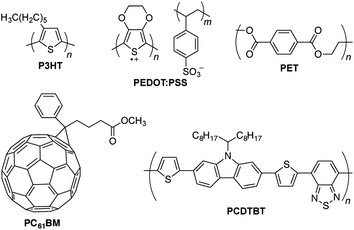 | ||
| Fig. 1 Chemical structures of materials discussed in the text. P3HT and PCDTBT are electron-donating semiconducting polymers; PC61BM (and PC71BM) is an electron-accepting small molecule semiconductor; PEDOT:PSS is a permanently conductive polymer often used as a transparent electrode; PET is a commodity polyester used as a transparent, flexible substrate. | ||
Organic solar cells typically comprise a heterostructure of two types of semiconductors with offset frontier molecular orbitals (Fig. 2).15 This heterostructure is sandwiched between a low-work-function electrode (which collects the electrons) and a high-work-function electrode (which collects the holes). This sandwich-like structure sits on a thin substrate (e.g., glass or polyethylene terephthalate, PET). Most, if not all, devices must be encapsulated to exclude water and oxygen.16 Upon the absorption of light, an electron is promoted in either the donor or the acceptor from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). Transfer of an excited electron in the LUMO of the donor to the LUMO of the acceptor produces free charge carriers. These carriers drift to opposite sides of the device, in part, because of the built-in electric field established by electrodes with different values of work function.4 This process is the basis of the photovoltaic effect in OPV and other “excitonic” cells, and is in contrast to the mechanism of conventional solar cells, in which charge carriers are generated directly upon the absorption of photons, and are segregated by the internal field established by a p–n junction.15,17
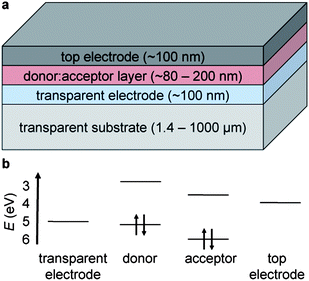 | ||
| Fig. 2 Schematic drawing and approximate energy levels of materials used in organic solar cells. (a) A transparent substrate (not drawn to scale) supports a transparent electrode, a semiconducting layer comprising an electron donor and an electron acceptor, and a top electrode. In part (b), the energy levels of the electrodes represent the values of work function, while the energy levels of the organic donor and acceptor represent the frontier molecular orbitals (highest occupied and lowest unoccupied molecular orbital, HOMO and LUMO). In the “conventional” geometry represented in the figure, the work function of the transparent electrode is higher than that of the top electrode; in the “inverted” geometry, the polarity is reversed. | ||
Organic photovoltaic cells are often cited for their potential to fulfill roles in unique applications such as wearable electronics,18 portable energy sources in the developing world,19 artificial retinas,20 and power-generating polarizing filters.21 Many of these applications are specifically targeted to the consumer market rather than to utility-scale generation of power. Considering the rate at which once-state-of-the-art consumer electronic devices are rendered obsolete by new models, it may be more important to minimize environmental impact than is it to maximize the lifetime—or even performance—of some devices for some applications.
1.3 Energy cannibalism
Intensive research efforts in organic photovoltaic materials and devices have produced cells with efficiencies around ten percent2 and projected lifetimes around ten years.22 Organic solar cells, despite recent setbacks,23 seem poised to garner attention of global leaders in business, investment, and policy. One aspect of the discussion about renewable energy that is often absent is the embodied energy and environmental impact of building a global infrastructure for sustainable energy, or so-called energy cannibalism.24 The production phase of the life cycle of high-technology products such as microchips25 and photovoltaic devices26 is famously energy intensive. The embodied energy of 1 kg of polycrystalline silicon of the kind needed for conventional silicon photovoltaic cells is approximately 1 GJ.25 This energy represents about 200 kg of carbon dioxide emissions, and the lifetime of carbon dioxide in the atmosphere is approximately 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years.27 The environmental costs of building an infrastructure for renewable energy are thus significant, and it is important to make the right choices not only with respect to the specific technologies that will make up the renewable infrastructure—i.e., wind, solar, geothermal—but also in considering the materials we use in these technologies.
000 years.27 The environmental costs of building an infrastructure for renewable energy are thus significant, and it is important to make the right choices not only with respect to the specific technologies that will make up the renewable infrastructure—i.e., wind, solar, geothermal—but also in considering the materials we use in these technologies.
1.4 Scale and costs of organic solar cells
Rigorous analysis of the environmental implications of thin-film photovoltaic technologies is an important—though nascent—field.28 Members of the research community have already targeted many of the components of an OSC for the reduction of cost. Reduced cost often correlates with increased “greenness,” as rare materials with high energies of production and possibly high toxicities are replaced with more abundant, environmentally benign alternatives.29 Networks of silver30–32 or copper33–35 nanowires, carbon nanotubes,36–38 graphene and reduced graphene oxide sheets,37,39,40 patterned metallic grids,41 films of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS)42–44 are promising alternative materials for tin-doped indium oxide (ITO) in large-area modules. While vacuum deposition must give way to solution processing if devices are to be manufactured in a roll-to-roll manner, evaporated low-work-function (usually top) contacts such as aluminum can be replaced in some circumstances by solution-processed metallic nanoparticles,32 carbon nanotubes,45 or PEDOT:PSS modified to have a low work function by an additive.46 The cost and production energy per unit area of a substrate can be reduced substantially simply by making it extremely thin.10 It is likely that advances in barrier materials will permit a reduction in cost by reducing the thickness of the encapsulant required.47 The most chemically sophisticated component of an OSC, however, is the organic active layer. While these layers are thin (typically 80 to 200 nm) and synthesized primarily from commodity petrochemicals, organic semiconductors have, in general, not been the target of studies designed to reduce the overall cost and environmental impact of OSCs.In 2006, Lewis and Nocera commented that a solar energy conversion system should cost no more than $10 per square meter in order to be competitive with fossil fuels for primary energy.48 This value is approximately equal to the cost of carpet per unit area, or ten times the cost of paint. Organic semiconductors—π-conjugated polymers and small molecules—are brightly colored, processable from solution, and thus are superficially similar to paint. Does the comparison extend to price? A 200 nm film of one of the structurally simplest conjugated polymers, poly(3-hexylthiophene) (P3HT, Fig. 1), over an area of 1 m2 costs $120, based on current catalog prices. This price per unit area is already an order of magnitude too expensive. The prices of low-bandgap materials with greater structural complexity than P3HT such as PCDTBT are roughly five times this price, or $600 m−2. The cost of PC71BM (the organic acceptor used in most of the devices with the highest efficiencies) of $500–$1000 m−2 makes use of this material at scale especially problematic. While one can expect eventually to negotiate prices for an organic semiconductor from a contract manufacturing organization (CMO) or to manufacture it in-house for 1–2 orders of magnitude less than the lowest catalog price,29,49 one still obtains costs of the organic active layer that are already dangerously close to Lewis' and Nocera's allotment of $10 m−2, before accounting for the costs of the electrodes, substrate, encapsulant, wiring, support racks, inversion, permitting, and land.
