 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-inspired NADH regeneration by carbon nitride photocatalysis using diatom templates†
Jian
Liu
* and
Markus
Antonietti
Department of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: Jian.Liu@mpikg.mpg.de
First published on 18th March 2013
Abstract
In this report, the bio-inspired photocatalytic regeneration of NADH employing graphitic carbon nitride photocatalysis with a diatom frustule structure was presented. The functional structure was constructed from green and sustainable diatomite (diatom earth) by an “incipient wetness impregnation” method. By using [Cp*Rh(bpy)H2O]2+ as an electron mediator, the NADH yield can reach nearly 100%, with the enzymatically active 1,4-NADH being the only product. Furthermore, even without an electron mediator, a direct electron transfer between graphitic carbon nitride and β-NAD+ was possible and NADH could still be regenerated. Diatom-based carbon nitride showed a substantial improvement in regeneration efficiency compared with mpg-C3N4 which has a higher specific surface area. This was ascribed to the diatom structure, with its enhanced light trapping and scattering promoting high photocatalytic efficiency. Furthermore, abundant species of diatoms in the earth offer more possibilities to construct an artificial photosynthetic system using diatom frustules with excellent light-to-energy conversion efficiency.
Broader contextEnzyme catalysis shows high efficiency and regioselectivity, and the interest of applying enzymes for organic synthesis is increasing. One factor that limits the practical application of enzymatic catalysis in organic reactions is the high cost of enzyme-specific cofactors. Enzyme-catalyzed regeneration of NAD(P)H (e.g. alcohol dehydrogenase and formate dehydrogenase) has been well established in the past years, but this procedure is complicated, as it usually involves the simultaneous presence of enzymes and substrates. In view of increasing environmental and sustainability considerations, seeking a green, sustainable, economic, and efficient regeneration method for NADH is indispensable. Learning from natural photosynthesis, photosystem I, an integral membrane protein complex, uses light energy to mediate electron transfer and the reduction of nicotinamide adenine dinucleotide phosphate (NADP) to NADPH occurs, storing the light energy in chemical “currency”. Therefore, imitating the function of PSI, the utilization of light energy for the reduction of NAD+ to NADH is very promising, cost effective and inherently sustainable. |
Introduction
Diatoms are unicellular photosynthetic organisms and can be found in nearly every aquatic habitat on earth. They are responsible for approximately one-fifth of the global photosynthesis yield.1 As is well known, the diatom features unique frustule architectures with hierarchical structures ranging from micrometric to nanometric scales, which are thought to contribute to their high photosynthesis efficiency.2–4 Therefore, imitating the structures of diatom frustules and reconstructing the frustules with photoactive materials will be promising for the construction of an artificial photosynthetic system for light energy conversion purposes.Photosystem I (PSI), an integral membrane protein complex in photosynthesis, uses light energy to mediate electron transfer from plastocyanin to ferredoxin and then to ferredoxin–NADP reductase, where the ultimate reduction of nicotinamide adenine dinucleotide phosphate (NADP) to NADPH occurs, storing the light energy in chemical “currency”, as NAD(P)H carries hydrogen in a biologically convertible form (Scheme 1). The resultant NAD(P)H is indeed used in many biochemical conversions, most prominently as a hydride donor for the biosynthesis of carbohydrates in the Calvin cycle of the dark reactions and also as a NAD(P)+/NAD(P)H cofactor in many other enzymatic redox reactions in metabolic processes.
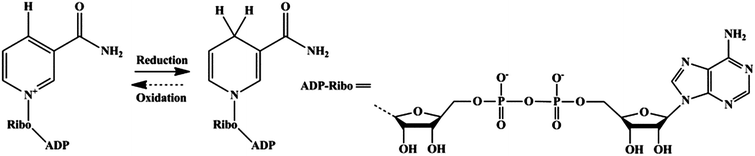 | ||
| Scheme 1 Redox equilibrium between β-NAD+ and NADH. | ||
Enzymes show high efficiency and regioselectivity, and interest in applying enzymes for the complex synthesis, e.g. of chiral organic compounds, is increasing.5 One factor that limits the practical application of enzymatic catalysis in organic reactions is the high cost of enzyme-specific cofactors. Enzyme-catalyzed regeneration of NAD(P)H (e.g. alcohol dehydrogenase and formate dehydrogenase) has been well established in the past few years, but this procedure is complicated, as it usually involves the simultaneous presence of enzymes and substrates.6 Non-enzymatic electrochemical and newly proposed photocatalytic methods in the recent years can also serve the purpose of NADH regeneration.7,8 However, those non-enzymatic methods usually have major drawbacks, i.e. a rare metal-chelating electron mediator or toxic methylviologen are usually prerequisites for the known NADH regeneration routes.9–13 In view of increasing environmental and sustainability considerations, seeking a green, sustainable, economic, and efficient regeneration method for NADH is indispensable.
