 Open Access Article
Open Access ArticleSources of activity loss in the fuel cell enzyme bilirubin oxidase†
Kulveer
Singh
ab,
Trevor
McArdle
b,
Patricia R.
Sullivan
a and
Christopher F.
Blanford
*b
aInorganic Chemistry Laboratory, Department of Chemistry, South Parks Road, Oxford, OX1 3QR, UK
bSchool of Materials and Manchester Institute of Biotechnology, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: christopher.blanford@manchester.ac.uk
First published on 20th June 2013
Abstract
Electrochemical quartz crystal microbalance measurements with energy dissipation monitoring (E-QCM-D) show that the catalytic lifetime of the O2-reducing multicopper oxidase bilirubin oxidase from Myrothecium verrucaria, adsorbed on or covalently attached to a organothiol-modified gold electrode, is shortened from days to hours when the applied potential is cycled to below a critical potential (0.6 V vs. SHE at pH 6.0)
Broader contextThe multicopper oxidases are a family of copper-containing enzymes that includes laccases and bilirubin oxidases. They have been studied as efficient replacements for platinum and its compounds to catalyse the four-electron reduction of O2 to H2O in low-temperature, enzymatically catalysed fuel cells. Enzymes' specificity, selectivity against inhibition, and ability to function at near-neutral pH values mean they can generate power in living plants or animals. One of the barriers to the practical use of redox enzymes in fuel cells—as well as biosensors and industrially useful enzymatic transformations—is the proteins' slow loss in activity with time. We simultaneously measured electrocatalytic activity, adsorbed mass of enzyme, and the stiffness of this protein adlayer and concluded that structural rearrangements and water loss are the primary culprits in activity loss in these cathodes. The effect was most pronounced when we simulated a changing power load by cycling the potential. Covalent attachment did not mitigate activity loss. Enzyme-coated cathodes that receive their electrons directly from an electrode, rather than via redox mediators, will need additional stabilisation before they will be durable enough for miniature fuel cells from which a sustained power output is required. |
Immobilised oxidoreductases show a loss in electrocatalytic activity with time, colloquially termed ‘film loss’, which limits their applicability to technological applications such as heterogeneous biocatalysis, biosensor development and fuel cells.1–4 We used the multicopper oxidase (MCO) bilirubin oxidase from Myrothecium verrucaria (MvBOx) as a model protein to dissect the mechanisms of film loss, using an electrochemical quartz crystal microbalance with dissipation measurement (E-QCM-D). This technique allows us to simultaneously measure the hydrated mass of the enzyme adsorbed on the sensor surface, its mechanical coupling to the sensor and its catalytic competence.
MCOs have been advanced as catalysts for the four-electron reduction of O2 to water, a key reaction in low-temperature fuel cells.5 Bilirubin oxidases carry out this reaction at near-neutral pH values, at a lower overpotential than Pt,6,7 and can produce currents limited only by transport of O2 into solution.6
Many methods exist for preparing oxidoreductases for heterogeneous electrocatalysis, including uses in enzyme-catalysed fuel cells.2 A common and relatively straightforward method is via direct adsorption on graphite electrodes.8 The roughness and heterogeneous surface chemistry of abraded graphite supports many enzymes for analytical protein film electrochemistry,9 but complicates any analysis of activity loss mechanisms. Immobilising enzymes in redox-active polymers,6 within porous electrodes10 or on high surface-area dispersions11,12 creates a reservoir of surplus enzyme that can mask underlying activity loss. In this work, the electrode is the surface of the QCM sensor, a flat, polished gold surface modified with the short-chain thiol 3-mercaptopropionic acid (3MPA) to produce a chemically homogeneous surface for stable adsorption of MvBOx and efficient direct electron transfer to it.13,14
An E-QCM has been used previously to follow the adsorption and electrocatalysis of MvBOx on bare gold sensors,15 but their work was carried out in static conditions with dilute enzyme solutions, did not include dissipation analysis, and presented adlayer masses equivalent to ∼10 layers of enzyme. Moreover, we have found the simultaneous energy dissipation analysis, which they could not do, is crucial to understanding the mechanisms of activity degradation, particularly when the applied potential is not constant. Also, Olejnik et al. used an E-QCM to study the activity and lifetime of covalently bound laccase, a related MCO, but they measured activity only under constant applied potential.16
Fig. 1 shows a typical adsorption of MvBOx on a 3MPA-modified gold-coated sensor. Buffer saturated with O2 was passed continually through the measurement cell except at ca. 15 min when the enzyme was injected. For thin, rigid and evenly distributed layers, the mass adsorbed on a QCM sensor is proportional to the change in the resonant frequency of the crystal's shear vibration modes;17 in this work the proportionality constant equals −17.9 ng cm−2 Hz−1 for the fundamental frequency and −2.55 for the seventh harmonic, which was used for its sensitivity to changes close to the surface and insensitivity to variations in sensor mounting (see ESI†). The 300 Hz drop in the resonant frequency of the seventh harmonic (Fig. 1A) is consistent with the formation of a hydrated enzyme monolayer of protein on the sensor surface (see ESI†). At the same time, the energy dissipation of the crystal increased to a maximum of about 3 ppm (Fig. 1B).
