 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Parameters affecting electron transfer dynamics from semiconductors to molecular catalysts for the photochemical reduction of protons†
Anna Reynala,
Fezile Lakadamyalib,
Manuela A. Grossb,
Erwin Reisnerb and
James R. Durrant*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk
bChristian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
First published on 5th June 2013
Abstract
The aim of this work is to use transient absorption spectroscopy to study the parameters affecting the kinetics and efficiency of electron transfer in a photocatalytic system for water reduction based on a cobalt proton reduction catalyst (CoP) adsorbed on a nanocrystalline TiO2 film. In the first approach, water is used as the proton and electron source and H2 is generated after band gap excitation of TiO2 functionalised with CoP. The second system involves the use of a sacrificial electron donor to regenerate the TiO2/CoP system in water at neutral pH. The third system consists of CoP/TiO2 films co-sensitised with a ruthenium-based dye (RuP). In particular, we focus on the study of different parameters that affect the kinetics of electron transfer from the semiconductor to the molecular catalyst by monitoring the lifetime of charge carriers in TiO2. We observe that low catalyst loadings onto the surface of TiO2, high excitation light intensities and small driving forces strongly slow down the kinetics and/or reduce the efficiency of the electron transfer at the interface. We conclude that the first reduction of the catalyst from CoIII to CoII can proceed efficiently even in the absence of an added hole scavenger at sufficiently high catalyst coverages and low excitation densities. In contrast, the second reduction from CoII to CoI, which is required for hydrogen evolution, appears to be at least 105 slower, suggesting it requires efficient hole scavenging and almost complete reduction of all the adsorbed CoP to CoII. Dye sensitisation enables visible light photoactivity, although this is partly offset by slower, and less efficient, hole scavenging.
Broader contextThe photochemical reduction of water into H2 offers the possibility of harvesting sunlight and storing this energy in the form of chemical bonds. Rapid progress is currently being made in the development of synthetic and bio-inspired molecular catalysts for proton reduction, which can also be integrated in heterogeneous photocatalytic systems through the functionalisation of semiconductors such as TiO2. The development of efficient hybrid photocatalytic H2 evolution systems involves not only the study of its catalytic activity, but also the analysis of the electron transfer kinetics and the control of the main recombination pathways. Molecular catalysts for H+ reduction require the accumulation of two electrons in one single catalyst, which is potentially a key limiting factor of their efficiency. In this paper, we study three different photochemical systems based on TiO2 functionalised with a molecular cobalt catalyst and a ruthenium photosensitiser. In particular, we address the kinetics of the two-electron transfer reactions needed for the H2 production by monitoring the charge carriers (electrons and holes) of the semiconductor by transient absorption spectroscopy. |
Introduction
The development of photocatalytic systems for the hydrogen generation from water splitting is receiving renewed attention as a renewable fuel synthesis strategy.1,2 The photochemical reduction of water into H2 offers the possibility of harvesting sunlight and storing its energy in the form of a chemical bond. Most of these approaches combine a photoactive element that harvests solar energy with a catalytic component to catalyse the two electron reduction of protons to molecular hydrogen.3,4 In this regard, rapid progress is being made in the development of molecular catalysts for H+ reduction, including Ni and Co complexes and dinuclear Fe or Ni–Fe hydrogenase mimics.4–8 However, studies integrating such H2 evolution electrocatalysts into photochemical systems are more limited to date,9–13 and have moreover largely focused on overall system performance, rather than elucidation of the underlying electron transfer kinetics that are critical in determining the efficiency of the system. In this paper, we focus on the electron transfer dynamics in a series of model systems based upon mesoporous, nanocrystalline TiO2 films functionalised with a cobalt based H+ reduction catalyst, and thereby elucidate how these charge carrier dynamics impact the proton reduction efficiency.Electron transfer dynamics between molecular photosensitisers and TiO2 has been widely studied in the context of dye sensitised solar cells.14–16 Such devices are based on the electron injection from the photosensitiser into the TiO2 semiconductor. In contrast, H+ reduction by molecular catalysts immobilised on TiO2 requires the reverse electron transfer reaction – from the TiO2 conduction band to the molecular catalyst. This reverse electron transfer reaction is an undesired recombination pathway in dye sensitised solar cells. The parameters required to optimise the efficiency of this reaction have received relatively little attention to date, and are therefore the subject of this paper.
Heterogeneous photochemical systems for proton reduction require semiconductors with a conduction band above the reduction potential of water reduction (−0.42 V vs. NHE at pH 7). TiO2, the semiconductor studied herein, is one of the most widely used semiconductors for such studies due to the appropriate position of its conduction and valence bands, its high stability and availability, ease of fabrication as high surface area nanocrystalline films and particles and its low cost.17,18 However the efficiency of TiO2 photocatalysts alone for proton photoreduction is typically relatively low, attributed to poor visible light absorption, fast recombination of photogenerated charge carriers, and the low driving force for the H+ reduction reaction, resulting in slow H+ reduction kinetics.19–22 A common strategy to address these limitations, for both TiO2 and alternative semiconductors, is the functionalisation of the semiconductor with a catalyst capable of accelerating the H+ reduction and increasing the selectivity for H2 evolution.23 A particular consideration with the use of molecular catalysts for solar driven fuel synthesis is that they typically require multiple oxidation/reduction steps, which contrasts with the single electron–hole pairs generated by photon absorption in bare semiconductors. Therefore hydrogen generation typically requires photogenerated charge carriers to drive a double reduction reaction of a single molecular catalyst,11,24 and this requirement is potentially a key factor limiting the system's performance. This contrasts with the use of inorganic catalysts (such as Pt nanoparticles) where separate electrons injected into the nanoparticle are likely to be able to come together to drive hydrogen generation.