Learning from natural photosynthesis, the utilization of light energy for the reduction of NAD+ is very promising, cost effective and inherently sustainable. The benefits of photocatalysis should ideally not be counterbalanced by toxic metals or the use of rare elements, such as ruthenium or iridium. The exploration for the replacement of rare metal chelated mediators or direct electron transfer (DET) between the photocatalyst and NAD+ is regarded as a key factor, but is very difficult. Photoinduced electron transfer is well established in modern photochemistry and long-lived charge separation states for artificial photosynthesis can occur via π–π stacking when the molecules are bound to polymers, micelles and biomolecules.14 Using electron rich aromatic scaffolds with electronic properties similar to DNA or nucleobases might enable the DET between a photocatalyst and NAD+ and consequently might serve the purpose of light induced substrate regeneration. Polymeric carbon nitride (simplified as g-C3N4 in the following context), a “molecular fossil” material but with (photo)catalytic functional properties revealed only in recent years, could in principle fulfil these conditions.15
Therefore, a carbon nitride material with a diatom frustule structure was constructed to act as a high efficiency artificial photosynthetic system for NADH regeneration. We present here a novel visible light driven photocatalytic method imitating the function of PSI for the regeneration of 1,4-NADH from NAD+ by diatom frustule structured graphitic C3N4 (DE-g-C3N4 in short). In the presence of [Cp*Rh(bpy)H2O]2+ as an electron mediator, it will be shown that nearly 100% of NADH can be regenerated. Triethanolamine (TEOA) is employed as a sacrificial electron donor. The oxidized state of TEOA loses H+ during its decomposition path to glycolaldehyde and di(ethanol)amine. g-C3N4 can however also strongly bind with β-NAD+ due to the π–π stacking interaction between the adenine subunit of β-NAD+ and the electron-rich, layered configurations of g-C3N4, as illustrated in Fig. 1. This charge transfer complex is the pre-state for the photoinduced electron transfer between carbon nitride and β-NAD+, which in principle works without a further mediator. As a proof-of-principle, it will be shown that in the mediator-free case, the regeneration yield of NADH can get up to about 50%, while the problems blocking further improvements could be identified. To the best of our knowledge, the work represents the first report about mediator-less metal-free photocatalytic regeneration of NADH from β-NAD+ and describes – with the involvement of an electron mediator – the highest conversion yield up to now.
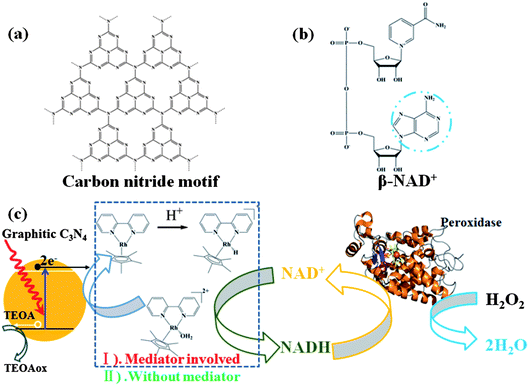 | ||
| Fig. 1 Schematic illustration of the interaction between g-C3N4 and β-NAD+. (a) Illustration of a perfect graphitic carbon nitride constructed from heptazine building blocks. (b) Molecular structure of β-NAD+. (c) Illustrated scheme of photocatalytic regeneration of NADH in the absence or presence of Mediator and H2O2 reduction mediated by Horseradish Peroxidase in the presence of photogenerated NADH. | ||
Results and discussion
DE-g-C3N4 was prepared through the “incipient wetness impregnation” method and obtained as yellow powder after polycondensation of cyanamide at 600 °C in a nitrogen atmosphere. SEM and confocal microscopy characterization results are illustrated in Fig. 2. It can be seen that the morphologies of the diatom frustule were well retained in the diatom–carbon nitride composite due to the favorable coating of carbon nitride onto the frustule. The confocal fluorescence characterization under 488 nm light excitation further confirms the excellent coating of carbon nitride onto the entire frustule. mpg-C3N4 as an internal standard was synthesized with commercial silica nanoparticles as a template and typical characterizations including TEM, FTIR and XRD are depicted in Fig. S1 of the ESI.