While small-scale reactions in the laboratory are sufficient for generating hundreds of milligrams of organic semiconductors for 1 cm2 devices, ultimately these materials must be produced at scales approaching the annual production of structurally simpler commodity polymers, or 2–3 orders of magnitude greater than those of top-selling small-molecule drugs of similar structural complexity. For example, if 10 TW of the 30 TW of power demanded in the year 2050 is to be generated by photovoltaics, and if organics account for 500 GW to 5 TW, then 10–100 kilotonnes of organic semiconductors will be required to cover the land area required, given an average solar flux of 200 W m−2, a module efficiency of 5%, a typical thickness of the active layer of 200 nm, and a density ∼1000 kg m−3. This extremely rough estimate assumes 100% yield of working modules, no waste in the coating processes, and infinite lifetime of devices. Both conjugated polymers and structurally complex drugs require synthetic sequences of 5–10 steps to produce. The multi-tonne synthesis of conjugated polymers will be a challenge in process chemistry with few precedents, and will influence the materials that could be seriously considered for installations that cover many square kilometers.
1.5 Green chemistry
The term green chemistry was coined by Paul Anastas of the Environmental Protection Agency.50 While the twelve principles of green chemistry50 are well known and practiced by process chemists in the fine chemical manufacturing (e.g., of pharmaceuticals), green chemistry is somewhat less well known to chemists in academia, where obtaining a specific target compound is of principal importance. In brief, a green process is tantamount to an efficient one. Such a process produces little waste, avoids protecting groups, uses catalysis in place of stoichiometric reagents, generates only environmentally benign byproducts, and uses energy efficiently (i.e., it has a small carbon footprint).50 Green chemistry represents a refocusing of strategy from treating or sequestering waste to preventing waste.51 It also represents a shift in risk management from limiting exposure to toxic substances to eliminating them to the extent possible.52In the pharmaceutical industry, drug discovery chemists benefit from a synergistic relationship with process chemists.29 A medicinal chemist working on the gram scale may design a route around inexpensive reagents identified or even prepared by the process department in anticipation of eventual scale-up29 (although the notion that discovery chemists ought to be designing their synthetic routes to anticipate the requirements of process chemists is not consensus53). One of the goals of this article is to identify promising, green approaches that might lower the overall cost and environmental impact of organic semiconductors. Whether or not an organic semiconductor can be produced in an environmentally benign way may influence the materials studied by device specialists and ultimately which materials are incorporated into manufacturing processes. A recent analysis by Osedach et al. on the “synthetic accessibility” of popular materials for OSCs reached a similar conclusion.54
1.6 Metrics for greenness
There are several approaches to evaluating the greenness of a synthetic sequence.55 None of these approaches, however, provide a no-compromise assessment. The atom economy or atom efficiency, introduced by Trost in 1991, is the ratio of the molecular weight of the product by the molecular weight of all substances formed.56 This approach is theoretical in that it assumes exactly stoichiometric quantities of all materials and 100% yield. Another metric, which takes into account the actual yield, solvents, and catalysts is the environmental factor (E-factor).57 This concept was developed by Sheldon after noting that the cost of disposing of waste in the fine chemical industry was comprising an increasing fraction of the revenue generated by the product.57 The E-factor is the ratio of the mass of waste to the mass of the desired product. The absolute mass of a waste product is, however, not always as important as is its environmental unfriendliness per unit mass.57 This fact led to the introduction of the EQ factor, in which the mass of each byproduct is scaled by a Q factor, which is an arbitrary number that scales with undesirability of the waste compound.57 While this approach suggests that a quantitative assessment of the greenness of a chemical synthesis is in principle possible, the arbitrariness of the Q factor invites a rigorously defined alternative. One alternative is the production energy of a chemical.58,59 The production energy is the part of the embodied energy of a good that represents the sum of the production energies of all of the starting materials and the additional energy required to form the product.25 This concept is used in life-cycle analysis of manufactured goods. Its attractiveness arises from the straightforward translation of production energy to carbon emissions: 0.2 kg carbon dioxide per MJ electricity given the average mix of fossil fuels and renewables used in the United States in 2013.60 Production energy also generally correlates with cost.59 The analysis by Williams et al., for example, led to the conclusion that a typical DRAM chip in 2002 cost more energy to produce than it used in its lifetime.25 The deficiency of production energy as a metric for greenness is that it only addresses one component of environmental friendliness—carbon emissions—and ignores the negative externalities of generation and disposal of toxic waste. Production energy is also not trivial to calculate, as it is dependent on details of manufacturing that can be difficult to find.Cost can in some circumstances be used as a proxy for environmental friendliness.29 The cost of producing a chemical entity (cost of goods, CoG) is influenced by the cost of the raw materials, labor, quality control, and disposal of waste.29 (In accounting terminology, cost of goods sold, COGS, includes shipping as well.) In the pharmaceutical industry, the target CoG for an advanced pharmaceutical intermediate is $1000–$3000 kg−1.29 Cost estimates can be carried out with more or less precision using commercial software or back-of-the-envelope methods. In general, cost increases with structural complexity. In favorable circumstances, cost can even be estimated by multiplying the number of rings, functional groups, stereocenters, heteroatoms, and regiocenters in a molecule by 100 to yield the cost per kilogram.53 Short of negotiating prices for all raw materials, one can estimate that it should be possible to obtain raw materials for 3–10% of the lowest published catalog price.49 An important drawback of using cost as a metric for environmental friendliness is that the costs of producing toxic waste are dependent on local regulations. Furthermore, the prices of raw materials, even if they could be obtained with accuracy for every possible synthetic route, are subject to market forces and thus only partially reflect scarcity, difficulty of extraction, and energy of production.