In this regard, we have recently reported an example of such a hybrid photocatalytic system for H2 evolution that involves the combination of anatase or mixed anatase/rutile (P25) TiO2 nanoparticles with a molecular ruthenium photosensitiser (RuP) and a cobaloxime H2 evolution catalyst (CoP) covalently attached to the semiconductor through phosphonic acid anchoring groups (Fig. 1).9,25 Double reduction of this catalyst, from CoIII to CoI, has been shown to enable proton reduction to molecular hydrogen, with an overall external quantum efficiency of the photocatalytic system of 1% under LED light irradiation at 465 nm (3 mW cm−2).9,25 In this paper, we employ this as a model system for exploring the parameters influencing the efficiency of electron transfer from the semiconductor to the molecular catalyst, including the impact of catalyst loading and light intensity. Our study employs three alternative approaches based upon this experimental system, as illustrated in Scheme 1. All three approaches employ nanocrystalline TiO2 films functionalised by CoP for the production of hydrogen in aqueous solution under N2. The first approach is based upon these materials alone, excluding any sacrificial electron donor, and therefore relying upon the ability of TiO2 holes to oxidise water to molecular oxygen as the source of electrons for proton reduction. In the second approach, a sacrificial electron donor (triethanolamine, TEOA) is used as a hole scavenger to remove photogenerated TiO2 holes. Finally, the third system involves the functionalisation of the TiO2 nanoparticles with a molecular ruthenium dye (RuP) and a cobalt catalyst anchored onto the surface, again using TEOA to regenerate the photosensitiser.
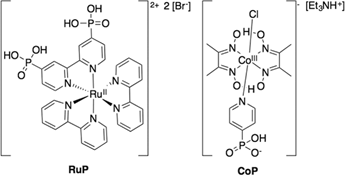
 | ||
| Scheme 1 Illustration of the three experimental systems studied herein, and the functional processes underlying their function comprising (a) TiO2 functionalised by a CoP proton reduction catalyst in water under UV excitation, with, (b) the addition of a sacrificial electron donor (D), and (c) the addition of a photosensitiser dye to enable visible light activity. The figure illustrates the two reduction potentials of the catalyst (CoIII/II and CoII/I, measured at pH 7) relevant for the H2 evolution reaction. Forward electron transfer processes are represented with solid black arrows, while recombination reactions are drawn with dashed grey arrows. | ||
Experimental section
Materials and methods
All starting reagents for the synthetic part of the work were purchased from commercial suppliers and used without any further purification. Chemicals used for the analytical part were of the highest available purity. (Et3NH)[CoIIICl(dmgH)2(pyridyl-4-hydrophosphonate)], (Et3NH)[CoP],9 and [RuII(2,2′-bipyridine)2(2,2′-bipyridine-4,4′-diylbis(phosphonic acid))]Br2, [RuP]Br2,26 were prepared as described previously. Triethanolamine hydrochloride buffer solutions (0.1 M) were adjusted to pH 7 upon the addition of 0.1 M NaOH.Anatase TiO2 nanoparticles and films were prepared as reported previously.27 Mesoporous nanocrystalline TiO2 films were prepared by the doctor blading technique from colloidal pastes. The resulting film thickness, determined by profilometry (Tencor Instruments), was 4 μm.
Preparation of dye sensitised TiO2 films with and without CoP
For approach (c), TiO2 films were sensitised with RuP by introducing the films into an aqueous RuP solution (0.1 mM) and monitoring the dye loading by UV-vis spectroscopy (see below). The sensitisation time for TiO2 films was optimised in order to adjust the MLCT absorption band (λ = 450 nm) to an absorbance of 0.6. TiO2 nanostructured films were co-sensitised with RuP and CoP by dipping the films for 48 h into a 5 mL aqueous solution containing CoP (16 nmol – corresponding to 80 CoP molecules per TiO2 particle) and RuP (8 nmol – corresponding to 40 RuP molecules per TiO2 particle).Loading TiO2 films with CoP
Two different methods were used to functionalise TiO2 films with CoP.28 Unless otherwise stated, monolayer coverage of CoP was achieved by dipping the films in a high concentration CoP aqueous solution (0.1 mM) for 12 h. For studies as a function of the number of CoP molecules anchored per TiO2 nanoparticle, the films were dipped into a low concentration CoP solution for a longer period of time (2 nmol, 8 nmol, 24 nmol, and 40 nmol, for 48 h). The amount of catalyst loaded was monitored by UV-vis spectroscopy after desorbing the catalyst from the nanoparticles by dipping the film into a 0.1 M NaOH solution.Spectroscopic characterisation
All functionalised films were kept in the fridge (4 °C) and in the dark to avoid degradation prior to measurement. Spectroscopic measurements on functionalised films were undertaken at room temperature with films immersed in 5 mL of water under N2 for approach (a), and with the addition of 5 mL of a 0.1 M TEOA solution buffered at pH 7 for approaches (b) and (c).The UV-vis spectra of the solutions were recorded using a quartz cuvette (one cm path length) on a Perkin Elmer Lambda 35 UV-Vis spectrophotometer.
The millisecond-second transient absorption decays were measured using a Nd:YAG laser (Big Sky Laser Technologies Ultra CFR Nd:YAG laser system, 6 ns pulse width). The third harmonic of the laser (corresponding to 355 nm) and a coupled OPO crystal fixed at 450 nm, were used to excite the band gap of TiO2 and the RuP, respectively. The laser intensity was adjusted using neutral density filters as appropriate, with experiments typically employing 350 μJ cm−2 unless otherwise stated and the frequency of the laser pulse was fixed to 1 Hz. A liquid light guide with a diameter of 0.5 cm was used to transmit the laser pulse to the sample. The probe light source was a 100 W Bentham IL1 tungsten lamp, and the probing wavelength was selected by using a monochromator (OBB-2001, Photon Technology International) placed prior to the sample. Several high pass and neutral density filters (Comar Instruments) were used to decrease the light arriving to the detector. Transient absorption data was collected with a Si photodiode (Hamamatsu S3071). The information was passed through an amplifier box (Costronics) and recorded using a Tektronics TDS 2012c oscilloscope (microsecond to millisecond timescale) and a National Instruments (NI USB-6211) DAQ card (millisecond to second timescale). The decays observed were the average between 500 and 1000 laser pulses. The data was processed using a home-built software based on Labview.