† Bulky g-C3N4 was fabricated from the thermal condensation of cyanamide and used as a second control sample. It must be mentioned that graphitic carbon nitride obtained in this work is not made up of ideally condensed carbon nitride, but is rather an incompletely condensed N-bridged “poly(tri-s-triazine)” polymer with heptazine as building blocks.16 Carbon nitride was found to activate a number of aromatic molecules through the π–π interactions between its electron rich scaffold and appropriate electron acceptors,17 and this is also true for the NAD+ molecule (Fig. 1a and b). The isoelectric point (IEP) of g-C3N4 is ca. 6.18 Not only the π–π stacking interactions, but also electrostatic interactions between charged g-C3N4 and NAD+ can serve the purpose of surface confinement of the NAD+ molecule. The interaction is expected to be strengthened further under illumination due to the increase of the Fermi-level of carbon nitride, i.e. the catalyst is getting even more negative and electron rich.19 | ||
| Fig. 2 Typical morphology characterizations of DE-g-C3N4. (a) Large area overview of the DE-g-C3N4, showing the well distributed diatom–carbon nitride composite. (b) The magnified image of the DE-g-C3N4, demonstrating the excellent coating of the single frustule with the carbon nitride material. (c and d) The microscopy investigations of DE-g-C3N4 with the inset showing the confocal fluorescence images under 488 nm excitation, further indicating the excellent g-C3N4 coating on the entire frustule. | ||
The photocatalytic experimental setup is illustrated in Fig. 3a. The blue light LED with an emission peak at 420 nm is employed as a light source. mpg-C3N4 was first employed to obtain the optimum pH conditions. [Cp*Rh(bpy)H2O]2+ (Cp* = pentamethylcyclopentadienyl, bpy = 2,2′-bipyridyl) (1H-NMR spectrum in Fig. S2 of ESI,† abbreviated as M in the following context) is a homogeneous redox catalyst that has been applied before for the photo- and electrochemical regeneration of NADH.20 Using M in our photocatalytic system enabled a practically complete conversion efficiency (see also the effect of different concentrations of M in Fig. S3 of ESI†). The absorbance spectra of the illuminated reaction media were monitored using the UV-Vis spectrophotometer every 15 or 30 min during a total of 2 h experimental time.
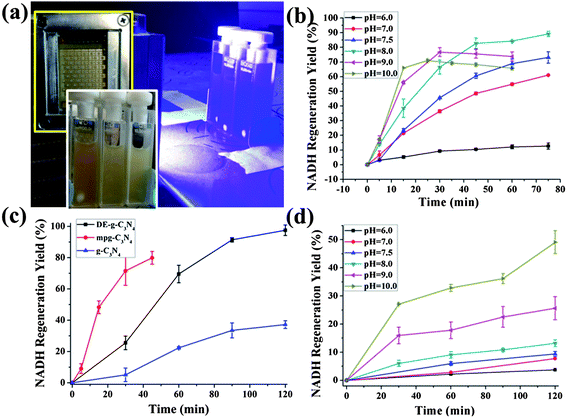 | ||
| Fig. 3 (a) Experimental setup for the photocatalytic regeneration of NADH. (b) The photogenerated NADH yield showed pH dependence in a series of phosphate buffered saline (PBS) buffer solutions of different pH. β-NAD+, 1 mM; M, 0.25 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH as indicated; mpg-C3N4, 2 mg. (c) Photocatalytic activity comparisons between DE-g-C3N4, mpg-C3N4, and g-C3N4 in regenerating NADH from a mediator involved system with a pH of 8. (d) The photogenerated NADH yield showed pH dependence in a series of PBS buffer solutions of different pH; β-NAD+, 1 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH as indicated; mpg-C3N4, 2 mg. | ||
The absorbance at 340 nm becomes saturated after only 45 minutes at close to the maximal possible level (Fig. S4 of ESI†). A quantum yield of 5.2 × 10−4 was obtained from the pH = 8 system after 30 min illumination. The pH dependence in the presence of M reveals a maximum activity at pH = 8 (Fig. 3b). Higher pH values still give higher initial reaction rates, but lower conversion efficiencies. According to the literature, the activity of M depends on the pH value of the buffer solution.20 Higher proton concentration would facilitate proton-driven oxidation of [Cp*Rh(bpy)H]+ yielding H2 while the NADH photogeneration is predominantly inhibited; at higher OH− concentration, [Cp*Rh(bpy)(OH)]+ is probably the predominant species and ligand exchange with TEOA becomes rate limiting. The optimum pH value for the mediator for maximal activity lies around 7.5 and matches with our experiment (pH = 8.0).