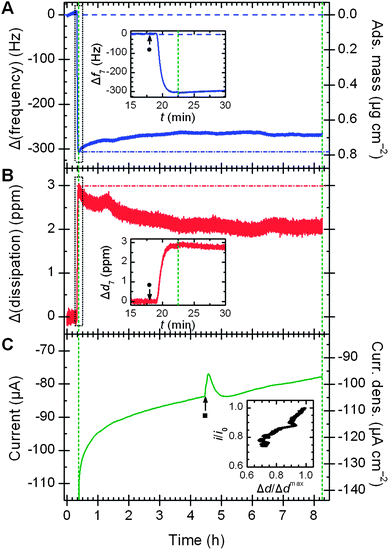 | ||
| Fig. 1 E-QCM-D response (7th harmonic) with time for bilirubin oxidase adsorbed non-covalently on a 3-mercaptopropionic acid layer: (A) frequency change, (B) energy dissipation change, and (C) O2 reduction current. Insets in (A) and (B) highlight sensor response after adding 25 μl 24 mg ml−1 enzyme (●). Green dashed lines show when a potential of 0.21 V vs. SHE was applied. Inset in (C) shows the correlation between dissipation value and electrocatalytic activity. Conditions: 0.1 M sodium phosphate pH 6.0 (pre-saturated with O2), 25 °C, 0.1 ml min−1, projected electrode area 0.80 cm2. Flow rate of O2 lowered for 10 min at ■. | ||
Dissipation is the sum of all energy losses in the system per oscillation cycle.18 Adsorption of proteins contributes towards this dissipated energy and this change can give insight into conformation changes during protein monolayer formation.19 Softer films experience more inelastic collisions than more rigid films. The formation of softer films, therefore, produces higher Δd values.
The surface of the QCM sensor also serves as a working electrode to quantify the catalytic activity of the protein film and any resistance to electron transfer from the sensor to the proteins' metal centres. The current is proportional to the areal density of the enzyme on the surface and its turnover frequency (see ESI†), thus providing a sensitive method to monitor catalytic activity at the same time as changes in mass and stiffness of the adlayer.
To probe how enzyme activity changes with time under conditions approximating constant load in a fuel cell, a potential of 0 V vs. Ag|AgCl|3 M NaCl was applied 7.5 min after the enzyme was injected and held constant for ca. 8 h. The applied potential is low enough to drive enzymatically catalyzed O2 reduction, but too high for O2 to be reduced on bare gold. An initial steady-state current of −110 μA (Fig. 1C) was observed in comparison to near-zero reduction current in the presence of buffer saturated with argon (see ESI†). The current magnitude was up to 30 μA larger for the first 1.0–1.5 min while dissolved O2 near the sensor surface was depleted. The initial turnover frequency of the enzyme (kcat) under constant potential conditions is about 35 s−1 (see ESI†), about 7× lower than values presented for MvBOx in the absence of mass-transport limitations,7 but more than 10× higher than previous work using an EQCM setup.15 Over the course of the measurement, the current decayed to about 30% of its initial value.
Although the initial adsorbed mass of enzyme correlates with the initial magnitude of the catalytic current, subsequent changes in adsorbed mass with time do not parallel changes in catalytic activity. The trace in Fig. 1A is typical: the mass dropped by about an eighth over the first 2.5 h, then remained stable for the next 5 h at (0.68 ± 0.01) μg cm−2. In contrast, changes in energy dissipation with time show an almost linear correlation with catalytic activity (Fig. 1C inset). Like the current magnitude, the dissipation also drops by about 30% over time.‡ The changes of MvBOx on bare gold sensors are almost identical (see ESI†).