1.7 Strategies for reducing environmental impact
Despite advances in encapsulation and impressive demonstrations of long lifetimes of devices, organic solar cells will have finite lifetimes, and thus it is important to design components that are easily recycled or discarded.47 Elimination of fullerenes from bulk heterojunction cells is regarded as an important step in this direction,61,62 because fullerenes (and especially derivatized fullerenes) have uncertain effects on the environment when manufactured at the needed scale and then released by either degradation or disposal.47 Landi and coworkers performed detailed analyses of the energy intensity of the production of fullerenes59 and whole devices.58 Fullerenes comprise 18–30% of the cumulative production energy of small molecule and polymer-based cells.58 The production energy of PC71BM—the acceptor used in essentially all of the highest-performing research-scale OPV devices—is approximately 90 GJ kg−1,59 nearly two orders of magnitude greater than that of polysilicon (though polysilicon is used in quantities 2–3 orders of magnitude greater in silicon solar cells than PC71BM is used in organic solar cells). Conjugated polymers are significantly less energy intensive than are fullerenes, but the production energy and therefore cost of these materials increases by approximately an order of magnitude with each doubling of the number of synthetic steps.58 The production energy of P3HT is 1.9 GJ kg−1; while that of PCDTBT is 5.8 GJ kg−1.58 Both polymers, however, are substantially greener than is PCBM. Thus the polymer–polymer planar or all-polymer bulk heterojunction cell can be treated as an environmentally friendlier solution than the standard polymer–fullerene system, with the caveat that all-polymer systems are—usually far—less efficient. Fullerenes can also be replaced by semiconductor nanocrystals,63–65 though the environmental impact of these materials has not been evaluated and the toxicology of nanoparticles is highly dependent on both geometry and composition.47 Moreover, the degradation products of organic solar cells are not limited to the semiconducting components, but also to additives such as dithio- and diiodooctane66,67 and polydimethylsiloxane.68Any material produced in quantities over 1000 kg is, in the U.S., subject to rules set forth by the Toxic Substances Control Act (TSCA) of 1976.52 In most cases, new substances are evaluated for hazards by the premanufacture notification (PMN) process. Polymers are often exempted from PMN reporting if they satisfy certain criteria. For example, high molecular weight samples (>10 kDa) with a small fraction of unreacted monomers (<10% oligomers below 500 Da), no additives, and no reactive functional groups (such as allyl ethers, epoxides, cyanates, imines, and others) are generally exempted from the PMN process.52 A conjugated polymer by itself will generally be exempted, except that it is usually blended with a small molecule acceptor and one or more additives of small molecular weight. The class of polymers described by Wei et al.,69 based on the polyazine unit (see Section 2.5) would probably not be exempted from the PMN process. A concise resource on the environmental and regulatory aspects of polymers is the book by Anastas, Bickart, and Kirchhoff.52
1.8 Scope
The remainder of this Perspective identifies approaches to the synthesis of conjugated polymers that satisfy several of the criteria of green chemistry. Our purpose is not to provide an exhaustive list of examples of such chemistry, but rather to identify a few key developments in the field that seem especially suited to large-scale synthesis. Of all the materials that will eventually be incorporated in manufactured OSCs—including the substrate, transparent electrode, and encapsulant—we decided to focus on the organic semiconductors because of (1) our own expertise and (2) the fact that lowering the cost of these other components is already the subject of a significant research effort within the community. While we believe that molecular photovoltaic devices based on solution-processed small-molecules will play a significant role in the future of organic photovoltaics and have already achieved impressive efficiencies in the laboratory,70 we restrict our attention to conjugated polymers. This decision also reflects our expertise, and should not be regarded as an endorsement of one type of organic semiconductor over another.The materials, energy, and time put into workup and purification for any multistep chemical synthesis are considerable.29 Workup and purification includes quenching (i.e., neutralization of reactive species), extraction, chromatography, recrystallization, and distillation or sublimation.54 The analysis by Osedach et al. suggests that workup and purification contributes ∼50% of the cost for the laboratory-scale synthesis of P3HT and a considerably greater fraction of the cost (≤90%) for conjugated polymers with greater structural complexity.54 We selected some of the topics in Section 2, below, based in part on the facility of workup and purification: e.g., heterogeneous catalysis, direct heteroarylation polymerization, and water-forming polycondensation reactions. In the interest of space, however, we chose not to have a separate section on workup and purification.
It is entirely possible that a reaction that is in many ways “green” may not have a product that cannot be isolated in a sequence that is environmentally benign. Failure to purify a conjugated polymer completely can have deleterious effects on the electronic properties. In particular, particles from residual palladium catalysts have deleterious effects on electrical performance.71 On the other hand, polymerization strategies developed for polyphenylenevinylene (PPV) derivatives—such as the Wessling72 and Gilch73 methods—tend to be relatively green and are easy to workup. Polyphenylenevinylene derivatives, such as MEH-PPV,74 however, are no longer found in OSCs with the highest reported efficiencies, and thus we did not focus on PPV derivatives in this Perspective.
2 Green reaction methodology for organic solar cells
2.1 Metal-mediated cross-coupling reactions
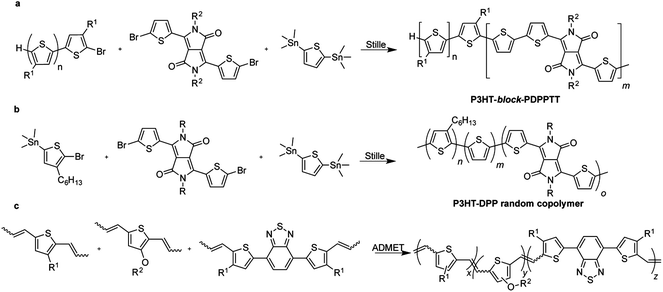 | ||
| Fig. 3 Examples of polymerizations mediated by transition metals. A palladium-catalyzed Stille reaction produces (a) block (ref. 86) or (b) random (ref. 87) copolymers of polythiophene and diketopyrrolopyrrole. (c) Ruthenium-catalyzed acyclic diene metathesis produces copolymers in which heterocylic moieties are linked through vinylene groups (ref. 91). | ||
While the Stille polymerization has risen to prominence in the field of conjugated polymers due to its broad scope and reliability, the reaction has several drawbacks that will increase in severity when this reaction is implemented at the scale required for utility-scale production of OSCs.92 Some of these issues are inescapable realities of condensation polymerizations and palladium catalysis—e.g., sensitivity to variations in stoichiometry and to exposure to air.93 Other factors may come into play on the scale of the pilot plant, such as contamination of metal reactor walls by tin,92 but are easy to overlook on the laboratory scale. Issues with the preparation and purification of stannane monomers and their polymerization raise a number of flags when running large-scale reactions, including the necessity of lithiating monomers in order to add trialkylstannyl groups, and the toxicity of organostannane compounds. To amplify both of these considerations, many researchers prefer to use the much more hazardous tert-butyl lithium in place of n-butyl lithium on lab scale for improved purity of monomers, and polymerizations of monomers bearing the trimethylstannane group are much more effective than those bearing the less toxic though seldom-used tributylstannane group.94–96 Chlorinated aromatic solvents, most commonly o-dichlorobenzene, are standard for the polymerization of these materials and happen to be both environmentally harmful and high-boiling, and thus require significant inputs of energy to recycle.29 Finally, the stoichiometric generation of trimethyltin bromide presents an added cost and environmental externality.92 It is telling that the Stille reaction is often omitted from prominent process29 and green chemistry51 textbooks, and that it most frequently appears in process chemistry journals when an alternative process is reported or when its problematic aspects are assessed.92
An alternative method for producing boronic esters has been the use of bis(pinacolato)diboron (BiPi) in a palladium-catalyzed borylation procedure of aryl bromides,99–101 This milder approach has proven especially useful in situations where lithiation is problematic, such as in the preparation of the benzothiadiazole diboronic ester. BiPi has also been used as an A2 monomer in polymerization reactions with dibromides as a B2 monomer (Fig. 4).102 The in situ borylation and the cross-coupling reaction can both be achieved by Pd(dppf)Cl2, and resulting polymers were of comparable size to the Stille-prepared analogs and exhibited much more well-defined, protic chain ends.102
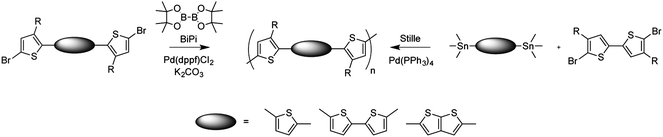 | ||
| Fig. 4 Use of bis(pinacolato)diboron (BiPi) in a mild form of polymerization by Suzuki cross-coupling (ref. 102). | ||
One promising advance in Suzuki methodology that has gained popularity in organic chemistry is the replacement of boronic esters with trifluoroborates. Trifluoroborate salts are robust, stable functional groups that are highly tolerant, stable to chromatography, and often readily recrystallize.98 Encouragingly, one example of a polymerization of a bis(trifluoroborate) has been reported for the preparation of an electron-accepting semiconducting polymer (Fig. 5).103 Additionally, polymerizations have been achieved by cascade Suzuki–Heck reactions with vinyl trifluoroborate salts,104–106 although the vinylene backbone motif has fallen out of favor in solar applications and these materials were studied in the context of electroluminescence. It is unclear whether the dearth of bis(trifluoroborate) monomers speaks to the recent development of this methodology or to its scope. Additionally, the installation of the trifluoroborate requires both lithiation and fluorination, and thus adds a step to those required to synthesize conventional boronic esters.