To analyze the influence of the sample architecture, the photocatalytic activity of DE-g-C3N4, g-C3N4 and mpg-C3N4 were compared and the results are illustrated in Fig. 3c. Equal amounts of carbon nitride content (3 mg) were employed for all three samples. In DE-g-C3N4, the carbon nitride composition accounts for about 20% of the total weight of the composite according to the TGA curve (Fig. S5 of ESI†), and 15 mg DE-g-C3N4 was used for the photocatalytic experiment. For DE-g-C3N4 in a mediator containing system (pH = 8), the regeneration efficiency is lower than that of the mpg-C3N4 at the beginning of the reaction but steadily increases with time. After 2 hours of illumination, DE-g-C3N4 gives the best regeneration yield (nearly 100%) while the mpg-C3N4 exceeds 80% yield, although in a shorter time. The surface area of DE-g-C3N4 is very low (∼5 m2 g−1) compared with mpg-C3N4 (286 m2 g−1). Bulky g-C3N4 gave the lowest regeneration efficiency and yield in the same reaction period, as expected, which is due to its extremely low surface area and its weak light-harvesting capability. Therefore, the superior behavior of DE-g-C3N4 can be ascribed to the introduced diatom structuring factor, which improves its coherent light trapping and scattering.4,21 The diatom frustule could be simplified as a two dimensional quasi-periodic or disordered media, which was proven to contribute to the broadband absorption enhancement for photovoltaic and photocatalytic applications.22–24 A Mie calculation was employed to roughly evaluate the light scattering behavior of the diatom frustule under 420 nm light irradiation and is illustrated in Fig. S6 of ESI.† The size and shape of the diatom frustule are considered to be the critical factors in determining the light scattering behavior.25 From the calculation results, the light scattering was predominately peaked in the forward direction. The light scattering function shows oscillatory behavior indicating the narrow size distribution of the diatom samples.25
In a second set of experiments, we tried to address the more challenging question of a potential mediator-free regeneration of NADH. Here, light-irradiated carbon nitride transfers the electrons directly to the bounded NAD+ molecule.26 To our surprise, a mediator-free regeneration was indeed possible, although with lower rates and lower maximal conversion. The optimum pH conditions for this process were analyzed employing mpg-C3N4 at first. The experimentally determined photocatalytic activities for different pH values are depicted in Fig. 3d. A reaction medium with pH = 10 gives the best conversion efficiency, and a quantum yield of 2.1 × 10−4 was achieved in the pH = 10 mediator-free reaction system after 30 min of light irradiation. The preference of alkaline media is presumably attributed to a spontaneously occurring side reaction, which is the liberation of H2 from carbon nitride, which peaks at neutral pH values.27 The lower rate for the direct charge transfer process (from 5.4 to 2.1 × 10−4) is typical for the transition from homogeneous to heterogeneous catalysis and indeed very promising. We can only speculate why the conversion settles at around 50% (see Fig. S7†), but a possible explanation would be that without a stronger redox agent (as the mediator), carbon nitride also catalyzes the back reaction, meaning an equilibrium between the two species is approached but not exceeded.