Results
The three experimental systems studied in this paper are illustrated in Scheme 1. We address the electron transfer kinetics measured for each system, respectively, and then compare and contrast these kinetics in our discussion. For approaches (a) and (b), all transient signals exhibited spectra indicative of their assignment to TiO2 electrons or holes (see ESI for typical spectra, Fig. S1†),19,29 with no signals directly from the CoP being apparent over the spectral range studied (460 nm–900 nm), consistent with the dominant optical absorption of CoP lying in the ultraviolet, and therefore masked by the strong ground state absorption of TiO2 in this region. Our study focuses on the yields and lifetimes of TiO2 holes (observed at 460 nm) and, of particular interest, electrons (observed at 900 nm), and their dependence upon experimental approach and catalyst loading. For approach (c), additional, spectrally distinct, optical signals from the RuP photosensitiser were observed, as detailed below.Band gap excitation of TiO2/CoP in water
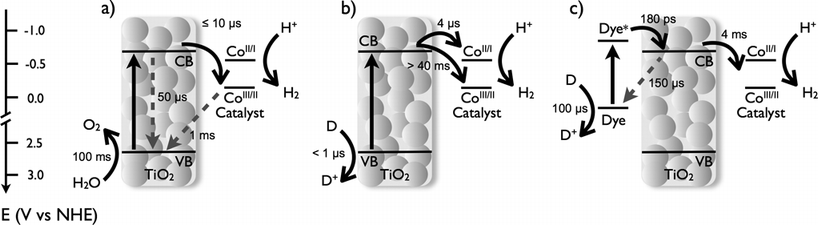
The lifetime of charge carrier species (electrons and holes) in bare and CoP monolayer-functionalised TiO2 films (140 CoP per TiO2 nanoparticle) was monitored by transient absorption spectroscopy after band gap excitation (λex = 355 nm). The decay of photoexcited electrons and holes of bare TiO2 in water under N2 show identical dynamics, exhibiting power law decay kinetics consistent with bimolecular (or ‘non-geminate’) recombination processes on the micro- to millisecond time scales (Fig. 2A).29–31 The exponent of this power law was ∼0.4, assigned previously to bimolecular recombination in the presence of charge trapping.32 Band gap excitation of TiO2 modified with a CoP monolayer should allow for electron transfer from the conduction band of the semiconductor to the catalyst, resulting in an accumulation of valence band holes and a fast depletion of conduction band electrons. Consistent with this expectation, the transient absorption signal corresponding to the photoexcited electrons in TiO2 show a lower signal amplitude for TiO2/CoP films when compared to bare TiO2 films (Fig. 2B), indicative of the electron transfer from the semiconductor to the catalyst that takes place in ≤10 μs. On the other hand, the signal amplitude of photoholes when the CoP is attached onto the surface of TiO2 is doubled in amplitude and longer-lived compared to that of bare TiO2 films, indicating an increased lifetime of holes in the TiO2 valence band. The decay dynamics of these holes in functionalised TiO2/CoP films (τ1/2 ∼ 1 ms) is slow compared to that observed in the absence of CoP, but faster than the oxidation of water in the presence of irreversible electron scavengers, such as Ag+, that suppress the electron–hole recombination.29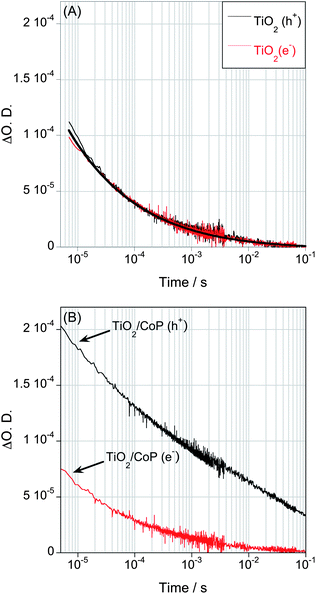 | ||
| Fig. 2 Transient absorption decays of photoelectrons and photoholes after band gap excitation of a bare TiO2 (A), and TiO2/CoP films (B) in water and under a N2 atmosphere. The excitation wavelength was 355 nm (350 μJ cm−2), the decay of photo-excited electrons was monitored at 900 nm, and the decay of photoholes was probed at 460 nm. | ||
In this simple model system, the primary process competing with electron transfer from the TiO2 to the CoP is the electron–hole recombination in TiO2. This recombination is a bimolecular process, and therefore dependent upon the charge carrier densities in the film. In order to address this competition further, we undertook transient kinetic measurements as a function of excitation density (Fig. 3 and Fig. S2, see ESI†). The excitation density from 50 μJ cm−2 to 1 mJ cm−2 corresponds to approximately 0.5 to 10 photons absorbed per TiO2 nanoparticle (c.f. 10 to 200 CoP molecules per photon). We have previously shown for bare TiO2 films that increasing the excitation densities generate more electron–hole pairs and the recombination becomes faster.29,33 The signal of photoexcited electrons of TiO2/CoP also shows faster decays with increasing laser intensity (Fig. 3A), consistent with electron–hole recombination in TiO2 being a competing reaction with the electron transfer to CoP. The decays of photoholes show a more complex behaviour as a function of excitation density, exhibiting a biphasic decay at higher excitation densities (Fig. 3B). A faster phase (μs–ms) increases in amplitude and becomes faster with increasing excitation density. The similarity between this behavior and that observed for the TiO2 electron signal allows us to assign this fast hole decay phase to electron–hole recombination. The slow phase (half time ∼100 ms) is assigned to holes left in the TiO2 valence band following electron transfer to the CoP; its decay dynamics are assigned to hole recombination with reduced CoP catalyst and/or water oxidation on the hundreds of milliseconds timescale (Fig. S3, see ESI†).