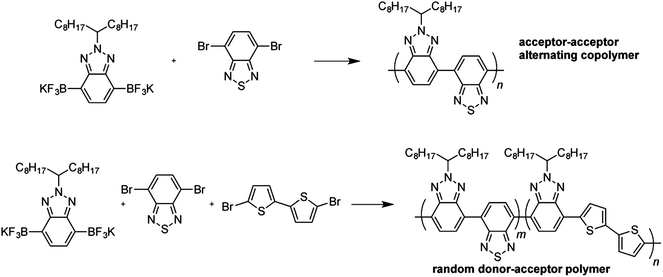 | ||
| Fig. 5 Use of trifluoroborate salts in Suzuki cross-coupling polymerization reactions (ref. 103). | ||
2.2 Heterogeneously catalyzed polymerizations
Another strategy that is popular in process chemistry and has found some use in the preparation of conjugated polymers is the implementation of supported catalysts (Fig. 6). Advanced products such as PdEnCat have proven highly effective cross-coupling catalysts that can vastly simplify removal of catalyst and thus reduce contamination of the products, but when simplicity and low cost are the goals, palladium on carbon (Pd/C) is preferred. Ligand-free Suzuki, Stille, and Heck polymerizations have been carried out using Pd/C.107 The use of this supported catalyst was found to decrease residual palladium as measured by atomic absorption spectroscopy from thousands to tens of parts per million.107 Its broad applicability to different cross-coupling reactions is notable, especially considering the comparative popularity of the Stille polymerization over the Suzuki. In order to supplant the Stille reaction, an alternative polymerization must offer substantial advantages.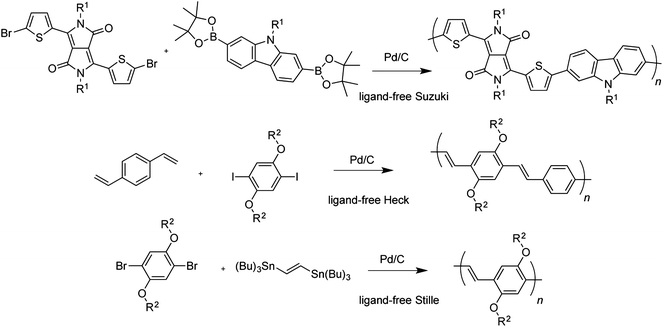 | ||
| Fig. 6 Heterogeneously catalyzed Suzuki, Heck, and Stille reactions applied to the synthesis of conjugated polymers (ref. 107). | ||
2.3 Direct heteroarylation polymerization
The direct use of aryl C–H bonds as cross-coupling partners has been an important subject of focus in the past decade, and the scope of such reactions has been vastly expanded.108–110 Early use of C–H activation in the preparation of organic semiconductors included the use of thiophene homocoupling to prepare symmetric function monomers,111 as well as oligothiophenes and other oligomers.112–114 To replace the Stille reaction, however, methodology to copolymerize difunctional monomers into alternating polymers is required.108,115,116 This methodology is referred to as direct heteroarylation polymerization (DHAP), and may hold more potential for decreasing the amount of toxic waste produced as byproduct in conjugated polymer synthesis than any other single method (Fig. 7).117–119 In its simplest form, DHAP is carried out with one dibromide monomer and one monomer with activated protons. This simplicity invites comparison to the typical Stille polymerization, which typically requires dibromination of both monomers, followed by lithiation and stannylation (two equivalents each) of one of those monomers. At minimum, assuming 100 percent yield at each step, DHAP saves two steps, two equivalents of trimethyltinchloride, and either two equivalents of n-BuLi or four equivalents of t-BuLi. The use of a diprotic monomer in place of a distannane also means that both monomers can be exhaustively purified; purification leads to ease in matching the stoichiometry precisely and obtaining polymers with high molecular weight.117–119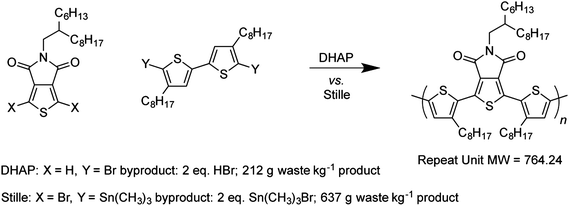 | ||
| Fig. 7 Direct heteroarylization polymerization (DHAP) produces less waste by mass than Stille coupling to produce the same polymer, and requires fewer steps for the synthesis of the monomers (ref. 118). | ||
The scope of DHAP is still being explored and expanded, but already several high-performing polymers have been synthesized using the method. Polymers based on DPP,117 thieno[3,4-c]pyrrole-4,6-dione (TPD),118 dithienosilole and dithienogermole monomers,119 cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and benzothiadiazole (BT)120 have all been reported using various optimized conditions, which usually employ the Herrmann–Beller catalyst. In each case, the molecular weight of the polymer synthesized is comparable or greater than that of the polymer synthesized from the same repeat units by Stille, though in the case of electron-rich segments with terminal thiophenes, it is necessary to functionalize the beta-position of that monomer.119
DHAP has also been used to polymerize the A–B monomer 2-bromo-3-hexylthiophene to synthesize the well-known material regioregular P3HT.121 At a loading of one mole percent of Herrmann catalyst and two mole percent phosphine ligand, P3HT with 98% regioregularity and Mn ∼ 30 kDa could be prepared in 99% yield in THF—a substantially more atom-economical route than the conventional Grignard metathesis (GRIM) polymerization which produces stoichiometric MgBr2.122 DHAP has also been used in place of Stille in the type of semi-random copolymerizations shown in Fig. 3b, with A2, B2, and A–B monomers.123 There is no reason the scope of DHAP on A–B monomers must stop with these impressive demonstrations: monobromination of oligothiophenes and other short sequences of heterocycles is typically straightforward and high-yielding.113
2.4 Conjugated polymers comprising biologically derived materials
While the ideal material for a green photovoltaic cell might be natural product, either farmed or derived from a culture broth, there have been only a few rare examples in which natural materials have been directly useful in OSCs. In light of the availability of materials such as Tyrian purple—a dibromated indigo—it is somewhat surprising that natural dyes have not been more extensively used as organic semiconductors.124 Unfortunately, though indigo itself has proven to be an acceptable semiconductor for use in organic field-effect transistors (OFETs),125 in order to make useful polymers for OSCs, chemists turned to its synthetic isomer, isoindigo (Fig. 8).126–128 Other materials derived from natural products are used frequently as dyes and have been used in OFETs, though only a few examples of such applications exist.129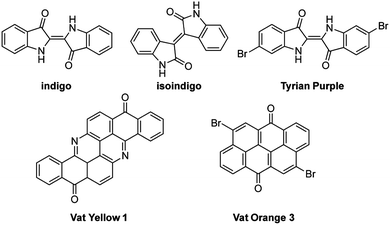 | ||
| Fig. 8 Structures of small molecule dyes discussed in the text. | ||
Some fully natural materials, such as indigo, aurin, and beta-carotene, have been used in organic electronics.129–131 Though they are excellent absorbers they occur in such low abundance that their biological production is not a feasible source for manufacturing. Other products are chemically modified derivatives of natural products, or are synthetic equivalents made from cheap, abundant petrochemicals. Examples of this class include perylene diimides, Vat Yellow and Vat Orange dyes. These naturally derived materials hold promise as being relatively environmentally benign after the device is discarded or recycled.132
One monomer stands apart in that it is derived in large part from biomass derivatives, and also finds application in some of the best-performing small-molecule and polymer active layer materials: diketopyrrolopyrrole flanked by two furan rings (DPPF).83 DPPF, derived from furfural and dialkylsuccinate, is largely derived from biomass prior to alkylation. Furfural is one of the most readily available, low-cost biorenewable materials on the market, with annual production at approximately 250000 tonnes.133 Similarly, approximately 15000 tonnes of succinic acid is produced each year. Though most of this material is petrochemical in origin, biorenewable succinate comprises a growing part of total production.134–136 The potential to derive DPPF from natural sources, on top of its greater solubility over that of DPPT, make DPPF an extremely attractive option for green production of OSCs. Indeed, DPPF is typically alkylated with ethylhexyl chains, which are much smaller than the octyldodecyl chains on similar DPPT polymers. The substitution of DPPT with DPPF thus comes with two advantages: the alkylating agent is less expensive and commercially available, and the ratio of semiconducting backbone to solubilizing “grease” is increased.