Horseradish peroxidase (HRP) mediated H2O2 reduction was employed to utilize the photocatalytic regenerated NADH.28 For a mediator containing system, 100 μl reaction solution (1 mM of NAD+/NADH in total) was immediately transferred to the incubation reaction cell with 44 U of HRP in 2.9 ml of a phosphate buffer (pH = 7.5) (see reaction media of pH = 8 in Fig. 4a). The photogenerated NADH was used to reduce H2O2 to H2O mediated by HRP, as illustrated in Fig. 1c. The characteristic absorbance at 403 nm ascribed to heme as a prosthetic group verified the existence of HRP in the reaction. After the addition of 50 μl 0.3 M of H2O2, the absorbance was redshifted to 420 nm, which is thought to be due to the formation of the complex [HRP–H2O2].29 After incubation for 20 min (avoiding light), the NADH absorbance at 340 nm decreased apparently as a result of consumption by H2O2 reduction mediated by HRP, while the reference NADH solution with only H2O2 showed almost no difference in absorbance intensity after the same duration. According to the absorbance change before and after the HRP reaction (ΔA = 0.104, calculated from the blue and pink plots in Fig. 4a), the concentration of consumed NADH is calculated to be 0.82 mM, which was comparable with the value obtained from the absorbance spectra (0.88 mM, calculated from the red plot in Fig. 4a). Due to the more efficient electron transfer between mpg-C3N4 and the electron mediator, the DET and the coupled partial generation of 1,6-NADH is significantly reduced. Therefore, 1,4-NADH will be the only product (see 1H-NMR spectra in Fig. S8 of ESI†). This leads to a higher availability for the enzymatic reaction and a higher consistence between optical and enzymatic concentration determination.
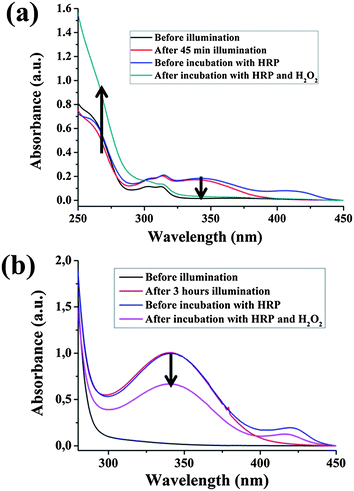 | ||
| Fig. 4 (a) Absorbance changes of NADH accompanying the HRP mediated H2O2 reduction. NADH regeneration: β-NAD+, 1 mM; M, 0.25 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH = 8; mpg-C3N4, 2 mg; HRP reaction: 44 U horseradish peroxidase in 2.9 ml pH = 7.5 0.1 M PBS buffer aqueous solution, 100 μl NADH reaction solution, 0.3 M H2O2 50 μl, incubation at room temperature. (b) Absorbance changes of NADH accompanying the HRP mediated H2O2 reduction. NADH regeneration: β-NAD+, 12 mM; TEOA, 15 w/v%; PBS buffer, 0.1 M, pH = 10; mpg-C3N4, 3 mg. HRP reaction: 44 U horseradish peroxidase in 2.9 ml pH = 7.5 0.1 M PBS buffer aqueous solution, 100 μl NADH reaction solution, 0.3 M H2O2 50 μl, incubation at room temperature. | ||
The HRP mediated H2O2 reduction could be accomplished by photogenerated NADH in a mediator free system too (Fig. 4b). 12 mM NAD+ was employed to regenerate NADH by direct carbon nitride photocatalysis in a pH = 10 reaction solution. After the illumination, 100 μl reaction solution was immediately transferred to the incubation reaction cell with 44 U of HRP in 2.9 ml of a phosphate buffer (pH = 7.5). According to the absorbance change before and after HRP reaction (ΔA = 0.3291), the concentration of NADH consumed in the enzymatic reaction is calculated to be 1.58 mM, which is lower than the value obtained from the absorbance spectra (4.7 mM), as illustrated in Fig. 4b. The data inconsistency is thought to be due to the fact that the obtained NADH consists of enzymatically active 1,4-NADH and enzymatically inactive 1,6-NADH (NMR spectra in Fig. S9 of ESI†). According to the literature, 1,6-NADH can be, for instance, generated through a bimolecular reaction between NAD+ and photogenerated 1,4-NADH.30 The alternative enzymatic assay discussed below will give additional evidence.
As a second quantification, NADH regenerated from mpg-C3N4 was also determined by a standard NAD+/NADH microplate assay experiment (see standard curve in Fig. S10 of ESI†). The overall NAD+/NADH concentration for the assay is 10 μM. In the mediator involved system, the regenerated NADH in the pH = 8 system (8.6 μM, as determined from the assay kit) matched well with the value obtained from the extinction spectra (9.5 μM). The photocatalytic regenerated NADH in the mediator involved system was almost completely sensed by the assay. Again, the assay does not sense all NADH created in the mediator free system (pH = 10), because not all the NADH regenerated in mediator free system is enzymatically active. The quantified NADH from the mediator free system by the assay kit is 1.1 μM, compared with 4.0 μM from the extinction spectra calculation.