Thus, the term ‘film loss’ is not strictly accurate here: the slow loss of activity in MvBOx must be due to some non-desorptive process like structural changes or copper loss. The strong correlation between adlayer stiffness (inversely related to Δd) and activity implicates structural changes as the primary source of activity loss. The effect of applied potential on protein structure also appears in the plot of Δd7vs. Δf7 (see ESI†), which normalises dissipation to adsorbed mass to quantify viscoelastic losses.19,20 The magnitude of ∂d7/∂f7 is more than two times higher when a potential is applied: the protein layer stiffens substantially under applied potential.
To simulate the effect of a variable load on an enzymatic biofuel cell, complementary E-QCM-D measurements were recorded with the potential cycling (Fig. 2). In contrast to the relatively stable mass when the applied potential is held constant, cycling the potential causes an immediate drop in adsorbed mass (Fig. 2A): the mass typically drops by 20–25% over ca. 15 min. The catalytic current degrades simultaneously, falling by about a third in the same period (Fig. 2B).§ As in the constant potential cases, the correlation between activity and dissipation (Fig. 2A inset) was nearly linear, albeit with a non-zero intercept.
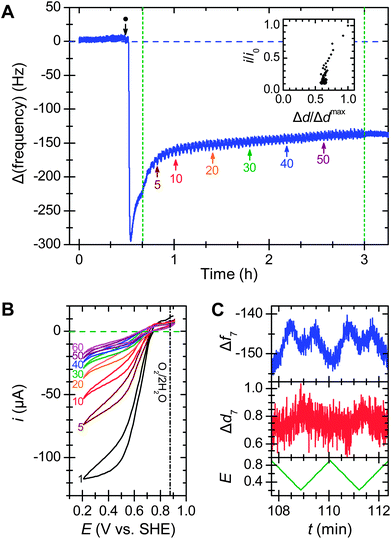 | ||
| Fig. 2 Effect of potential cycling on bilirubin oxidase adsorbed on a 3-mercaptopropionic acid layer. (A) Frequency response from the addition of 24 mg ml−1MvBOx (●) followed by 60 potential cycles between 0.91 and 0.11 V vs. SHE at 10 mV s−1 (green dashed lines); arrows mark the start of numbered cycles. Inset shows the relationship between enzyme activity (current at the lowest potential for that scan) and energy dissipation for each scan, normalized to values from scan 1. (B) Selected scans from the cyclic voltammetry. (C) Frequency (Hz) and dissipation (ppm) responses with applied potential (V vs. SHE) for scans 30 and 31. Conditions: O2-saturated 0.1 M sodium phosphate buffer pH 6.0 at 25 °C flowing at 0.1 ml min−1 with continuous O2 bubbling; 7th harmonic. | ||
If the scan range is kept above 0.6 V vs. SHE, the pattern of mass and activity loss is like that for chronoamperometry. Decreasing the scan rate to 1 mV s−1 led to the same mass loss but only about half the activity loss. Changing the step size had no significant effect on either current loss or mass loss (see ESI†).
After about 10–20 potential cycles, the frequency trace stabilises, but the O2 reduction activity approaches zero. A QCM measures hydrated mass and about 45% of the protein's mass is entrained water (see ESI†) that contributes to frequency changes of the sensor,19–21 suggesting that the enzyme adlayer extensively dehydrates during potential cycling. However, in this case some desorption cannot be ruled out.
When the potential was cycled, both the frequency and dissipation traces showed periodic changes that were slightly out of phase with the applied potential (Fig. 2C). The frequency traces showed multiple peaks and a maximum peak-to-peak amplitude of ca. 9 Hz (0.02 μg cm−2), about 3% of an enzyme monolayer. The varying potential or electric field near the surface may repeatedly distort the adsorbed protein, causing it to repeatedly expel mass in the form of water or ions, and hastening its denaturation. This hypothesis is consistent with STM observations on single molecules of another MCO, laccase from Streptomyces coelicolor, that showed different heights for reduced and oxidised enzyme on a thiol-modified gold electrode.14
To control for the possibility that diminishing activity is due to enzyme desorption, MvBOx was covalently linked to a thiol-modified gold crystal via an amide linkage between an activated carboxylate on the surface and one of the lysine side chains on the protein's exterior (Fig. 3), identical to EDC–NHS-type coupling reactions. After uncoupled enzyme was washed away, the potential was cycled. Unlike the adsorbed samples, the mass dropped by only 6% in the first 7 cycles then stabilised between 0.68 and 0.70 μg cm−2. Despite this, the catalytic activity still dropped off rapidly (Fig. 3A inset), diminishing by 80% in less than 1 h. The activity versus dissipation trace (Fig. 3B inset) displayed the same characteristic correlation, again suggesting that structural changes are responsible for activity loss, even if they are not accompanied by a large mass loss.