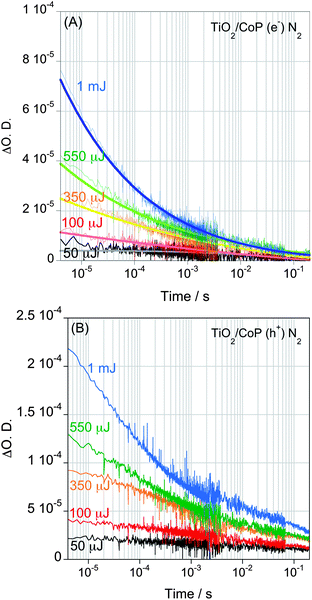 | ||
| Fig. 3 Transient absorption decays of photoexcited electrons (A) and holes (B) of a TiO2/CoP film in water under N2 after band gap excitation (λex = 355 nm) with different laser pulse intensities, ranging from 50 μJ cm−2 to 1 mJ cm−2. The decay of photo-excited electrons was proved at 900nm, while holes were monitored at 460 nm. | ||
Fig. 4 plots the amplitudes of the two TiO2 decay phases observed for TiO2/CoP films as a function of excitation densities. It is apparent that the fast decay phase, assigned to TiO2 electron–hole recombination, is only observed for excitation densities greater than ∼100 μJ cm−2. In contrast, the long-lived photoholes, assigned to TiO2 holes left in the valence band following electron transfer to CoP, exhibits a strongly sub-linear increase in yield with excitation density. This sub-linear behaviour is consistent with electron–hole recombination increasingly competing with electron transfer to CoP as the excitation density is increased. At low excitation densities, the charge carrier recombination becomes dominated by electron transfer to CoP and the accumulation of h+ in the TiO2 valence band.
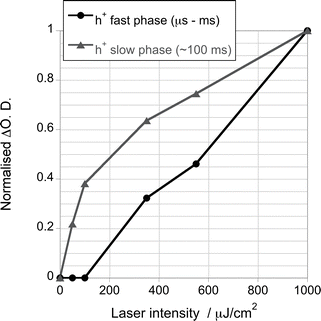 | ||
| Fig. 4 Signal amplitude of generated photoholes of a TiO2/CoP film after band gap excitation (λex = 355 nm), assigned to the recombination of charge carriers in TiO2 (h+ fast phase) and to the holes accumulated at the valence band of TiO2 after electron transfer to CoP (h+ slow phase), using different excitation densities. | ||
Band gap excitation of TiO2/CoP in the presence of a sacrificial electron donor
In the previous section, we have addressed the kinetic competition between electron–hole recombination within the TiO2 versus transfer to CoP. It has been widely reported that molecular hole scavengers can be effective in suppressing this competing recombination reaction. The electron transfer reactions from the TiO2 to the CoP catalysts were therefore studied when immersing the films into a 0.1 M TEOA aqueous solution as hole scavenger (pH 7, under N2). As shown in Fig. 5, for bare TiO2 films the addition of a sacrificial hole scavenger results in a very substantial increase in the electron lifetime to ∼1 s, indicating that TEOA very efficiently suppresses electron–hole recombination losses. In the presence of TEOA, no TiO2 hole signal could be observed, indicating hole scavenging by TEOA in <1 μs. In the further presence of CoP, the electron decay is substantially accelerated. Due to the absence of electron–hole recombination, this acceleration can be directly assigned to electron transfer from the semiconductor to the molecular catalyst.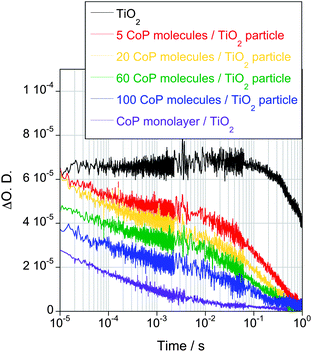 | ||
| Fig. 5 Transient absorption decays corresponding to TiO2 photoexcited electrons in bare TiO2 and TiO2-functionalised films with different quantities of CoP under N2 and in the presence of a 0.1 M TEOA aqueous solution at pH 7 as sacrificial electron donor (λex = 355 nm, λprobe = 900 nm, Eex = 350 μJ cm−2). | ||
We now turn to the influence of CoP loading upon the kinetics of electron transfer from TiO2 to CoP. With decreasing amounts of CoP (ranging from 140 to 5 molecules per TiO2 particle), the photoinduced electron decay slows down significantly, and becomes biphasic at low coverages (Fig. 5). The decay at early timescales (μs–ms) is assigned to the electron transfer from the semiconductor to the molecular catalyst, while the slower phase corresponds to the decay observed in the absence of catalyst (most probably corresponding to electron transfer into the electrolyte, such as reduction of residual oxygen in the degassed solution). From these data, the half-time for electron transfer from TiO2 to CoP was estimated, as shown in Fig. 6 and Fig. S4, see ESI.†34 It is apparent that this electron transfer half-time varies substantially (by five orders of magnitude) with CoP coverage.