The overall results including the concentrations of NADH obtained from the UV-Vis spectroscopy, the HRP enzymatic reaction and assay analysis are shown in Table 1. As discussed, the mediated electron transfer obviously creates a biologically viable NADH while this occurs only partly in the direct electron transfer process. Improving the selectivity of the DET as well as blocking a potential hydrogen evolution from NADH by the heterogeneous catalysis are however questions which could be addressed by catalyst design, i.e. adjusting the surface chemistry and the electronic states of the photocatalyst.
| UV-Vis | HRPc reaction | Assay | |||
|---|---|---|---|---|---|
| NAD+/NADH (μM) | NADH (μM) | NADH (μM) | NAD+/NADH (μM) | NADH (μM) | |
a Light reaction with a mediator (M, 0.25 mM).
b The concentration of original NAD+ solution (1 mM) was diluted in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 before light reaction.
c The NADH consumed in the HRP enzymatic reaction.
d For comparison, the consumed NADH concentration was converted from 0.82 mM after decreasing 100 times.
e For comparison, the consumed NADH concentrations were converted from 1.58 mM and 0.11 mM after decreasing about 1200 times and 100 times, respectively. :100 before light reaction.
c The NADH consumed in the HRP enzymatic reaction.
d For comparison, the consumed NADH concentration was converted from 0.82 mM after decreasing 100 times.
e For comparison, the consumed NADH concentrations were converted from 1.58 mM and 0.11 mM after decreasing about 1200 times and 100 times, respectively.
|
|||||
| pH = 8a | 10b | 9.5 | 8.2d | 12.0 | 8.6 |
| pH = 10 | 10b | 4.0 | 1.3/1.1e | 2.6 | 1.1 |
However, all the enzyme reactions prove that a photochemical regeneration of the cofactor is possible, i.e. enzymatic and artificial photosynthesis systems can work hand-in-hand to build up complex molecules. Using NAD+, the energy can be first fixed in “biological coins”. This opens up conceptual pathways towards a real convergent technology, where enzyme factories are fuelled by sunlight via synthetic energy transduction, transferring the synthesis of molecules to chemical environments far from the biological ones.
Conclusion
In conclusion, bio-inspired photocatalytic regeneration of NADH employing graphitic carbon nitride photocatalysis with a diatom frustule structure was presented. The functional structure was constructed from green and sustainable diatomite (diatom earth) by an “incipient wetness impregnation” method. By using [Cp*Rh(bpy)H2O]2+ as an electron mediator, the NADH yield can reach nearly 100%, with enzymatically active 1,4-NADH being the only product. Diatom-based carbon nitride showed a substantial improvement in regeneration efficiency compared with mpg-C3N4 which has a higher specific surface area. This was ascribed to the diatom structure, with its enhanced light trapping and scattering promoting high photocatalytic efficiency. Beyond the current work, selecting single cases from the various species of diatoms offers further possibilities to construct artificial photosynthetic systems with high light-to-energy conversion efficiency.Excitingly, even without an electron mediator, a direct electron transfer between graphitic carbon nitride and β-NAD+ was possible, presumably due to the π–π stacking between the heptazine building blocks of the graphitic carbon nitride photocatalyst and adenine subunit of the NAD+. At pH = 10, the regeneration rates were still comparable, but the yield of NADH in the direct case saturated at about 50% conversion efficiency after two hours of illumination, which is potentially due to the catalytic activation of both reactant and product. In addition, only part of the heterogeneously generated NADH is enzymatically active 1,4-NADH. This limited conversion as well as a redox selectivity towards 1,4-NADH are certainly points needing improvement, which can be addressed in the near future by adjusting the carbon nitride composition and structure to address the problems identified here.