 | ||
| Fig. 3 Effect of potential cycling on (A) frequency response and (B) dissipation response for bilirubin oxidase covalently bound via an amide linkage to a thiol-modified QCM sensor. After addition of 25 μL of 24 mg ml−1MvBOx (●), buffer flow stopped (▽) to allow adsorption and reaction with surface layer (reaction adapted from the literature)22 then resumed (▼) for ca. 10 min before voltammetry. Potential cycled 26 times between 0.91 and 0.31 V vs. SHE (green dashed lines). Arrows indicate start of selected cycles and correspond to numbering in top inset. Lower inset relates activity and dissipation as in Fig. 2. Other conditions as in Fig. 2. | ||
Covalent attachment did not stabilise the catalytic activity. It also decreased the efficiency of electron transfer from the electrode to the enzyme. The position of Ecat, a characteristic potential defined by the maximum of the change in current with potential on a swept-potential voltammogram, shifts by −0.1 V, meaning that the covalently attached enzyme needs a greater driving force (∼10 kJ mol−1) to drive electrocatalysis compared to the adsorbed sample. The increased charge-transfer resistance is also evident in the shape of the voltammograms in the inset of Fig. 3A: a more linear, ohmic response dominates the sigmoid shape characteristic of electrochemical–chemical (EC) reactions (compare to Fig. 2B).
Reactive dye labelling measurements identified three reactive lysine side chains on MvBOx through which the enzyme could couple to the surface (see ESI†). All of these groups position both of the enzyme's electron acceptors more than 2 nm from the electrode surface, too far for interfacial electron transfer rate compatible with biochemical processes.23
Controls using catalytically inactive bovine serum albumin (BSA) on 3MPA-layers showed stable frequency and dissipation traces when the potential was cycled.
The structures of the high-potential MCOs used for fuel cell cathodes are similar,24 so the patterns of activity loss are likely to be the similar for all three-domain MCOs. For example, laccase from Trametes versicolor (TvL) showed similar patterns of mass and activity loss, though the magnitudes differed (ESI†). The observed behaviour depends on protein structure and electrode composition, however. For example, covalent attachment by amide linkage stabilised a [NiFe] hydrogenase over 2 h in response to potential steps up to 250 mV,25 and covalently bound [FeFe] hydrogenases showed activity decay time constants ranging from hours to days depending on immobilisation method.1 in activity that was attributed to fast desorption and slower denaturation processes.
Also, an enzyme can be stabilised by its immediate environment: surface modifications and buffered polyelectrolytes let some fuel cell enzymes retain some activity for months.3,26 The current goal, therefore, is to relate extended catalytic stability to enzyme structure and the enzyme's interface with the electrode and other surroundings.
Conclusions
We applied E-QCM-D as a real-time probe of the potential-dependent stability of enzymes used as catalysts in fuel cell cathodes. The catalytic activity of MvBOx drops rapidly when the applied potential is cycled to simulate variable loads on a fuel cell cathode. Although this activity loss was accompanied by some loss of mass from the adlayer, covalent attachment did not improve catalytic stability and also resulted in lower power output because of longer distances for interfacial electron transfer. Catalytic activity decreased even when the adlayer's mass remained stable, such as when the applied potential was constant. Decreasing catalytic activity always correlated with increasing layer stiffness, implicating structural rearrangements over desorption as the primary mechanism of activity loss. These observations suggest that without additional treatment or overpotential limits, MCO-coated cathodes may be unsuitable for long-term power generation in enzymatically catalysed fuel cells.Acknowledgements
The authors thank Hannah Roberts and Ivana Sardzikova for assistance with protein digestion and mass spectrometry, Tom McNamara for protein purification, Robert A.W. Dryfe for valuable discussions, and the mechanical and electronics workshop at the University of Oxford's Department of Chemistry for help with design and construction of the E-QCM-D insert and ozone generator. The authors gratefully acknowledge funding from the UK's Engineering & Physical Sciences Research Council (EP/G00434X/2).Notes and references
- C. Baffert, K. Sybirna, P. Ezanno, T. Lautier, V. Hajj, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Anal. Chem., 2012, 84, 7999–8005 CrossRef CAS.