An interesting but seemingly seldom-used way to incorporate biomass into conjugated polymers is as the solubilizing alkyl chains, which are either straight-chain or which contain at least one branch. These alkylating agents are typically produced from alcohols, and their prices increase with size. Branched chains, produced by the Guerbet process at high temperatures, typically cost approximately three times more than straight chains of equal molecular weight. Therefore a savings in cost can be achieved simply by using simpler straight chain solubilizing groups. The substitution of branched sidechains with linear ones is not necessarily deleterious to the photovoltaic efficiency of devices.81,82,84 A strategy that utilizes inexpensive, abundant materials, high yielding reactions, and produces highly soluble polymers for low-temperature solution processing is usually favorable. One such strategy is the use of natural products as alkylating agents. In one example, farnesol is used as a starting material, readily reduced and halogenated, and used to alkylate DPPT (Fig. 9).137
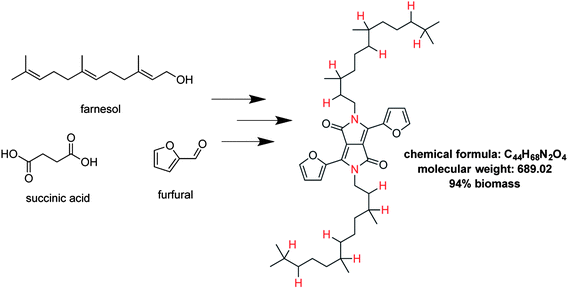 | ||
| Fig. 9 Proposed DPPF unit alkylated with a farnesol derivative derives 94% of its molecular weight from biomass. All atoms except those labeled in red are derived from natural materials. | ||
2.5 Water-forming polycondensation reactions
Condensation reactions involving the loss of the carbonyl oxygen atom in the form of water—as in the condensation of aldehydes with amines or hydrazine to form imines or hydrazones—or the loss of water and carbon dioxide—as in the Knoevenagel condensation—are some of the most ubiquitous reactions in organic chemistry and are useful in the synthesis of organic semiconductors because their products contain unsaturated bonds (Fig. 10). Such reactions are also green because they are typically efficient, give off water or carbon dioxide as byproducts, requires either no catalyst or simple acid catalysis, and occur between relatively easily installed functional groups. One of the simplest uses has been in the synthesis of quinoxaline,138,139 thienopyrazine,140 thiazole, and thiazolothiazole units,141 all of which have found use in low bandgap polymers for solar energy collection and use in OFETs. | ||
| Fig. 10 Water-forming condensation reactions as tools for synthesizing polymers and monomers. (a) DPPT unit bearing two aldehydes polymerizes with hydrazine to yield a fully conjugated ambipolar polyazine. (b) Imine-substitution on CDT monomers can tune the properties of resulting polymers. (c) Alternating polymers formed from polymerization of bis(cyanomethyl)benzene and oligothiophene-dicarbaldehydes. (d) Heterocycles formed by condensation: thienopyrazine (TP), quinoxaline (QU), thiazolothiazole (TzTz), and bithiazole (BTz). The red bonds represent bonds formed by condensation reactions. | ||
These materials, however, should not automatically be treated as greener alternatives to conventional monomers: the synthesis of quinoxaline begins with the deconstruction of dibromobenzothiadiazole. Thienopyrazine requires several steps to product, requiring nitration of thiophene and subsequent reduction to form the diaminothiophene starting material. Thiazole-based materials require the use of expensive dithiooxamide. Such considerations must be balanced in choosing materials for synthesis on a large scale. A specific reaction should be used not simply because it is green, but because it leads to a holistically efficient synthesis of the target material even after the steps taken to produce the starting materials have been taken into account.
An especially successful example of a water-evolving polycondensation reaction is the recent report of a conjugated polyazine based on the DPPT chromophore (Fig. 10).69 In this case, the DPPT unit was functionalized with aldehydes, which were then condensed with hydrazine. The resulting polymer had impressive characteristics, including ambipolar transport with electron and hole mobilities ∼0.4 cm2 V−1 s−1. (We note that one caveat of polymers containing imine linkages or other possibly reactive moieties when produced at the tonne scale is that they will probably invite increased regulatory scrutiny, as discussed in Section 1.7.) In another example of polycondensation involving loss of a carbonyl oxygen atom, alternating copolymers of oligothiophene and cyano-substituted polyphenylenevinylene (CN-PPV) were produced from aldehyde-terminated thiophene oligomers and alkoxy-substituted biscyanomethylbenzene.142 The properties of the polymers could be tuned by modifying the length of the thiophene oligomers. In a similar vein, complimentary bis(cyanomethyl)fluorene and fluorene dicarbaldehyde can be used to produce cyano-substituted poly(fluorenevinylene), which demonstrated improved electroluminescence over the poly(fluorenevinylene) without cyano substitutions, which was prepared by the Gilch polymerization.143
In some cases, a condensation reaction can be used to access monomers that have chemical structures that are not necessarily accessible by metal-mediated cross-coupling reactions. For example, condensation was used in the polymerization of some dioxythiophene, thiophene, and phenylene monomers, but it was also used to prepare a bis(dioxythieno)cyanovinylene monomer, which could then be polymerized by Yamamoto or electropolymerization.144 Condensation reactions for monomer synthesis and polymerization provided a library of new eletrochromic materials. In another example of novel monomer synthesis, Azoulay and coworkers prepared a series of imine-substituted cyclopentadithiophene monomers.145 Not only did this synthesis provide a handle for tuning the electronic properties of resulting materials, but the imine substitution provided solubility which otherwise would have necessitated additional synthetic steps to reduce and alkylate the bridgehead position of the cyclopentadithiophenone starting material.