Experimental section
Materials
Cyanamide, triethanolamine (TEOA), rhodium(III) chloride hydrate, 2,2′-bipyridyl, 1,2,3,4,5-pentamethylcyclopentadiene and β-nicotinamide adenine dinucleotide phosphate sodium salt hydrate (β-NAD+) were purchased from Sigma-Aldrich (St. Louis, MO). The stock solution of 25 mM of β-NAD+ was dispensed as a 1 ml package and stored at −80 °C. The Amplite™ Fluorimetric NAD+/NADH Ratio Assay Kit (15263) was ordered from the AAT Bioquest, Inc.Synthesis
DE-g-C3N4 was synthesized through an “incipient wetness impregnation” method. 0.5 g of cyanamide and 1 g of Celatom® FW-14 diatomite were mixed evenly in the mortar and transferred into the crucibles and heated with the lid on under N2 atmosphere to 600 °C (4 hours) and kept at the temperature for another 4 hours. The obtained yellow powder composite is directly used in the photocatalytic experiment.mpg-C3N4 was synthesized as follows: 5 g of cyanamide and 12.5 g of Ludox-HS 40 colloidal silica suspension (1:1of solid ratio) were mixed together until the complete dissolution of cyanamide was achieved. The mixed solution was then placed into an oil bath at 100 °C and stirred to remove water. The obtained white solid was ground in a mortar, and then transferred to a crucible and heated with the lid on under air at 2.3 °C min−1 up to 550 °C (4 hours) and kept at 550 °C for another 4 hours. The resultant yellow powder was treated with 4 M NH4HF2 solution and stirred for 48 hours (be cautious!). The dispersion was then filtered; the precipitate was thoroughly rinsed with deionized water and ethanol. After the filtering procedure, the yellow powder was dried under vacuum at 60 °C overnight. g-C3N4 was obtained through the thermal condensation of cyanamide at 550 °C (4 hours) under air and used as control sample.
[Cp*Rh(bpy)H2O]2+ was synthesized as follows: RhCl3·H2O was refluxed in methanol with one equivalent of 1,2,3,4,5-pentamethylcyclopentadiene for 24 h. The resulting red precipitate was filtrated and suspended in methanol. On the addition of two equivalents of 2,2-bipyridine, the suspension cleared up immediately and a yellowish solution was formed. [Cp*Rh(bpy)Cl]Cl was precipitated by the addition of diethyl ether into the obtained yellowish solution. Stock solutions (100 mM) were prepared in water and stored at room temperature avoiding direct light exposure. [Cp*Rh(bpy)Cl]Cl readily hydrolyzes to [Cp*Rh(bpy)(H2O)]2+.
Characterization
XRD measurements were performed on a D8 Diffractometer from Bruker instruments (Cu Kα radiation, λ = 0.154 nm) equipped with a scintillation counter. N2 sorption experiments were done with a Quantachrome Autosorb-1 at liquid nitrogen temperature. TEM images were taken on a Phillips CM200 FEG (Field Emission Gun), operated at an acceleration voltage of 200 kV. SEM measurement was performed on a LEO 1550 Gemini instrument. The UV-Vis absorbance spectra were recorded on a T70 UV/Vis spectrophotometer. The optical density at 570 nm wavelength was recorded with an Absorbance Microplate Reader (BioTek Synergy HT).NADH regeneration and NAD+/NADH assay
In a typical regeneration procedure, the reaction medium was composed of NAD+ (1 mM), TEOA (15 w/v%), phosphate buffer (100 mM) and mpg-C3N4 or DE-g-C3N4 (2 or 3 mg). The pH value of the reaction medium can be adjusted by the addition of dilute HCl or dilute NaOH solution. The reaction system was placed into a quartz reactor equipped with a stirring bar and illuminated with an LED lamp (wavelength = 420 nm, OSA Opto Light GmbH). The distance between reactor and LED lamp was fixed at 5.5 cm. Before the illumination, the reaction solution was placed in a dark environment for 25 minutes to achieve adsorption–desorption equilibrium. During the illumination, the concentration of NADH was estimated by measuring the absorbance of the diluted reaction system at 340 nm. NAD+ has peak absorption at a wavelength of 260 nm, with an extinction coefficient of 16900 M−1 cm−1. NADH has peak absorption at 340 nm with an extinction coefficient of 6220 M−1 cm−1.
The NAD+/NADH assay was performed following the assay protocol provided by the manufacturer (AAT Bioquest). The assay kit is based on the alcohol dehydrogenase's micro cycling reaction. The NAD+ cycling enzyme mix in the assay kit specifically recognizes NAD+/NADH in an enzyme cycling reaction. Tetrazolium dye (MTT) is reduced by NADH in the presence of phenazine ethosulfate (PES). The intensity of the reduced product color measured at 570 nm is proportional to the NAD+/NADH concentration in the photocatalytic medium. The NADH generated from mediator-less (pH = 10) and mediator-involved (pH = 8) systems were diluted 1:100 with a PBS buffer (pH = 7.4) and then quantified using the assay. Thus, the overall NAD+/NADH concentration for the assay is 10 mM (1/100 times dilution from 1 mM NAD+ original solution after light reaction).
Horseradish peroxidase catalyzed reaction
100 μl of freshly prepared photogenerated NADH reaction solution was transferred to 2.9 ml PBS buffer with 44 U Horseradish peroxidase (pH = 7.5). 50 μl of 0.3 M H2O2 aqueous solution was added into the above solution and incubated for 20 min in the dark. The extinction spectra were recorded for every step.Quantum yield calculation
The quantum yield for the NADH production was calculated according to the following formula:
Acknowledgements
J.L. acknowledges the support of Alexander von Humboldt Foundation. J.L. thanks Dr Mingzhu Li of ICCAS for helpful discussion and Dr Hongqiang Wang of MPIKG for experimental assistance. J.L. thanks Dr Guozhang Wu and Dr Jiang Zhang from MPIMP in Potsdam for assistance in microplate reader usage. J.L. is also grateful for Dr Regine JAHN for providing the diatom samples.References
- F. E. Round, R. M. Crawford and D. G. Mann, Diatoms: biology and morphology of the genera, Cambridge University Press, 1990 Search PubMed.
- T. Fuhrmann, S. Landwehr, M. El Rharbi-Kucki and M. Sumper, Appl. Phys. B: Lasers Opt., 2004, 78, 257 CrossRef CAS.
- S. Yamanaka, R. Yano, H. Usami, N. Hayashida, M. Ohguchi, H. Takeda and K. Yoshino, J. Appl. Phys., 2008, 103, 074701 CrossRef.
- C. Jeffryes, J. Campbell, H. Li, J. Jiao and G. Rorrer, Energy Environ. Sci., 2011, 4, 3930 CAS.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196 CrossRef CAS.
- K. Seelbach, B. Riebel, W. Hummel, M. R. Kula, V. I. Tishkov, A. M. Egorov, C. Wandrey and U. Kragl, Tetrahedron Lett., 1996, 37, 1377 CrossRef CAS.
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583 CrossRef CAS.
- J. Ryu, S. H. Lee, D. H. Nam and C. B. Park, Adv. Mater., 2011, 23, 1883 CrossRef CAS.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 1429 CrossRef CAS.
- Z. Jiang, C. Lü and H. Wu, Ind. Eng. Chem. Res., 2005, 44, 4165 CrossRef CAS.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 9417 CrossRef CAS.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367 CrossRef CAS.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef CAS.
- R. E. Holmlin, P. J. Dandliker and J. K. Barton, Angew. Chem., Int. Ed., 1997, 36, 2714 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467 CrossRef CAS.
- J. A. Singh, S. H. Overbury, N. J. Dudney, M. Li and G. M. Veith, ACS Catal., 2012, 2, 1138 CrossRef CAS.
- M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19, 167 CrossRef.
- J. Toster, K. S. Iyer, W. Xiang, F. Rosei, L. Spiccia and C. L. Raston, Nanoscale, 2013, 5, 873 RSC.
- K. Vynck, M. Burresi, F. Riboli and D. S. Wiersma, Nat. Mater., 2012, 11, 1017 CAS.
- C. Wang, S. Yu, W. Chen and C. Sun, Scientific Reports, 2013, 3, 1025 Search PubMed.
- J. Liu, G. Liu, M. Li, W. Shen, Z. Liu, J. Wang, J. Zhao, L. Jiang and Y. Song, Energy Environ. Sci., 2010, 3, 1503 CAS.
- A. Gogoi, A. K. Buragohain, A. Choudhury and G. A. Ahmed, J. Quant. Spectrosc. Radiat. Transfer, 2009, 110, 1566 CrossRef CAS.
- H. Zhou, Z. Zhang, P. Yu, L. Su, T. Ohsaka and L. Mao, Langmuir, 2010, 26, 6028 CrossRef CAS.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS.
- R. Baron, O. Lioubashevski, E. Katz, T. Niazov and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 1572 CrossRef CAS.
- R. K. Banerjee, J. Biol. Chem., 1989, 264, 9188 CAS.
- S. E. Godtfredsen, M. Ottesen and N. R. Andersen, Carlsberg Res. Commun., 1979, 44, 65 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee40696b |
| This journal is © The Royal Society of Chemistry 2013 |