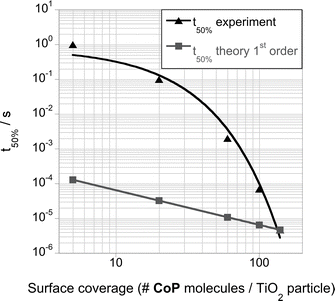 | ||
| Fig. 6 Half-time electron transfer (t50%), calculated as the full width at half maximum, for the μs – ms timescale phase of the photoexcited electrons decay of TiO2/CoP films after band gap excitation (λex = 355 nm) with different surface coverage of CoP, measured under a N2 purged 0.1 M TEOA solution at pH 7. | ||
For a simple bimolecular recombination kinetic model, the half-time (t50%) for electron transfer from TiO2 to CoP should vary linearly with CoP coverage, corresponding to the grey line in Fig. 6. It is apparent that the observed dependence on CoP coverage is much greater than this simple model prediction. In order to address this surprising result, we investigated the dependence of these kinetics upon laser excitation density for a fixed (monolayer) catalyst coverage, again in the presence of TEOA (Fig. 7). Rather strikingly, the TiO2 electron decay is substantially delayed as the laser intensity is increased. This is the inverse of the behavior observed in the absence of TEOA (Fig. 3), where higher laser intensity resulted in faster decays. This observation suggests that at higher light fluxes, electron transfer from TiO2 to CoP is impeded.
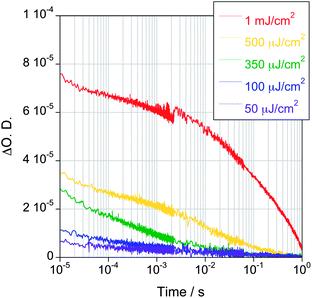 | ||
| Fig. 7 Transient absorption decays corresponding to photoexcited electrons in TiO2/CoP films sensitised with a monolayer of catalyst measured at different excitation densities ranging from 50 μJ cm−2 to 1 mJ cm−2 (λex = 355 nm, λprobe = 900 nm). The films were kept under N2 and in the presence of a 0.1 M TEOA at pH 7. | ||
The studies shown in both Fig. 5 and 7 vary the ratio of photogenerated electron density per CoP (by varying either the catalyst coverage, Fig. 5, or electron density, Fig. 7). In order to address this effect further, we compared decays measured at different catalyst coverages, but with laser intensity modulated in order to obtain matched ratios of electrons per CoP (see ESI Fig. S5†). It is apparent that decays measured at matched electron/CoP ratios showed very similar kinetics for a range of CoP coverages. This suggests that the key determinant of the observed rate of electron transfer from TiO2 to CoP is this ratio of photoinduced electron density per CoP molecules. As we discuss below, this behavior most probably is associated with the accumulation of singly reduced CoP (i.e.: CoII) species under conditions of high electron generation rates per CoP, with electron transfer from TiO2 to CoII being substantially slower than that to the unreduced CoIII state.
Dye sensitised TiO2/CoP in the presence of a sacrificial electron donor
The functionalisation of wide band gap semiconductors with a molecular dye shifts the absorption spectrum of the photocatalytic system from UV to visible light, allowing light harvesting of a broader range of the solar spectrum. In addition, this interface also enhances charge separation without the addition of sacrificial species, through the oxidation of the molecular photosensitiser after electron injection into the TiO2. In this kind of system, the function of the semiconductor has a different role when compared to non-dye sensitised systems: TiO2 does not act as a light harvesting antenna, but acts as an electron transport layer between the dye and the catalyst. The kinetics and efficiency of dye electron injection into the TiO2 and dye regeneration by the sacrificial electron donor of this three-component system were previously reported,25 and in this paper we will focus on the study of the electron transfer kinetics from the semiconductor to the molecular catalyst.The excitation of RuP with visible light (λex = 450 nm) leads to the injection of electrons to the conduction band in the TiO2. In the presence of the sacrificial electron donor TEOA, transient absorption spectroscopy measurements of RuP/TiO2 monitored at λprobe = 690 nm (corresponding to RuP+ maximum absorption wavelength), show a biphasic transient absorption decay, in which the faster component (∼200 μs) is assigned to electron transfer from TEOA to RuP+ (analogous to hole scavenging), and the slow component (∼300 ms) is assigned to the presence of long-lived electrons in the TiO2 (Fig. S6†). We note this dye regeneration timescale is at least 200 fold slower than the analogous scavenging of TiO2 holes by TEOA discussed above. It is also of similar magnitude to the recombination timescale of TiO2 electrons with RuP+ ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 – suggesting that kinetic competition between this recombination and dye regeneration may be a significant loss factor under the conditions studied. Fig. 8 shows the analogous data probing at 900 nm, and therefore dominated by TiO2 electron absorption. Again a biphasic decay is observed, with the electron signal showing a 50% decay on the timescale of dye regeneration and dye cation/TiO2 electron recombination, indicating that under these conditions, hole scavenging of RuP sensitised TiO2 by TEOA is significantly less efficient than for TiO2 holes generated by direct UV excitation.
25 – suggesting that kinetic competition between this recombination and dye regeneration may be a significant loss factor under the conditions studied. Fig. 8 shows the analogous data probing at 900 nm, and therefore dominated by TiO2 electron absorption. Again a biphasic decay is observed, with the electron signal showing a 50% decay on the timescale of dye regeneration and dye cation/TiO2 electron recombination, indicating that under these conditions, hole scavenging of RuP sensitised TiO2 by TEOA is significantly less efficient than for TiO2 holes generated by direct UV excitation.