3 Outlook
We began this Perspective by describing several aspects of the fabrication of organic solar cells that might have a deleterious effect on the environment. We then identified green chemistry—applied to the synthesis of semiconducting polymers—as one way to mitigate environmental externalities of manufacturing organic solar cells. Green chemistry is, of course, not the only avenue the community can pursue to lessen the environmental impact of organic solar modules. Replacing fullerenes with conjugated polymers or small molecules, engineering thinner substrates and barrier films, and investigating solution-processable replacements for transparent electrodes based on rare-earth elements will drastically reduce the production energy, waste, and toxicity of manufacturing these devices.It seems increasingly likely that organic photovoltaics will play a role in meeting the future global demand for energy. The objective of this Perspective was to anticipate the need to synthesize organic semiconductors at a scale much greater than that used in laboratories in order to cover land areas on the order of perhaps thousands of square kilometers. It seems that whether or not a particular organic semiconductor can be made in an environmentally benign way would influence the choice of a researcher as to whether the material is—or is not—worth pursuing. We believe that cross-talk between device specialists and synthetic chemists interested in process development will support the “double bottom line” possible with organic solar cells: the potential to produce clean energy using devices that are manufactured according to green practices.
Acknowledgements
This work was supported by startup funds from the University of California, San Diego and by the Air Force Office of Scientific Research (AFOSR) Young Investigator Program, grant number FA9550-13-1-0156.References
- B. Kippelen and J. L. Bredas, Energy Environ. Sci., 2009, 2, 251–261 CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- REN 21: Renewables 2012 Global Status Report, 2012 Search PubMed.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- F. C. Krebs, T. Tromholt and M. Jorgensen, Nanoscale, 2010, 2, 873–886 RSC.
- S. I. Na, B. K. Yu, S. S. Kim, D. Vak, T. S. Kim, J. S. Yeo and D. Y. Kim, Sol. Energy Mater. Sol. Cells, 2010, 94, 1333–1337 CrossRef CAS.
- N. Espinosa, M. Hosel, D. Angmo and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117–5132 CAS.
- J. Y. Lee, S. T. Connor, Y. Cui and P. Peumans, Nano Lett., 2010, 10, 1276 CrossRef CAS.
- C. M. Amb, M. R. Craig, U. Koldemir, J. Subbiah, K. R. Choudhyry, S. A. Gevorgyan, M. Jorgensen, F. C. Krebs, F. So and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2012, 4, 1847–1853 CAS.
- M. Kaltenbrunner, M. S. White, E. D. Glowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. G. Mei and Z. N. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS.
- D. J. Lipomi, B. C.-K. Tee, M. Vosgueritchian and Z. N. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS.
- D. J. Lipomi and Z. N. Bao, Energy Environ. Sci., 2011, 4, 3314–3328 CAS.
- L. Bony, S. Doig, C. Hart, E. Maurer and S. Newman, Achieving Low-Cost Solar PV: Industry Workshop Recommendations for Near-Term Balance of System Cost Reductions, Rocky Mountain Institute, Snowmass, CO, 2010.
- B. A. Gregg, J. Phys. Chem. B, 2003, 107, 4688–4698 CrossRef CAS.
- M. Jorgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef.
- B. A. Gregg, in Organic Photovoltaics: Mechanisms, Materials, and Devices, ed. S. S. Sun and N. S. Sariciftci, CRC Press, Boca Raton, FL, 2005, ch. 6, pp. 139–159 Search PubMed.
- F. C. Krebs, M. Biancardo, B. Winther-Jensen, H. Spanggard and J. Alstrup, Sol. Energy Mater. Sol. Cells, 2006, 90, 1058–1067 CrossRef CAS.
- F. C. Krebs, T. D. Nielsen, J. Fyenbo, M. Wadstrom and M. S. Pedersen, Energy Environ. Sci., 2010, 3, 512–525 CAS.
- D. Ghezzi, M. R. Antognazza, R. Maccarone, S. Bellani, E. Lanzarini, N. Martino, M. Mete, G. Pertile, S. Bisti, G. Lanzani and F. Benfanati, Nat. Photonics, 2013 DOI:10.1038/nphoton.2013.34.
- R. Zhu, A. Kumar and Y. Yang, Adv. Mater., 2011, 23, 4193–4198 CrossRef CAS.
- C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupre, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS.
- P. Clarke, in EE Times, London, 2012 Search PubMed.
- J. M. Pearce, Int. J. Nucl. Governance, Econ. Ecol., 2008, 2, 113–130 CrossRef CAS.
- E. D. Williams, R. U. Ayres and M. Heller, Environ. Sci. Technol., 2002, 36, 5504–5510 CrossRef CAS.
- P. Zhai and E. D. Williams, Environ. Sci. Technol., 2010, 44, 7950–7955 CrossRef CAS.
- D. J. Jacob, Introduction to Atmospheric Chemistry, Princeton University Press, Princeton, NJ, 1999 Search PubMed.
- H. C. Kim, V. Fthenakis, J.-K. Choi and D. E. Turney, J. Ind. Ecol., 2012, 16, S110–S121 CrossRef CAS.
- N. G. Anderson, Practical Process Research and Development: A Guide for Organic Chemists, Academic Press, Waltham, MA, 2012 Search PubMed.
- Z. B. Yu, Q. W. Zhang, L. Li, Q. Chen, X. F. Niu, J. Liu and Q. B. Pei, Adv. Mater., 2011, 23, 664–668 CrossRef CAS.
- L. B. Hu, H. S. Kim, J. Y. Lee, P. Peumans and Y. Cui, ACS Nano, 2010, 4, 2955–2963 CrossRef CAS.
- C. Girotto, B. P. Rand, S. Steudel, J. Genoe and P. Heremans, Org. Electron., 2009, 10, 735–740 CrossRef CAS.
- H. Wu, L. B. Hu, M. W. Rowell, D. S. Kong, J. J. Cha, J. R. McDonough, J. Zhu, Y. A. Yang, M. D. McGehee and Y. Cui, Nano Lett., 2010, 10, 4242–4248 CrossRef CAS.
- A. R. Rathmell, S. M. Bergin, Y. L. Hua, Z. Y. Li and B. J. Wiley, Adv. Mater., 2010, 22, 3558–3563 CrossRef CAS.
- M. G. Kang, H. J. Park, S. H. Ahn and L. J. Guo, Sol. Energy Mater. Sol. Cells, 2010, 94, 1179–1184 CrossRef CAS.
- D. J. Lipomi, M. Vosgueritchian, B. C.-K. Tee, C. H. Fox, J. A. Lee and Z. N. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS.
- L. B. Hu, D. S. Hecht and G. Gruner, Chem. Rev., 2010, 110, 5790–5844 CrossRef CAS.