- X.-Y. Yang, G. Tian, N. Jiang and B.-L. Su, Energy Environ. Sci., 2012, 5, 5540–5563 CAS.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2009, 140, 319–335 RSC.
- H. C. Angove, J. A. Cole, D. J. Richardson and J. N. Butt, J. Biol. Chem., 2002, 277, 23374–23381 CrossRef CAS.
- M. T. Meredith and S. D. Minteer, Annu. Rev. Anal. Chem., 2012, 5, 157–179 CrossRef CAS.
- N. Mano, J. L. Fernandez, Y. Kim, W. Shin, A. J. Bard and A. Heller, J. Am. Chem. Soc., 2003, 125, 15290–15291 CrossRef CAS.
- L. dos Santos, V. Climent, C. F. Blanford and F. A. Armstrong, Phys. Chem. Chem. Phys., 2010, 12, 13962–13974 RSC.
- F. A. Armstrong, H. A. O. Hill, B. N. Oliver and N. J. Walton, J. Am. Chem. Soc., 1984, 106, 921–923 CrossRef CAS.
- C. F. Blanford and F. A. Armstrong, J. Solid State Electrochem., 2006, 10, 826–832 CrossRef CAS.
- U. Salaj-Kosla, S. Pöller, W. Schuhmann, S. Shleev and E. Magner, Bioelectrochemistry, 2013, 91, 15–20 CrossRef CAS.
- K. Murata, K. Kajiya, N. Nakamura and H. Ohno, Energy Environ. Sci., 2009, 2, 1280–1285 CAS.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- M. Tominaga, M. Ohtani and I. Taniguchi, Phys. Chem. Chem. Phys., 2008, 10, 6928–6934 RSC.
- V. Climent, J. Zhang, E. P. Friis, L. H. Østergaard and J. Ulstrup, J. Phys. Chem. C, 2012, 116, 1232–1243 CAS.
- Y. Kamitaka, S. Tsujimura, T. Ikeda and K. Kano, Electrochemistry, 2006, 74, 642–644 CrossRef CAS.
- P. Olejnik, B. Palys, A. Kowalczyk and A. M. Nowicka, J. Phys. Chem. C, 2012, 116, 25911–25918 CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- M. Rodahl, F. Höök, C. Fredriksson, C. A. Keller, A. Krozer, P. Brzezinski, M. Voinova and B. Kasemo, Faraday Discuss., 1997, 107, 229–246 RSC.
- F. Höök, M. Rodahl, B. Kasemo and P. Brzezinski, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12271–12276 CrossRef.
- U. B. Jensen, E. E. Ferapontova and D. S. Sutherland, Langmuir, 2012, 28, 11106–11114 CrossRef CAS.
- B. Ge, T. Meyer, M. J. Schöning, U. Wollenberger and F. Lisdat, Electrochem. Commun., 2000, 2, 557–561 CrossRef CAS.
- C. C. Page, C. C. Moser, X. X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS.
- C. J. Rodgers, C. F. Blanford, S. R. Giddens, P. Skamnioti, F. A. Armstrong and S. J. Gurr, Trends Biotechnol., 2010, 28, 63–72 CrossRef CAS.
- A. A. Hamdan, B. Burlat, O. Gutiérrez-Sanz, P.-P. Liebgott, C. Baffert, A. L. De Lacey, M. Rousset, B. Guigliarelli, C. Léger and S. Dementin, Nat. Chem. Biol., 2013, 9, 15–17 CrossRef CAS.
- C. M. Moore, N. L. Akers, A. D. Hill, Z. C. Johnson and S. D. Minteer, Biomacromolecules, 2004, 5, 1241–1247 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods; calculations; images of bare sensor surface; controls with bare gold, azide-inhibited MvBOx, BSA and TvL; variations in scan parameters and oxygenation; plots of Δd7vs. Δf7. See DOI: 10.1039/c3ee00043e |
| ‡ The dissipation trace for enzyme loadings greater than about 0.6 mg (10 nmol) initially spikes but returns to a stable baseline in about 10 min, indicative of loosely bound protein (ESI†). The value of Δdmax is taken from the post-spike baseline. |
| § Reduction current magnitudes above 110 μA produced CV scans with a flattened shape indicative of mass-transport polarisation, so the current at 0.4 V vs. SHE on the reductive sweep was used for activity loss calculations. |
| This journal is © The Royal Society of Chemistry 2013 |