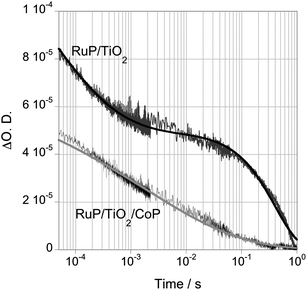 | ||
| Fig. 8 Transient absorption decays of TiO2 films functionalised with CoP and/or RuP, in the presence of 0.1 M TEOA at pH 7 (λex = 450 nm, λprobe = 900 nm). The decay of RuP/TiO2 is fitted to a combination of a stretched exponential equation and a monoexponential equation, and the decay of TiO2 functionalised with RuP and CoP is fitted to a stretched exponential equation. | ||
The co-attachment of CoP to the dye sensitised TiO2 nanoparticles (approximately 80 CoP and 40 RuP molecules per TiO2 particle) results in an acceleration of the slow phase of the decay, assigned to long lived TiO2 electrons, from ∼300 ms to ∼4 ms. This decay halftime is similar to that observed for CoP/TiO2 films in the absence of RuP and at a similar CoP coverage, indicating that the presence of co-attached RuP does not significantly impede electron transfer from TiO2 to CoP.
Discussion
The surface functionalisation of semiconductor based photoelectrodes with molecular catalysts is an attractive strategy for solar driven fuel synthesis. This study focuses on the parameters determining the efficiency of electron transfer from the semiconductor to the molecular catalyst, and their potential impact on the overall solar to fuel conversion efficiency, addressing these kinetics for a model system comprising of a CoP proton reduction catalyst attached to a nanocrystalline, mesoporous TiO2 photoelectrode.Competing charge recombination reactions
From a thermodynamic point of view, photoexcited electrons in TiO2 could be used for the reduction of protons to hydrogen, while the holes that remain in the valence band could be used for water oxidation. However, in practice rapid recombination (nano- to micro-second) of photogenerated electrons and holes, and slow water oxidation/reduction kinetics prevents significant H2 and O2 evolution from TiO2 photoelectrodes alone.20 Even in the presence of TEOA, which increases the lifetime of TiO2 electrons to 1 s, negligible hydrogen generation is observed, indicating that the rate constant for proton reduction by TiO2 electrons must be substantially slower than 1 s−1 (at least under pH 7).One strategy to accelerate the kinetics of proton reduction to molecular hydrogen, and to aid the spatial separation of electrons and holes, is the addition of a chemical co-catalyst, such as the CoP employed herein. Such co-catalysts have been shown to substantially increase the photocatalytic activity of this semiconductor towards H2 evolution.23,25 Our studies show that the electron transfer from the TiO2 to the cobalt catalyst (CoP) indeed substantially retards electron–hole recombination. Fig. 2 indicates hole recombination with reduced CoP is at least four orders of magnitude slower than recombination with TiO2 conduction band electrons. However such lifetime enhancement is only useful if the electrons are able to transfer to the co-catalyst in the first place. The efficiency of this transfer depends on the rate constant for electron transfer from TiO2 to the catalyst relative to any competing pathways – primarily electron recombination with photogenerated holes.
In the presence of monolayer catalyst coverage, electron transfer from TiO2 to CoP can proceed reasonably fast (half time of ∼ few microseconds). The competing electron–hole recombination reaction depends on density of electrons and holes, and therefore upon the incident light flux. Both processes exhibit highly dispersive kinetics (i.e.: spread over a broad range of timescales), indicative of the effects of charge trapping and/or inhomogeneous catalyst binding to the TiO2 surface. For low pulsed laser intensities, of most relevant to conditions of continuous solar irradiation, and monolayer catalyst coverage, electron–hole recombination is sufficiently slow to allow photogenerated electrons to avoid electron–hole recombination and efficiently transfer to the CoP. At lower catalyst coverages, or higher light fluxes, the efficiency of catalyst reduction is progressively reduced.
Two electron reduction of CoP
The catalytic mechanism of cobaloximes is reported as a sequential two electron transfer process that involves the sequential reduction of CoIII species to CoII and CoI, with the reduction potentials of the reactions being −0.15 V and −0.55 V vs. NHE, respectively.25,35,36 It should be noted that the non-innocent character of diimines could also result in a ligand-based reduction product (CoIIL˙−) instead of CoI after two single-electron reduction steps of CoP. From a thermodynamic point of view, the first electron transfer has a larger driving force, and therefore should be faster than the second reduction reaction. Under photoexcitation conditions, we therefore expect the accumulation of CoII species, with the efficiency of molecular hydrogen generation being critically dependent upon achieving the second photoreduction of CoP catalyst to CoI.The discussion in the preceding section was concerned with the efficiency of single electron transfers from TiO2 to CoP, but did not address this requirement for double reduction of CoP to yield molecular hydrogen. Herein, we have addressed this issue by two sets of experiments which varied the ratio of the number of photogenerated electrons per catalyst molecule: (1) varying the amount of catalyst anchored onto the surface for a fixed laser intensity, and (2) modifying the electron density in the film by using different excitation densities for a fixed catalyst coverage (Fig. 5 and 7). Both experiments were done in the presence of TEOA sacrificial electron donor to avoid electron–hole recombination losses, and maximise the lifetime of photogenerated electrons. In both cases, increasing the ratio of photogenerated electrons per catalyst molecule resulted in a very large delay of the electron transfer timescale (by ≥104). Increasing the ratio of photoelectrons/CoP can be expected to increase the proportion of adsorbed CoP being singly reduced to CoII. Thus, this delay can be most easily understood as resulting from an increased accumulation of CoII, with electron transfer from TiO2 to CoII being at least 104 fold slower than to CoIII. We also note the possibility that our observation of slow electron transfer to CoP for high electron density/catalyst ratios could be associated with the accumulation of CoIII–H states at the surface of TiO2 or the slow H2 release. However, these processes have been reported to be relatively fast in an analogous cobaloxime catalyst (μs–ms),37 indicating that the performance of CoP is most likely to be limited by the kinetics of the second electron transfer when attached to TiO2 nanoparticles.
The more negative reduction potential for the CoII–CoI couple relative to CoIII–CoII reduces the thermodynamic driving force for electron transfer from the TiO2 conduction band. Using a standard Marcus analysis, assuming a TiO2 conduction band edge of −0.6 eV vs. NHE,38,39 and a value for the reorganisation energy λ of 1.0 eV,40 the 0.4 eV difference in reduction potential can be estimated to result in 105 slower electron transfer rate constant. This difference in rate constant is consistent with our conclusion that our experimental observation of ≥104 slower electron transfer under conditions of high photoelectron/CoP ratios, where accumulation of CoII is expected. Thus, our observations of a kinetically slower step attributed to the second electron reduction of CoP can now explain the relatively low external quantum efficiencies (1%) observed for H2 evolution with this photocatalytic system.25
We note that the reduction potential for CoII–CoI is close to the TiO2 conduction band edge, thereby potentially reducing not only the electron transfer rate constant but also the equilibrium constant for this transfer. A complete analysis of this equilibrium constant would require consideration of the TiO2 Fermi energy and the impact of subsequent proton double reduction and the generation of molecular hydrogen. In particular the latter effect is likely to provide a ‘sink’ effectively pull the equilibrium forwards. A quantitative analysis of these equilibrium effects is beyond the scope of this study.
Our observation of a > 104 delay of electron transfer lifetime under conditions of high photoelectron/CoP ratio indicates that under these conditions, almost all the CoP is in its CoII state. In the experiments reported herein, we have employed pulsed laser excitation, at a repetition frequency of 1 Hz, at pulse energies corresponding to ∼0.5–10 absorbed photons per TiO2 particle. These laser intensities are too low to reduce all the adsorbed CoP to CoII from a single laser pulse, indicating this reduction from the accumulated effect of multiple laser pulses. Given our 1 Hz repetition frequency, this implies the lifetime of CoII species in the presence of TEOA is ≫1 s.
There are two key conclusions from our analysis above. Firstly, as electron transfer to CoIII is at least 4 orders of magnitude faster than to CoII, significant reduction of CoII to CoI, enabling hydrogen generation, will only be possible once almost all the adsorbed CoP has been reduced to CoII. This has significant implications for the optimum catalyst loading, CoII lifetime and light flux. The second conclusion is that efficient hydrogen generation will require the generation of TiO2 electrons with sufficient lifetime to reduce not only CoIII but also CoII. This lifetime requirement is relatively facile for CoIII reduction – at monolayer CoP coverage electron transfer from TiO2 to CoIII exhibits a half time of ∼4 μs, and can thus compete effectively with electron–hole recombination even in the absence of sacrificial electron donors, at least under reasonably low excitation conditions. However electron transfer to CoII is kinetically much slower (≫ 40 ms), and therefore places a much more severe requirement on the TiO2 lifetime required to enable efficient electron transfer to the catalyst. It also increases the potential for long-lived photogenerated electrons to undergo unwanted side reactions (such as the reduction of the products of TEOA oxidation), thereby reducing the efficiency of hydrogen generation.
The role of hole scavengers and dye sensitisation
The above discussion has all focused on data collected in the presence of the hole scavenger TEOA. Such hole scavengers are typically added to remove photogenerated holes from semiconductor photocatalysts so as to enable photoreduction reactions such as the proton reduction reaction reported herein. The TEOA achieves this very efficiently, scavenging holes in <1 μs, resulting in the generation of long lived conduction band electrons with lifetimes of ∼1 s in the absence of CoP.It is striking that for the system studied herein, in the presence of a CoP monolayer, and employing low excitation densities, single electron transfer from TiO2 to CoP can proceed efficiently even in the absence of TEOA. In other words, for a sufficiently fast electron transfer rate constant to the adsorbed catalyst (measured as ∼3 × 105 s−1 herein for monolayer coverage), this electron transfer can compete reasonably effectively with electron–hole recombination in the TiO2. Nevertheless the addition of TEOA is necessary to achieve measureable yields of hydrogen generation.9,25,41 This can be understood as resulting from two factors. Firstly efficient electron transfer to the catalyst requires near monolayer catalyst coverage, which might be difficult to achieve in the suspension systems typically employed for hydrogen generation measurements. Secondly, and critically, hydrogen generation requires not a single but a double reduction of CoP. The second reduction reaction (CoII to CoI) is kinetically at least 104 slower than the first reduction, and therefore is unable to compete with electron–hole recombination in the absence of TEOA. Moreover this double reduction of CoP requires almost complete accumulation of singly reduced CoP (CoII) on the TiO2 surface. In the absence of TEOA, these reduced CoP species would act as recombination sites for photogenerated holes (indeed even in the presence of TEOA, this recombination may be a significant loss pathway).
An attractive way to achieve visible light photoactivity is to employ a molecular sensitiser, such as the RuP dye. The addition of such dyes indeed enables visible light driven hydrogen generation.42 However it should be noted that the use of such a sensitiser significantly reduces the thermodynamic driving force available for the hole scavenging or ‘regeneration’ reaction. It is likely that the much slower kinetics for scavenging of ‘holes’ from RuP relative to TiO2 (100 μs versus <1 μs respectively) is associated with less oxidising potential of the RuP cations relative to the TiO2 valence band. This is offset partly by slower recombination kinetics for RuP+/TiO2 (e−) versus TiO2 (h+)/TiO2 (e−) – half times of ∼150 μs versus 50 μs respectively at matched excitation densities. Nevertheless the substantially slower hole scavenging kinetics for the RuP/TiO2 system results in this hole scavenging achieving (for the system studied herein) a quantum efficiency of only ∼50%. This represents an additional efficiency loss not present when employing direct TiO2 band gap excitation, although under solar irradiation, this loss is more than offset by the enhanced light harvesting. Additionally, the less efficient hole scavenging by TEOA in the dye sensitised system also contributes in decreasing the yield of the slower electron transfer to the molecular catalyst, resulting in a smaller H2 evolution yield.
The use of sacrificial electron donors is clearly undesirable for any technological application, which would require these donors to be replaced by reversible redox couples or hole transport to an external circuit. It is apparent from the above discussion that any such replacement will be very challenging. If a reversible redox couple is employed, the oxidised state of this couple is likely to compete with singly reduced CoP as acceptors for TiO2 electrons. If hole transport to an external circuit is employed, this will have to be very rapid to prevent hole accumulation in the semiconductor and subsequent recombination with either TiO2 electrons or single reduced CoP species. As a consequence, such replacements are only likely to be viable in more complex system architectures, including in particular the use of heterojunctions or additional oxidation co-catalysts.
Conclusions
In this work we explain in detail the kinetics of electron transfer processes that take place in three different molecule-functionalised systems for the photochemical H2 evolution, with special emphasis on the parameters that affect the electron transfer from the TiO2 to the molecular catalyst. The molecular catalyst employed is a cobaloxime proton reduction catalyst, which requires a two-electron reduction from CoIII to CoI for hydrogen generation. We find that the kinetics of electron transfer from TiO2 to CoP, their competition with electron–hole recombination and the requirement for hole scavenging are very different for the first electron reduction from CoIII to CoII versus the second from CoII to CoI. At sufficiently high catalyst coverages, and low excitation densities, the first reduction can proceed efficiently even in the absence of added hole scavenger. In contrast, the second reduction reaction appears to be at least 105 slower, suggesting it requires efficient hole scavenging and almost complete reduction of all adsorbed CoP to CoII for it to proceed. Dye sensitisation enables visible light photoactivity, although this is partly offset by slower, and less efficient, hole scavenging by TEOA.These results were obtained with a single semiconductor/molecular catalyst system. However, they address an issue likely to be critical to many potential implementations of such semiconductor/molecular catalyst systems, namely the requirement for photogenerated electrons and holes to drive multiple oxidation/reduction reactions on a single catalyst. Addressing this challenge is likely to be critical in developing such hybrid systems for solar driven fuel production.
Acknowledgements
Financial support from EPSRC (EP/H046380/1 to J.D., EP/H00338X/2 to E.R.), the ERC (project Intersolar to J.D.), the Christian Doppler Research Association (Austrian Federal Ministry of Economy, Family and Youth and National Foundation for Research, Technology and Development) and the OMV Group (to E.R.), the Spanish Ministry of Education (EX2010-0479 to A.R.) and the European Commission Marie Curie CIG (PCIG10-GA-2011-303650 to A.R.) are gratefully acknowledged. The authors thank Dr Xiaoe Li for assistance in TiO2 preparation.Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- H. Krassen, S. Ott and J. Heberle, Phys. Chem. Chem. Phys., 2011, 13, 47–57 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 CAS.
- S. Losse, J. G. Vos and S. Rau, Coord. Chem. Rev., 2010, 254, 2492–2504 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef CAS.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- J. M. Gardner, M. Beyler, M. Karnahl, S. Tschierlei, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 19322–19325 CrossRef CAS.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472–16475 CrossRef CAS.
- F. Wen, X. Wang, L. Huang, G. Ma, J. Yang and C. Li, ChemSusChem, 2012, 5, 849–853 CrossRef CAS.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS.
- N. A. Anderson and T. Lian, Coord. Chem. Rev., 2004, 248, 1231–1246 CrossRef CAS.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- A. Reynal, A. Forneli and E. Palomares, Energy Environ. Sci., 2010, 3, 805–812 CAS.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, J. Phys. Chem. B, 2001, 105, 7258–7262 CrossRef CAS.
- A. J. Cowan, J. Tang, W. Leng, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2010, 114, 4208–4214 CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS.
- S. Sato and J. M. White, J. Phys. Chem., 1981, 85, 592–594 CrossRef CAS.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 CAS.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2009, 132, 1060–1065 CrossRef.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS.
- S. A. Trammell, J. A. Moss, J. C. Yang, B. M. Nakhle, C. A. Slate, F. Odobel, M. Sykora, B. W. Erickson and T. J. Meyer, Inorg. Chem., 1999, 38, 3665–3669 CrossRef CAS.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- B. O'Regan, L. Xiaoe and T. Ghaddar, Energy Environ. Sci., 2012, 5, 7203–7215 CAS.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS.
- Y. Tamaki, A. Furube, M. Murai, K. Hara, R. Katoh and M. Tachiya, Phys. Chem. Chem. Phys., 2007, 9, 1453–1460 RSC.
- M. Murai, Y. Tamaki, A. Furube, K. Hara and R. Katoh, Catal. Today, 2007, 120, 214–219 CrossRef CAS.
- J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15374 CrossRef CAS.
- R. Katoh, M. Murai and A. Furube, Chem. Phys. Lett., 2008, 461, 238–241 CrossRef CAS.
- t50% was calculated considering that the maximum electron density in TiO2 was achieved when no CoP was anchored onto the surface.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS.
- O. Pantani, E. Anxolabéhère-Mallart, A. Aukauloo and P. Millet, Electrochem. Commun., 2007, 9, 54–58 CrossRef CAS.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774–16776 CrossRef CAS.
- G. Rothenberger, D. Fitzmaurice and M. Graetzel, J. Phys. Chem., 1992, 96, 5983–5986 CrossRef CAS.
- B. Enright, G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1994, 98, 6195–6200 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252–11262 CrossRef CAS.
- T. R. N. Kutty and M. Avudaithai, Mater. Res. Bull., 1988, 23, 725–734 CrossRef CAS.
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional transient absorption data and Marcus theory equation. See DOI: 10.1039/c3ee40961a |
| This journal is © The Royal Society of Chemistry 2013 |