- C. Feng, K. Liu, J. S. Wu, L. Liu, J. S. Cheng, Y. Y. Zhang, Y. H. Sun, Q. Q. Li, S. S. Fan and K. L. Jiang, Adv. Funct. Mater., 2010, 20, 885–891 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS.
- M. G. Kang, T. Xu, H. J. Park, X. G. Luo and L. J. Guo, Adv. Mater., 2010, 22, 4378–4383 CrossRef CAS.
- M. Vosgueritchian, D. J. Lipomi and Z. N. Bao, Adv. Funct. Mater., 2012, 22, 421–428 CrossRef CAS.
- D. J. Lipomi, J. A. Lee, M. Vosgueritchian, B. C.-K. Tee, J. A. Bolander and Z. N. Bao, Chem. Mater., 2012, 24, 373–382 CrossRef CAS.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Muller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- M. P. Ramuz, M. Vosgueritchian, P. Wei, C. G. Wang, Y. L. Gao, Y. P. Wu, Y. C. Chen and Z. N. Bao, ACS Nano, 2012, 6, 10384–10395 CrossRef CAS.
- Y. H. Zhou, C. Fuentes-Hernandez, J. W. Shim, J. Meyer, A. J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T. M. Khan, H. Sojoudi, S. Barlow, S. Graham, J. L. Bredas, S. R. Marder, A. Kahn and B. Kippelen, Science, 2012, 336, 327–332 CrossRef CAS.
- Y.-S. Zimmerman, A. Schaffer, C. Hugi and K. Fent, Environ. Int., 2012, 49, 128–140 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20142 CAS.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS.
- M. Lancaster, Green chemistry: an introductory text, The Royal Society of Chemistry, Cambridge, UK, 2002 Search PubMed.
- P. T. Anastas, P. H. Bickart and M. M. Kirchhoff, Designing safer polymers, Wiley-Interscience, New York, 2000 Search PubMed.
- C. M. Henry, Chem. Eng. News, 2002, 80, 53–66 Search PubMed.
- T. P. Osedach, T. L. Andrew and V. Bulovic, Energy Environ. Sci., 2013, 6, 711–718 CAS.
- D. J. C. Constable, C. Jiminez-Gonzalez and A. Lapkin, in Green chemistry metrics, ed. A. Lapkin and D. Constable, Blackwell Publishing Ltd, West Sussex, UK, 2009, ch. 6, pp. 228–247 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- A. Anctil, C. W. Babbitt, R. P. Raffaelle and B. J. Landi, Prog. Photovolt: Res. Appl., 2012 DOI:10.1002/pip.
- A. Anctil, C. W. Babbitt, R. P. Raffaelle and B. J. Landi, Environ. Sci. Technol., 2011, 45, 2353–2359 CrossRef CAS.
- U. E. P. Agency, Clean energy calculations and references, accessed 24 January 2013, 2013.
- T. Liu and Al. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 715–726 CrossRef CAS.
- F. A. Bokel, P. K. Sudeep, E. Pentzer, T. Emrick and R. C. Hayward, Macromolecules, 2011, 44, 1768–1770 CrossRef CAS.
- E. Pentzer, F. A. Bokel, R. C. Hayward and T. Emrick, Adv. Mater., 2012, 24, 2254 CrossRef CAS.
- J. Weickert, R. B. Dunbar, H. C. Hesse, W. Wiedemann and L. Schmidt-Mende, Adv. Mater., 2011, 23, 1810–1828 CrossRef CAS.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS.
- J. Peet, A. J. Heeger and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1700–1708 CrossRef CAS.
- K. R. Graham, J. G. Mei, R. Stalder, J. W. Shim, H. Cheun, F. Steffy, F. So, B. Kippelen and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1210–1215 CAS.
- W. Hong, B. Sun, H. Aziz, W. T. Park, Y. Y. Noh and Y. N. Li, Chem. Commun., 2012, 48, 8413–8415 RSC.
- L. Y. Lin, Y. H. Chen, Z. Y. Huang, H. W. Lin, S. H. Chou, F. Lin, C. W. Chen, Y. H. Liu and K. T. Wong, J. Am. Chem. Soc., 2011, 133, 15822–15825 CrossRef CAS.
- F. C. Krebs, R. B. Nyberg and M. Jorgensen, Chem. Mater., 2004, 16, 1313–1318 CrossRef CAS.
- R. W. Lenz, C.-C. Han, J. Stenger-Smith and F. E. Karasz, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 3241–3249 CrossRef CAS.
- H. Becker, H. Spreitzer, K. Ibrom and W. Kreuder, Macromolecules, 1999, 32, 4925–4932 CrossRef CAS.
- M. M. Alam and S. A. Jenekhe, Chem. Mater., 2004, 16, 4647–4656 CrossRef CAS.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS.
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- W. W. Li, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2012, 134, 13787–13795 CrossRef CAS.
- J. H. Hou, M. H. Park, S. Q. Zhang, Y. Y. Yao, L. M. Chen, J. H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Fréchet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS.
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS.
- D. Venkataraman, S. Yurt, H. Venkatraman and N. Gavvalapalli, J. Phys. Chem. Lett., 2010, 1, 947–958 CrossRef CAS.
- S. Y. Ku, M. A. Brady, N. D. Treat, J. E. Cochran, M. J. Robb, E. Kramer, M. L. Chabinyc and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 16040–16046 CrossRef CAS.
- P. P. Khlyabich, B. Burkhart, C. F. Ng and B. C. Thompson, Macromolecules, 2011, 44, 5079–5084 CrossRef CAS.
- B. Burkhart, P. P. Khlyabich, T. Cakir Canak, T. W. LaJoie and B. C. Thompson, Macromolecules, 2011, 44, 1242–1246 CrossRef CAS.
- B. Burkhart, P. P. Khlyabich and B. C. Thompson, J. Photonics Energy, 2012, 2, 021002 CrossRef.
- B. Burkhart, P. P. Khlyabich and B. C. Thompson, ACS Macro Lett., 2012, 1, 660–666 CrossRef CAS.
- J. C. Speros, B. D. Paulsen, B. S. Slowinski, C. D. Frisbie and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 986–990 CrossRef CAS.
- J. A. Ragan, J. W. Raggon, P. D. Hill, B. P. Jones, R. E. McDermott, M. J. Munchhof, M. A. Marx, J. M. Casavant, B. A. Cooper, J. L. Doty and Y. Lu, Org. Process Res. Dev., 2003, 7, 676–683 CrossRef CAS.
- V. Farina, V. Krishnamurthy and W. J. Scott, in Organic Reactions, John Wiley & Sons, Inc., 2004 Search PubMed.
- F. Babudri, A. Cardone, L. Chiavarone, G. Ciccarella, G. M. Farinola, F. Naso and G. Scamarcio, Chem. Commun., 2001, 1940–1941 RSC.
- F. Babudri, A. Cardone, G. M. Farinola, F. Naso, T. Cassano, L. Chiavarone and R. Tommasi, Macromol. Chem. Phys., 2003, 204, 1621–1627 CrossRef CAS.
- Q. B. Liu and D. J. Burton, Org. Lett., 2002, 4, 1483–1485 CrossRef CAS.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS.
- G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS.
- C. Kleeberg, L. Dang, Z. Y. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS.
- K. B. Woody, B. J. Leever, M. F. Durstock and D. M. Collard, Macromolecules, 2011, 44, 4690–4698 CrossRef CAS.
- H.-Y. Chen, C.-T. Chen and C.-T. Chen, Macromolecules, 2010, 43, 3613–3623 CrossRef CAS.
- F. Brouwer, J. Alma, H. Valkenier, T. P. Voortman, J. t. Hillebrand, R. C. Chiechi and J. C. Hummelen, J. Mater. Chem., 2011, 21, 1582–1592 RSC.
- J. K. Lee, M. C. Gwinner, R. Berger, C. Newby, R. Zentel, R. H. Friend, H. Sirringhaus and C. K. Ober, J. Am. Chem. Soc., 2011, 133, 9949–9951 CrossRef CAS.
- R. Grisorio, P. Mastrorilli, C. F. Nobile, G. Romanazzi and G. P. Suranna, Tetrahedron Lett., 2005, 46, 2555–2558 CrossRef CAS.
- R. Grisorio, P. Mastrorilli, C. F. Nobile, G. Romanazzi, G. P. Suranna, G. Gigli, C. Piliego, G. Ciccarella, P. Cosma, D. Acierno and E. Amendola, Macromolecules, 2007, 40, 4865–4873 CrossRef CAS.
- R. Grisorio, C. Piliego, P. Fini, P. Cosma, P. Mastrorilli, G. Gigli, G. P. Suranna and C. F. Nobile, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6051–6063 CrossRef CAS.
- S. Y. Liu, H. Y. Li, M. M. Shi, X. L. Hu, W. Q. Li, L. Fu and H. Z. Chen, Macromolecules, 2012, 45, 9004–9009 CrossRef CAS.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013 DOI:10.1021/ar3003305.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS.
- A. R. Murphy, J. S. Liu, C. Luscombe, D. Kavulak, J. M. J. Fréchet, R. J. Kline and M. D. McGehee, Chem. Mater., 2005, 17, 4892–4899 CrossRef CAS.
- K. Masui, H. Ikegami and A. Mori, J. Am. Chem. Soc., 2004, 126, 5074–5075 CrossRef CAS.
- M. Takahashi, K. Masui, H. Sekiguchi, N. Kobayashi, A. Mori, M. Funahashi and N. Tamaoki, J. Am. Chem. Soc., 2006, 128, 10930–10933 CrossRef CAS.
- C.-Y. Liu, H. C. Zhao and H.-H. Yu, Org. Lett., 2011, 13, 4068–4071 CrossRef CAS.
- D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS.
- A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS.
- J.-R. Pouliot, L. G. Mercier, S. Caron and M. Leclerc, Macromol. Chem. Phys., 2013, 214, 453–457 CrossRef CAS.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068–2071 CrossRef CAS.
- L. G. Mercier, B. R. Aich, A. Najari, S. Beaupre, P. Berrouard, A. Pron, A. Robitaille, Y. Tao and M. Leclerc, Polym. Chem., 2013 10.1039/C3PY21138J.
- S. Kowalski, S. Allard and U. Scherf, ACS Macro Lett., 2012, 1, 465–468 CrossRef CAS.
- Q. F. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- A. E. Rudenko, C. A. Wiley, S. M. Stone, J. F. Tannaci and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3691–3697 CrossRef CAS.
- M. J. Robb, S. Y. Ku, F. G. Brunetti and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1263–1271 CrossRef CAS.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef CAS.
- J. G. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS.
- R. Stalder, J. G. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- G. B. Zhang, Y. Y. Fu, Z. Y. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414–1420 CrossRef CAS.
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, G. Schwabegger, M. Ullah, R. Schwoediauer, A. Mumyatov, M. Bodea, J. W. Fergus, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Org. Electron., 2010, 11, 1974–1990 CrossRef CAS.
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, L. Shmygleva, Y. Kanbur, G. Schwabegger, M. Bodea, R. Schwödiauer, A. Mumyatov, J. W. Fergus, V. F. Razumov, H. Sitter, N. S. Sariciftci and S. Bauer, Adv. Funct. Mater., 2010, 20, 4069–4076 CrossRef CAS.
- M. Irimia-Vladu, N. S. Sariciftci and S. Bauer, J. Mater. Chem., 2011, 21, 1350–1361 RSC.
- S. W. Hwang, H. Tao, D. H. Kim, H. Y. Cheng, J. K. Song, E. Rill, M. A. Brenckle, B. Panilaitis, S. M. Won, Y. S. Kim, Y. M. Song, K. J. Yu, A. Ameen, R. Li, Y. W. Su, M. M. Yang, D. L. Kaplan, M. R. Zakin, M. J. Slepian, Y. G. Huang, F. G. Omenetto and J. A. Rogers, Science, 2012, 337, 1640–1644 CrossRef CAS.
- F. W. Lichtenthaler, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- M. McCoy, Chem. Eng. News, 2009, 87, 22 Search PubMed.
- M. McCoy, Chem. Eng. News, 2012, 90, 12 Search PubMed.
- M. M. Bomgardner, Chem. Eng. News, 2012, 90, 10–15 Search PubMed.
- Z. L. Wu, A. Y. Li, B. H. Fan, F. Xue, C. Adachi and J. Y. Ouyang, Sol. Energy Mater. Sol. Cells, 2011, 95, 2516–2523 CrossRef CAS.
- T. Yamamoto, Z. H. Zhou, T. Kanbara, M. Shimura, K. Kizu, T. Maruyama, Y. Nakamura, T. Fukuda, B. L. Lee, N. Ooba, S. Tomaru, T. Kurihara, T. Kaino, K. Kubota and S. Sasaki, J. Am. Chem. Soc., 1996, 118, 10389–10399 CrossRef CAS.
- Q. Peng, J. Xu and W. X. Zheng, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3399–3408 CrossRef CAS.
- C. H. Chen, C. H. Hsieh, M. Dubosc, Y. J. Cheng and C. S. Hsu, Macromolecules, 2010, 43, 697–708 CrossRef CAS.
- M. Yang, B. Peng, B. Liu, Y. P. Zou, K. C. Zhou, Y. H. He, C. Y. Pan and Y. F. Li, J. Phys. Chem. C, 2010, 114, 17989–17994 CAS.
- F. Wang, Y. S. Liu, X. J. Wan, J. Y. Zhou, G. K. Long and Y. S. Chen, Macromol. Chem. Phys., 2010, 211, 2503–2509 CrossRef CAS.
- Y. G. Jin, J. M. Ju, J. W. Kim, S. G. Lee, J. Y. Kim, S. H. Park, S. M. Son, S. H. Jin, K. H. Lee and H. S. Suh, Macromolecules, 2003, 36, 6970–6975 CrossRef CAS.
- B. C. Thompson, Y. G. Kim, T. D. McCarley and J. R. Reynolds, J. Am. Chem. Soc., 2006, 128, 12714–12725 CrossRef CAS.
- J. D. Azoulay, Z. A. Koretz, B. M. Wong and G. C. Bazan, Macromolecules, 2013, 46, 1337–1342 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |