Organic dye-sensitized solar cells with a cobalt redox couple: influences of π-linker rigidification and dye–bath solvent selection†
Ning
Cai
,
Renzhi
Li
,
Yinglin
Wang
,
Min
Zhang
and
Peng
Wang
*
State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: peng.wang@ciac.jl.cn; Fax: +86 431 852 629 53; Tel: +86 431 852 629 52
First published on 1st November 2012
Abstract
The rigidification of π-conjugated linkers represents a viable strategy towards the energy-level engineering of organic push–pull photosensitizers in dye-sensitized solar cells. In this paper we deploy 4-hexylphenyl substituted cyclopenta[1,2-b:5,4-b′]dithiophene[2′,1′:4,5]thieno[2,3-d]thiophene as the π-linker of a D-π-A dye, which displays an improved molar absorption coefficient and a red-shifted absorption peak in contrast to its model dye with the 2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene segment. The energy-gap reduction is concomitant with negative and positive shifts of ground-state and excited-state redox potentials, which however do not exert an adverse impact on the net charge separation yield at the titania/dye/electrolyte interface, probably due to the formation of a favored microstructured dye assembly. Furthermore, the dye load amount can be tuned by changing the bath solvent and has a significant influence on some key photovoltaic features such as the photocurrent and photovoltage, the latter of which is dissected via the joint charge extraction and photovoltage decay experiments. The charge recombination lifetime could be roughly rationalized by analyzing the damping of signals on emitted electrons from titania in the X-ray photoelectron spectroscopy measurements.
Broader contextThe development of exotic conjugated organic materials plays an important role in enhancing the performance of new generation optoelectronic devices and the stepwise efficiency improvement of dye-sensitized solar cells has been mainly supported by the design of powerful dye molecules. We herein report a bulky organic push–pull photosensitizer featuring the cyclopenta[1,2-b:5,4-b′]dithiophene[2′,1′:4,5]thieno[2,3-d]thiophene π-linker, which displays an enhanced molar absorption coefficient and a red-shifted absorption peak with respect to its reference dye with the 2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene linker. It is found that the net charge separation yield is not impacted by the energy-gap narrowing via the π-linker rigidification, which is likely related to a three-dimensional configuration owing to the tethering of four hexylphenyl segments attached to the conjugated backbone. The effects of bath solvent correlated dye load amounts are also scrutinized in terms of light-harvesting, interfacial energetic and kinetic parameters as well as their joint contribution to photovoltaic performance. |
1 Introduction
The dye-sensitized solar cell (DSC) as a prospective photovoltaic technology to accomplish the transformation of solar light to electricity at an affordable price has attracted a great deal of research passion on both new material synthesis and insightful physical analysis.1–6 As one of the most critical functional components in DSCs, the photosensitizer is of unfailing research focus and substantial efforts have been devoted to the exploration of metal-free organic dyes in the past decade, primarily owing to the abundance of raw materials and flexibility of molecular design.7–16 Aside from the electron donor and acceptor segments in a push–pull dye, the conjugated π-linker is of paramount importance in tuning cell performance. In this respect, a large number of structurally versatile thiophene-based organic dyes have been reported17–43 since the initial incorporation of vinylthiophene and bithiophene moieties in the coumarin DSC photosensitizers by Arakawa and his coworkers.44In the previous work we introduced the cyclopentadithiophene (CPDT) segment in place of 2,2′-dithiophene (DT) in a push–pull dye45 and the related dye modifications46–48 have achieved several new benchmarks of iodine-free DSCs.48–51 In this contribution, we extended our studies on this line by employing cyclopenta[1,2-b:5,4-b′]dithiophene[2′,1′:4,5]thieno[2,3-d]thiophene (CPDTTT)52,53 as the conjugated linker, in combination with a hydrophobic triphenylamine electron-donor and a hydrophilic cyanoacrylic acid electron-acceptor, to construct the C243 dye as presented in Fig. 1A. To further assess the merit of π-linker rigidification in terms of light absorption and electrochemical properties, we also prepared its counterpart C242 featuring the 2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene (DTTT) bridge as a reference.
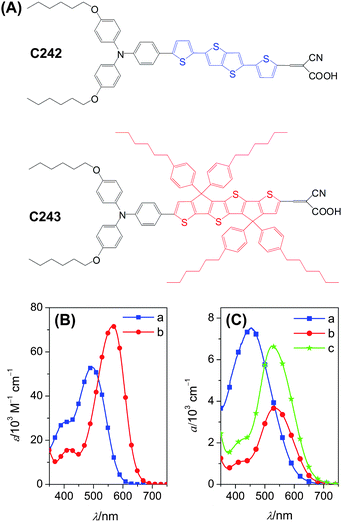 | ||
Fig. 1 (A) Dye structures. (B) Electronic absorption spectra of (a) C242 and (b) C243 in THF. (C) Absorption spectra of titania films coated with (a) C242 from THF, (b) C243 from THF and (c) C243 from the binary solvent of THF and MeCN (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The absorption of a titania film has been subtracted for clarity of presentation. :1). The absorption of a titania film has been subtracted for clarity of presentation. | ||
On account of the flexible aliphatic chains tethered on the backbone, C243 has a high solubility in the dye-bath solvent tetrahydrofuran (THF), leading to a low grafting density of dye molecules on titania, which can be improved by using a mixed solvent of THF and acetonitrile (MeCN). The dye assemblies on the surface of titania, tuned by either dye structure or dye-bath solvent, can impact some crucial photovoltaic parameters such as photocurrent and photovoltage by altering the interfacial energetics and charge transfer kinetics, which will be systematically analyzed by joint photophysical and electrical measurements.
2 Experimental section
2.1 Materials
N,N-Dimethylformamide (DMF), phosphoryl trichloride, 1,2-dichloroethane, MeCN and THF were distilled before use. 2-Thiophene-boronic acid, palladium acetate, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos), N-bromosuccinimide (NBS), cyanoacetic acid, lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI), 4-tert-butylpyridine (TBP) and n-butyllithium (1.6 M in hexane) were purchased from Sigma-Aldrich. 2,5-Dibromothieno[3,2-b]thiophene54 and 4,4,5,5-tetramethyl-2-{4-[N,N-bis(4-hexyloxyphenyl)amino]phenyl}-1,3,2-dioxaborolane45 were synthesized according to the corresponding literature methods and the synthetic details of C242 and C243 are described in the ESI.†2.2 Electronic absorption and photoluminescence (PL) measurements
Static electronic absorption and PL spectra were recorded on an Agilent G1103A spectrometer and a PerkinElmer LS55 luminescence spectrometer, respectively. The emitted light was detected with a Hamamatsu R928 red-sensitive photomultiplier. Transient absorption experiments were carried out on a LP920 laser flash spectrometer equipped with a nanosecond tunable OPOLett 355II laser. Transient absorption spectra were recorded with an Andor iStar ICCD camera, and the absorption decay traces were measured with a fast photomultiplier tube and a TDS 3012C digital oscillograph. The PL decays were measured with a LifeSpec-II fluorescence spectrometer in conjunction with an EPL488 laser diode.2.3 Square-wave voltammetric measurements
The ground-state redox potentials of dyes dissolved in THF were measured on a CHI660C electrochemical workstation in conjunction with a three-electrode electrochemical cell composed of a platinum gauze counter electrode, silver-wire auxiliary electrode and a platinum ultramicroelectrode. All potentials were reported against the ferrocene/ferrocenium (Fc/Fc+) reference.2.4 Cell fabrication
A 2.4 μm thick, transparent layer of 22 nm sized TiO2 particles was first screen-printed onto FTO glass and further coated with a 3.5 μm thick second layer of scattering titania particles to produce a bilayer titania film, which was used later as the negative electrode of a DSC. The preparation procedures of TiO2 nanocrystals and paste for screen-printing were reported in a previous paper.55 The film thickness was monitored with a bench-top Ambios XP-1 stylus profilometer. After sintering at 500 °C and cooling to 80 °C, a circular titania electrode (∼0.28 cm2) was stained by immersing it overnight into a solution of 100 μM dye dissolved in THF or a binary solvent of THF and MeCN (volume ratio 1:1). The dye-coated titania electrode was then rinsed with MeCN and dried by air flow, and was further assembled with a thermally platinized FTO positive electrode by a 25 μm thick Surlyn (DuPont) hot-melt gasket and sealed up by heating. The internal space was perfused with an electrolyte with the aid of a vacuum back-filling system. Our cobalt electrolyte is composed of 0.25 M tris(2,2′-bipyridine)cobalt(II) di[bis(trifluoromethanesulfonyl)imide], 0.05 M tris(2,2′-bipyridine)cobalt(III) tris[bis(trifluoromethanesulfonyl)imide], 0.5 M TBP and 0.1 M LiTFSI in MeCN.
2.5 Transient photovoltage decay and charge extraction measurements
Transient photoelectrical experiments56 were performed with an Autolab-PGSTAT302N electrochemical workstation. The steady and perturbative lights on the photoanode side of a testing cell were supplied with white and red light-emitting diodes, respectively. We used the red light to generate a photovoltage perturbation near the open-circuit photovoltage of a testing cell under a certain white light and measured the voltage decay process thereafter. The modulated photovoltage by the red pulse of a testing cell was below 5 mV. Normally, the transient signal follows a mono-exponential decay, thus the electron lifetime can be obtained by fitting an exponential function to the photovoltage decay. The electron density was estimated by the charge extraction method. A testing cell was first kept at open circuit under white light and subsequently the white light was turned off upon switching the cell from open circuit to short circuit to record the resulting current transient, and the electron density was obtained by current integration.2.6 X-Ray photoelectron spectroscopy (XPS)
XPS spectra were recorded with an ESCALAB 250 spectrometer equipped with a hemispherical electron energy analyzer using the Al Kα radiation (hν = 1486.6 eV) and an energy step of 0.1 eV. The electron take-off angle was 90°. Photoelectron spectra were measured in the constant analyzer energy (CAE) mode. The spectra are energy calibrated by setting the Ti2p3/2 signal to 458.6 eV.57 The small binding energy shift owing to the dye adsorption can be due to a charge redistribution, a variation of the interface dipole and/or a change in the Fermi level position in the titania band gap.58,59 The charging and radiation effects during the measurements are negligible for all our spectra.3 Results and discussion
As illustrated in Schemes S1 and S2,† the synthesis of C242 and C243 in general followed our previously reported strategy45 of a Suzuki cross-coupling of pinacol bisarylaminoaryl boronates and bromine substituted aromatic aldehydes, and the widely employed Knoevenagel condensation to construct cyanoacrylic acid DSC dyes.60,61 The symmetric intermediates of DTTT 2 and carboxylated DTTT 10 were prepared in an excellent or good yield with the Suzuki–Miyaura reaction, by virtue of a very active phosphine ligand, SPhos, developed by Buchwald and his coworkers.62,63 Hexylphenyl substituted CPDTTT 12 was prepared with alcohol 11 as the starting compound via intramolecular Friedel–Crafts cyclization in the presence of a solid acid catalyst Amberlyst 15. The selective monoformylation of 2 and 12 were carried out by Vilsmeier–Haack reaction to produce aldehydes 3 and 13, which were further brominated with NBS to generate the key intermediates 4 and 14 for further dye preparations.The light-harvesting capacities at the molecular level were first compared by recording the UV-visible absorption spectra (Fig. 1B) of C242 and C243 in THF and the detailed parameters were compiled in Table 1. The C242 reference dye with the DTTT π-linker has a maximum absorption wavelength (λabsmax) of 495 nm, which is red-shifted to 565 nm upon employing the rigidified CPDTTT counterpart for C243. Moreover, the molar absorption coefficient at the maximum absorption wavelength (εabsmax) is augmented from 53.1 to 71.7 × 103 M−1 cm−1 during the π-linker rigidification. The bathochromic and hyperchromic effects of fusing two adjacent thiophene units with the cyclopentadienyl group in organic push–pull dyes are generally consistent with our previous comparisons on the other two dyes with the π-linkers of DT and CPDT,45 probably on account of the planarity of the conjugated segment, the electron-donating property of the bis(4-hexylphenyl)methyl group and the differentiated relative configuration of thiophene rings.
| Dye | λ absmax /nm | ε absmax /103 M−1 cm−1 | λ plmax /nm | E 0–0 /eV | E D/D+ /V | E D*/D+ /V |
|---|---|---|---|---|---|---|
| a The maximum absorption wavelength (λabsmax) and maximum molar absorption coefficient (εabsmax), and PL maximum wavelength (λplmax) were derived from the static electronic absorption and emission spectra in THF solutions. b The zero–zero transition energy (E0–0) was estimated from the intersection points of normalized absorption and emission spectra. c The ground-state redox potential (ED/D+) was reported with Fc/Fc+ as reference. d The excited-state redox potential (ED*/D+) was estimated by equation ED*/D+ = ED/D+ − E0–0/e without considering any entropy change during the light excitation. | ||||||
| C242 | 495 | 53.1 | 627 | 2.18 | 0.21 | −1.97 |
| C243 | 565 | 71.7 | 691 | 1.98 | 0.15 | −1.83 |
Owing to the poor solubility of C242 in the routine DSC dye-bath solvents, THF was our preliminary choice for the adsorption of dye molecules on a mesoporous titania film. In accord with the scenario of dye solution absorptions, the C243 coated titania film exhibits a red-shifted absorption peak compared to C242, as shown in Fig. 1C. However the titania film coated by C243 from THF has a much lower decadic absorption coefficient (a) than the C242 analogue, although the former dye has a much higher molar absorption coefficient in THF. This can be further rationalized by measuring the amount of dye molecules loaded from THF solutions onto our titania films, being 1.24 and 0.37 × 10−8 mol cm−2 μm−1 for C242 and C243, respectively. At the first glance, one may attribute the lower load amount of C243 to its larger lateral diameter if the coverage of both dyes on the surface of titania is close to unity or the same. To prove the validity of this assumption, we further selected the mixed THF and MeCN (volume ratio 1 :1) as the dye-bath solvent of C243 and have found that there is a remarkably improved absorption coefficient of C243-coated titania film, with a dye load amount of 1.03 × 10−8 mol cm−2 μm−1. The dye amount improvement of C243 upon the dye-bath solvent variation could be related to the augmentation of the adsorption–desorption equilibrium constant, which changes the molecular coverage on the surface of titania. The dye aggregation is considered not to be the reason in view of its three-dimensional structure.
To fabricate an efficient DSC, besides the light-harvesting yield of a dye-coated titania film, it is also of much pertinence that there are favorite energy-offsets of dye molecules with respect to the titania nanocrystals and redox electrolytes. Thereby we measured the ground-state redox potential (ED/D+) of C242 and C243 dissolved in THF, which can be accurately derived by averaging the anodic and cathodic peak potentials of square-wave voltammograms. As shown in Fig. 2, the C243 dye has a 60 mV negatively shifted ED/D+ in comparison with C242, suggesting an electron-richer feature of CPDTTT in contrast to DTTT. The zero–zero transition energies (E0–0) were estimated from the intersection points of normalized absorption and emission spectra (Fig. S1†) of a 200 μM dye solution, being 2.18 eV for C242 and 1.98 eV for C243. Hence we could further calculate the excited state redox potential (ED*/D+) by equation ED*/D+ = ED/D+ − E0–0/e without considering any entropy change during light excitation. The derived ED*/D+ values of C242 and C243 are −1.97 and −1.83 V with the Fc/Fc+ redox couple as reference.
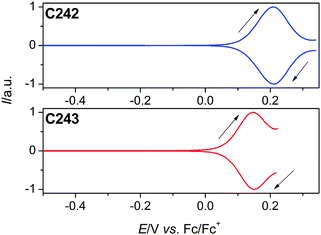 | ||
| Fig. 2 Normalized square-wave voltammograms of C242 and C243 in THF with 0.1 M EMITFSI as the supporting electrolyte. The potential scan direction is marked by a black arrow and all potentials are reported against the Fc/Fc+ redox couple. | ||
It is well documented that a modulation of molecular energy-levels can bring forth a variation in incident monochromatic photon-to-collected electron conversion efficiencies (IPCEs) by impacting the photocarrier generation yield.64–68 Here we first resorted to the time-correlated single photon counting (TCSPC) technique69–72 to roughly evaluate the yield of electron injection from the electronically excited dye molecules to titania nanocrystals in contact with a tris(2,2′-bipyridine)cobalt electrolyte. By use of a mesoporous alumina film coated with C242 from THF or C243 from THF or its mixture with MeCN, a control cell was first assembled and displayed a strong red light emission upon laser excitation at 488 nm (Fig. S2†). Considering the energetic retardance of electron injection at the dye/alumina interface, these PL decays can be ascribed to the radiative and non-radiative deactivations of excited-state dye molecules (D*). However, substitution of alumina with titania induced a remarkable PL quenching, indicative of the electron injection occurrence at the energy-offset dye/titania interface, which will be verified further by transient absorption measurements. Through comparing the integrals of areas beneath PL traces on titania and alumina, the electron injection yields (ϕinj) are calculated to be 92%, 93% and 96% for cells made from titania films coated with C242 from THF, C243 from THF and C243 from the binary solvent of THF and MeCN, respectively. Overall, replacing the conjugated π-linker of DTTT with CPDTTT in push–pull dyes to harvest low-energy photons does not result in a reduction of exciton dissociation yield. The comparable electron injection yields observed for two C243-coated titania films with considerably distinct load amounts have also suggested the merit of the four hexylphenyl substituents tethered on the two sp3 carbons of CPDTTT, restraining the π–π stacking of dye molecules and attenuating intermolecular exciton annihilation.45–51,73,74
We further recorded the transient absorption spectra of 2.1 μm thick, dye-coated titania films immersed in an inert electrolyte consisting of 0.5 M TBP and 0.1 M LiTFSI in MeCN. In sharp contrast to the static electronic spectra (Fig. 1C), strong absorptions (Fig. 3A) in the near-infrared region were probed for both dyes upon excitation with a nanosecond laser pulse. With the help of spectroelectrochemical measurements (Fig. S3†) of dye-coated titania films dipped in EMITFSI, we could assign the positive signals in the transient absorption spectra mainly to electronic transitions of oxidized dyes. An observation light at 765 nm was selected in our kinetic measurements with a photomultiplier tube. The excitation wavelengths were also intentionally chosen considering a ∼0.2 optical density of dye-coated titania films, which enables a similar distribution profile of charge carriers in our testing samples.
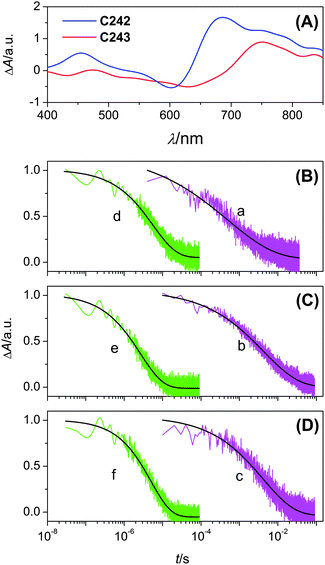 | ||
| Fig. 3 (A) Transient absorption spectra recorded at 200 ns upon nanosecond pulsed, 488 nm laser excitation of dye-coated titania films in contact with an inert electrolyte of 0.1 M LiTFSI and 0.5 M TBP in MeCN. (B, C and D) Kinetic absorption traces probed at 765 nm of 4.5 μm thick, titania films coated with (B) C242 from THF, (C) C243 from THF and (D) C243 from the binary solvent of THF and MeCN, in contact with the inert (a–c) and cobalt (d–f) electrolytes. Laser pulse fluence and excitation wavelength: (a) 20 μJ cm−2 at 639 nm; (b) 19 μJ cm−2 at 679 nm; (c) 19 μJ cm−2 at 688 nm; (d) 20 μJ cm−2 at 642 nm; (e) 19 μJ cm−2 at 683 nm; (f) 19 μJ cm−2 at 692 nm. Smooth lines are stretched exponential fittings over raw data obtained by averaging 400 laser shots. | ||
The slow charge recombination (traces a–c in Fig. 3) between injected electrons in titania and oxidized dye molecules was recorded with cells fabricated with the aforementioned inert electrolyte and 4.5 μm thick, dye-coated titania films. The absorption decays were fitted to a stretched exponential function ΔA ∝ A0exp[−(t/τ)α], where A0 is the pre-exponential factor, α is the stretching parameter and τ is the characteristic time. With the gamma function Γ(x), the average time (〈τ〉bet) of this charge recombination reaction was derived through 〈τ〉 = (τ/α)Γ(1/α).75 It is found that C242 has a 〈τ〉bet of 2 ms, while C243 with a rigidified π-linker features a general deceleration of back electron transfer from titania to oxidized dye molecules, with the 〈τ〉bet values of 9 and 7 ms, which do not depend too much on the significant dye-load amount variation stemming from the dye-bath solvent selection of THF or its mixture with MeCN. Since a reduced charge recombination rate could be related to a longer tunneling distance between titania electrons and triphenylamine holes,76 we may imagine that with respect to C242 there is a smaller tilt angle of the C243 dye molecules anchored on the surface of titania probably due to its three-dimensional steric hindrance, as illustrated in Scheme S3.† The dye load amount augmentation along with the change of bath solvent from THF to its mixture with MeCN could enhance the intermolecular interaction but only change the tilt angle of dye molecules slightly, resulting in a small fluctuation of charge recombination rate.
Furthermore, substituting the inert electrolyte with the electroactive cobalt electrolyte gave rise to evidently faster absorption decays (traces d–f in Fig. 3), indicative of swift reduction of oxidized dyes by cobalt(II) ions.77–79 Employing the same protocol as above, the mean reaction times (〈τ〉reg) of dye regeneration were acquired, being 7, 3 and 5 μs for titania films coated with C242 from THF, C243 from THF and C243 from the binary solvent of THF and MeCN, respectively. However, no positive correlation between ED/D+ and dye regeneration rates is found. According to the Marcus electron transfer theory,80 the dye regeneration reaction is involved in the changes of Gibbs free energy, reorganization energy and electronic coupling factor. Thereby we further calculated the reorganization energies of C242 and C243 with the DFT method, being a comparable value of 86 and 84 meV, respectively. Considering that even the dye-bath solvent change can lead to a variation of dye regeneration rate, it is likely that apart from the molecular-level character, the microstructure of dye assemblies on titania81,82 is worthy of much attention in the further development of DSC dyes as well as device optimization. Overall, in spite of dye and bath solvent dependent dual-channel charge transfer rate variation, the kinetic branch ratios of 〈τ〉bet to 〈τ〉reg exceed 280 for all these three dye assemblies in combination with the tris(2,2′-bipyridine)cobalt redox shuttle, suggesting the efficient retardation of charge recombination between injected electrons in titania and holes remained in oxidized dye molecules at the titania/dye/electrolyte interface.
Shown in Fig. 4A are the photocurrent action spectra of DSCs made from relatively thin (thickness, 2.4 + 3.5 μm) bilayer titania films coated with C242 from THF, C243 from THF and C243 from the binary solvent of THF and MeCN, in combination with a tris(2,2′-bipyridine)cobalt(II/III) electrolyte. The maximum IPCEs for both dyes can reach ∼80%, which are however not unity owing to the presence of a small portion of non-electron injecting dyes (Fig. S2†), except for the light absorption and scattering losses by the conducting glass. The red-shifted onset wavelength of photocurrent response and the lower IPCEs of THF cells with the C243 dye compared to C242 are generally consistent with absorption measurements (Fig. S4†) on 2.4 μm thick, dye-coated nanoporous titania films immersed in the cobalt electrolyte. On the basis of the exciton dissociation and long-range charge separation yields discussed above, it can be concluded that the C243 dye-bath replacement from THF to its mixture with MeCN improves the short-circuit photocurrent by enhancing the light-harvesting capacity of a thin titania film. In addition, the employment of a thicker titania film to increase the light absorption is not a wise strategy to attain a higher IPCE for C243, on account that the charge collection yield is problematic in that case.
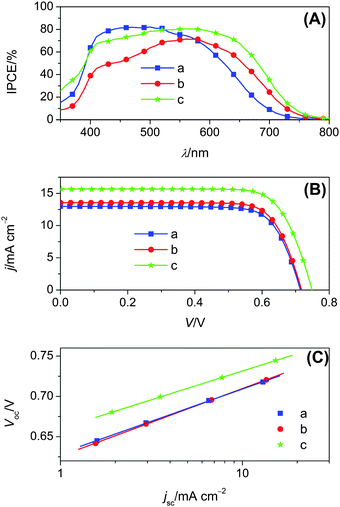 | ||
| Fig. 4 (A) Photocurrent action spectra. (B) j–V characteristics measured at an irradiation of 100 mW cm−2, AM1.5 sunlight. (C) Plots of open-circuit photovoltage as a function of short-circuit photocurrent density. Cells were made from 2.4 + 3.5 μm thick bilayer titania films coated with (a) C242 from THF, (b) C243 from THF and (c) C243 from the binary solvent of THF and MeCN, and were tested using a metal mask with an aperture area of 0.158 cm2. An antireflection film was adhered to the testing cell during measurements. | ||
As presented in Fig. 4B, we also recorded the photocurrent density–voltage (j–V) characteristics at the 100 mW cm−2, simulated AM1.5 conditions and collected the detailed photovoltaic parameters in Tables 2 and S1.† By use of THF as the dye-bath solvent, the C242 and C243 cells display power conversion efficiencies (η) of 7.0 and 7.5%, respectively, with the same open-circuit photovoltage (Voc) of 0.72 V. The small efficiency improvement is mainly related to the short-circuit photocurrent density (jsc) increase from 12.92 to 13.55 mA cm−2, because of a better light-harvesting of solar photons in the red spectral region by C243. Moreover, in agreement with the integral of IPCEs over the standard AM1.5 solar emission spectrum, the enhanced jsc of the C243 cell with the THF and MeCN mixture as dye-bath solvent remarkably contributes to the overall solar-to-electricity efficiency. With a jsc of 15.67 mA cm−2, a Voc of 0.75 V and a fill factor (FF) of 0.756, the C243 dye featuring the CPDTTT π-linker can yield an efficiency of 8.9%. While stability tests under the thermal and light-soaking stress need to be addressed in a future study by employing a low-volatility electrolyte, we observed a very small efficiency drop of our cobalt cells left on the lab bench for over two months.
| Dye | Bath solvent | j sc/mA cm−2 | V oc/V | FF | η/% |
|---|---|---|---|---|---|
| a The validity of our photovoltaic data is confirmed by comparing the calculated jscvia wavelength integration of the product of the standard AM1.5 emission spectrum (ASTM G173-03) and measured IPCE spectra with the experimental jsc, showing a less than 5% error. Also note that all our cells show a linear dependence of photocurrent on light intensity. | |||||
| C242 | THF | 12.92 | 0.72 | 0.756 | 7.0 |
| C243 | THF | 13.55 | 0.72 | 0.764 | 7.5 |
| C243 | THF + MeCN | 15.67 | 0.75 | 0.756 | 8.9 |
Assuming jsc is proportional to the photocarrier generation flux, we further measured j–V curves of these three cells at various light intensities and plotted Voc as a function of jsc in Fig. 4C. At a given jsc, the C243 cell made with the mixture of THF and MeCN as dye-bath solvent exhibits an augmented photovoltage. For a certain redox electrolyte in DSCs, it is well recognized that a rise or fall of Voc stems from a shift of electron quasi-Fermi-level (EF,n) in titania, which could be related to a variance in titania conduction band edge (Ec) and/or a fluctuation of electron density.5,6 At a given photocarrier generation flux, the electron density is controlled by the interfacial recombination of titania electrons with electron accepting species in an electrolyte and/or dye cations. Based on the aforementioned transient absorption experiments, we consider the recombination with tris(2,2′-bipyridine)cobalt(III) ions as the main channel. Thus, we further performed charge extraction and transient photovoltage decay measurements to understand the influences of dye assemblies on Voc. As depicted in Fig. 5A, the C242 and C243 cells made with the THF dye-bath solvent have a dissimilar extracted electron density (dn) at the same Voc or potential bias, showing that with respect to the C242 cell, there is an approximately 30 mV upward shift in the C243 cell of the titania conduction-band edge against the electrolyte Fermi-level. This could be understood as more TBP adsorbed on the surface of titania,83–85 which is not completely covered by the C243 dye in the case of THF as bath-solvent. However, the relatively higher conduction-band edge of the C243 cell does not lead to an improved Voc, owing to a shortened charge recombination lifetime (τn) at a given dn as presented in Fig. 5B. The augmentation of C243 dye coverage by use of the mixed bath solvent of THF and MeCN, exerts a clear influence on the conduction band-edge by a downward movement of about 30 mV (Fig. 5A), which is now comparable to that of the C242 cell, and slows down the interfacial charge recombination rates as illustrated in Fig. 5B. For a comparable dye load amount, it is likely that the three-dimensional bulky character of C243 relative to C242 plays a pivotal role in controlling the recombination rate.
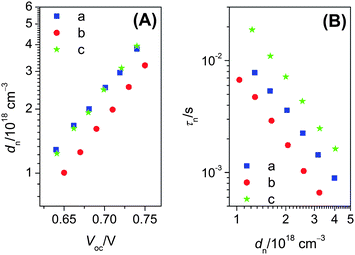 | ||
| Fig. 5 Plots of (A) extracted electron density in titania against open-circuit photovoltage and (B) electron lifetime as a function of extracted electron density for cells made with titania films coated by (a) C242 from THF, (b) C243 from THF and (c) C243 from the binary solvent of THF and MeCN. | ||
The mean thickness (d) of an organic coating on titania nanocrystals, formed by the chemical grafting of dye molecules, was further estimated via comparing the intensity (I) of the Ti2p3/2 signal originating from the substrate in XPS measurements.86,87 As presented in Fig. 6, there is a reduction of both Ti2p3/2 and Ti2p1/2 intensities for all dye-coated titania films in comparison to the bare substrate. This type of signal attenuation is ascribed to the partial blocking effect of dye molecules on the escape of photoelectrons from titania to vacuum. A very evident diminishment is noted for the dye-bath solvent alteration from THF to its mixture with MeCN, which can be easily understood according to the preceding load amounts of C243 molecules. In contrast, a further increment of dye load amount on titania along with the dye variation from C243 to C242 counterintuitively leads to more photoelectrons emitted from titania, verifying the molecular size effect in dye assemblies on the surface of titania. If the dye coating on titania is homogeneous, the Ti2p3/2 intensity from a dye-coated mesoporous titania film can be quantitatively described with a two-layer model88
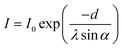
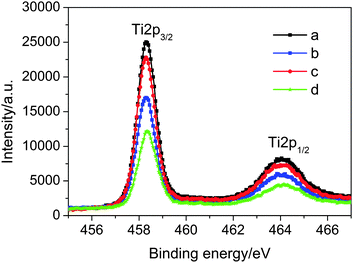 | ||
| Fig. 6 Ti2p3/2 and Ti2p1/2 photoelectron signals of titania films uncoated (a) and coated by (b) C242 from THF, (c) C243 from THF and (d) C243 from the binary solvent of MeCN and THF. | ||
4 Conclusions
To sum up, we have synthesized a new push–pull dye featuring the cyclopenta[1,2-b:5,4-b′]dithiophene[2′,1′:4,5]thieno[2,3-d]thiophene π-linker, which displays a red-shifted absorption peak and larger molar absorption coefficient compared to its model dye with 2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene. Time-resolved photoluminescence and absorption measurements have suggested that the net charge generation yield is not affected by the energy-gap narrowing via the π-linker rigidification, which is likely related to the tethering of four hexylphenyl groups on the conjugated backbone for a three-dimensional dye configuration. We further studied the bath solvent dependent dye-load amount and systematically analyzed its influence on light absorption, multi-channel charge transfer kinetics and conduction-band edge movement as well as their joint contribution to photovoltaic performance. The observed charge recombination lifetimes for three dye assemblies are roughly rationalized in accordance with the mean thicknesses of dye coatings through the photoelectron spectroscopy measurements on the damping of Ti2p signals. Our work has highlighted the future necessities of controlling the microstructure of dye assemblies on the surface of titania. Further investigations on this line are underway to take the full power of CPDTTT π-linker in metal-free photosensitizers.Acknowledgements
The National Science Foundation of China (no. 21203175, no. 91233206, no. 51125015, no. 50973105 and no. 51103146), the National 973 Program (no. 2011CBA00702), the National 863 Program (no. 2011AA050521) and the Key Scientific and Technological Program of Jilin Province (no. 10ZDGG012) are acknowledged for financial support. We are grateful to Dyesol for supplying the WER4-O scattering paste and to DuPont Packaging and Industrial Polymers for supplying the Surlyn and Bynel films.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115 RSC.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381 CrossRef CAS.
- B. O'Regan and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1799 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Séro and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083 RSC.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809 CrossRef CAS.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602 CrossRef CAS.
- J.-H. Yum, E. Baranoff, S. Wenger, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2011, 4, 842 CAS.
- J. N. Clifford, E. Martínez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635 RSC.
- M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145 RSC.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613 CrossRef CAS.
- Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC.
- K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476 CrossRef CAS.
- M. Velusamy, K. R. T. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Org. Lett., 2005, 7, 1899 CrossRef CAS.
- K. R. J. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Chem. Commun., 2005, 4098 RSC.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245 RSC.
- S.-L. Li, K.-J. Jiang, K.-F. Shao and L.-M. Yang, Chem. Commun., 2006, 2792 RSC.
- N. Koumura, Z. S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. De Anglis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS.
- Z.-S. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138 CrossRef CAS.
- K. R. J. Thomas, Y.-C. Lin, K.-M. Lee, K.-C. Ho, C.-H. Lai, Y.-M. Chen and P.-T. Chou, Chem. Mater., 2008, 20, 1830 CrossRef CAS.
- H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 9202 CrossRef CAS.
- H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 327 CrossRef CAS.
- G. Zhang, H. Bala, Y. Cheng, D. Shi, X. Lv, Q. Yu and P. Wang, Chem. Commun., 2009, 2198 RSC.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915 CrossRef CAS.
- S. Ko, H. Choi, M.-S. Kang, H. Hwang, H. Ji, J. Kim, J. Ko and Y. Kang, J. Mater. Chem., 2010, 20, 2391 RSC.
- L.-Y. Lin, C.-H. Tsai, K.-T. Wong, T.-W. Huang, L. Hsieh, S.-H. Liu, H.-W. Lin, C.-C. Wu, S.-H. Chou, S.-H. Chen and A.-I. Tsai, J. Org. Chem., 2010, 75, 4778 CrossRef CAS.
- H. Choi, I. Raabe, D. Kim, F. Teocoli, C. Kim, K. Song, J.-H. Yum, J. Ko, M. K. Nazeeruddin and M. Grätzel, Chem.–Eur. J., 2010, 16, 1193 CrossRef CAS.
- D.-Y. Chen, Y.-Y. Hsu, H.-C. Hsu, B.-S. Chen, Y.-T. Lee, H. Fu, M.-W. Chung, S.-H. Liu, H.-C. Chen, Y. Chi and P.-T. Chou, Chem. Commun., 2010, 46, 5256 RSC.
- J. Liu, R. Li, X. Si, D. Zhou, Y. Shi, Y. Wang, X. Jing and P. Wang, Energy Environ. Sci., 2010, 3, 1924 CAS.
- Y. Bai, J. Zhang, D. Zhou, Y. Wang, M. Zhang and P. Wang, J. Am. Chem. Soc., 2011, 133, 11442 CrossRef CAS.
- T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211 CrossRef CAS.
- D. Sahu, H. Padhy, D. Patra, J.-F. Yin, Y.-C. Hsu, J.-T. Lin, K.-L. Lu, K.-H. Wei and H.-C. Lin, Tetrahedron, 2011, 67, 303 CrossRef CAS.
- J. Kim, Y. Jo, W.-Y. Choi, Y. Jun and C. Yang, Tetrahedron Lett., 2011, 52, 2764 CrossRef CAS.
- J.-H. Chen, C.-H. Tsai, S.-A. Wang, Y.-Y. Lin, T.-W. Huang, S.-F. Chiu, C.-C. Wu and K.-T. Wong, J. Org. Chem., 2011, 76, 8977 CrossRef CAS.
- Y. Cui, Y. Wu, X. Lu, X. Zhang, G. Zhou, F. B. Miapeh, W. Zhu and Z.-S. Wang, Chem. Mater., 2011, 23, 4394 CrossRef CAS.
- M. Xu, M. Zhang, M. Pastore, R. Li, F. De Angelis and P. Wang, Chem. Sci., 2012, 3, 976 RSC.
- Y.-C. Chen, H.-H. Chou, M. C. Tsai, S.-Y. Chen, J. T. Lin, C.-F. Yao and K. Chen, Chem.–Eur. J., 2012, 18, 5430 CrossRef CAS.
- K. Do, D. Kim, N. Cho, S. Paek, K. Song and J. Ko, Org. Lett., 2012, 14, 222 CrossRef CAS.
- K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC.
- R. Li, J. Liu, N. Cai, M. Zhang and P. Wang, J. Phys. Chem. B, 2010, 114, 4461 CrossRef CAS.
- N. Cai, S.-J. Moon, L. Cevey-Ha, T. Moehl, R. Humphy-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, Nano Lett., 2011, 11, 1452 CrossRef CAS.
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591 CrossRef CAS.
- Y. Cao, N. Cai, Y. Wang, R. Li, Y. Yuan and P. Wang, Phys. Chem. Chem. Phys., 2012, 14, 8282 RSC.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N.-L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133, 18042 CrossRef CAS.
- H. N. Tsao, J. Burschka, C. Yi, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2011, 4, 4921 CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS.
- H. Bronstein, R. S. Ashraf, Y. Kim, A. J. P. White, T. Anthopoulos, K. Song, D. Kames, W. Zhang and I. McCulloch, Macromol. Rapid Commun., 2011, 32, 1664 CrossRef CAS.
- Y.-J. Cheng, C.-H. Chen, T.-Y. Lin and C.-S. Hsu, Chem.–Asian J., 2012, 7, 818 CrossRef CAS.
- T. Kunz and P. Knochel, Chem.–Eur. J., 2011, 17, 866 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336 CrossRef CAS.
- B. C. O'Regan, K. Bakker, J. Kroeze, H. Smit, P. Sommeling and J. R. Durrant, J. Phys. Chem. B, 2006, 110, 17155 CrossRef CAS.
- E. M. J. Johansson, M. Hedlund, H. Siegbahn and H. Rensmo, J. Phys. Chem. B, 2005, 109, 22256 CrossRef CAS.
- G. Liu, W. Jaegermann, J. He, V. Sundström and L. Sun, J. Phys. Chem. B, 2002, 106, 5814 CrossRef CAS.
- K. E. Lee, M. A. Gomez, T. Regier, Y. Hu and G. P. Demopoulos, J. Phys. Chem. C, 2011, 115, 5692 CAS.
- K. Hara, K. Sayama, Y. Ohga, A. Shinpo, S. Suga and H. Arakawa, Chem. Commun., 2001, 569 RSC.
- T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. A. Anderson, X. Ai, T. Q. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806 CrossRef CAS.
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871 CrossRef CAS.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS.
- A. Listorti, I. López-Duarte, M. V. Martínez-Díaz, T. Torres, T. DosSantos, P. R. F. Barnes and J. R. Durrant, Energy Environ. Sci., 2010, 3, 1573 CAS.
- D. Zhou, N. Cai, H. Long, M. Zhang, Y. Wang and P. Wang, J. Phys. Chem. C, 2011, 115, 3163 CAS.
- H. Long, D. Zhou, M. Zhang, C. Peng, S. Uchida and P. Wang, J. Phys. Chem. C, 2011, 115, 14408 CAS.
- J. Liu, D. Zhou, F. Wang, F. Fabregat-Santiago, S. G. Miralles, X. Jing, J. Bisquert and P. Wang, J. Phys. Chem. C, 2011, 115, 14425 CAS.
- K. Sunahara, A. Furube, R. Katoh, S. Mori, M. J. Griffith, G. G. Wallace, P. Wagner, D. L. Officer and A. J. Mozer, J. Phys. Chem. C, 2011, 115, 22084 CAS.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, R. D. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538 CrossRef CAS.
- S. E. Koops, P. R. F. Barnes, B. C. O'Regan and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 8054 CAS.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276 Search PubMed.
- Z. Ning, Q. Zhang, H. Pei, J. Luan, C. Lu, Y. Cui and H. Tian, J. Phys. Chem. C, 2009, 113, 10307 CAS.
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261 CAS.
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2011, 115, 2439 CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Grätzel, J. Nelson, X. Li, N. J. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225 CrossRef CAS.
- S. Pelet, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1791 CrossRef CAS.
- A. Morandeira, I. López-Duarte, B. O'Regan, M. V. Martínez-Díaz, A. Forneli, E. Palomares, T. Torres and J. R. Durrant, J. Mater. Chem., 2009, 19, 5016 RSC.
- A. Reynel, A. Forneli and E. Palomares, Energy Environ. Sci., 2010, 3, 805 Search PubMed.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679 CrossRef CAS.
- S. Mathew, H. Iijima, Y. Toude, T. Umeyama, Y. Matano, S. Ito, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, J. Phys. Chem. C, 2011, 115, 14415 CAS.
- L. Ellis-Gibbings, V. Johansson, R. B. Walsh, L. Kloo, J. S. Quinton and G. G. Andersson, Langmuir, 2012, 28, 9431 CrossRef CAS.
- G. Schlichthörl, S. Y. Huang, J. Sprague and A. J. Frank, J. Phys. Chem. B, 1997, 101, 8141 CrossRef.
- S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3480 CrossRef CAS.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808 CrossRef CAS.
- T. Marinado, M. Hahlin, X. Jiang, M. Quintana, E. M. J. Johansson, E. Gabrielsson, S. Plogmaker, D. P. Hagberg, G. Boschloo, S. M. Zakeeruddin, M. Grätzel, H. Siegbahn, L. Sun, A. Hagfeldt and H. Rensmo, J. Phys. Chem. C, 2010, 114, 11903 CAS.
- M. Hahlin, E. M. J. Johansson, S. Plogmaker, M. Odelius, D. P. Hagberg, L. Sun, H. Siegbahn and H. Rensmo, Phys. Chem. Chem. Phys., 2010, 12, 1507 RSC.
- M. F. Hochella and A. H. Carim, Surf. Sci., 1988, 197, L260 CrossRef CAS.
- P. J. Cumpson, Surf. Interface Anal., 2001, 31, 23 CrossRef CAS.
Footnote |
| † Electronic supplementary information available: Dye synthesis and other spectral data. See DOI: 10.1039/c2ee23592g |
| This journal is © The Royal Society of Chemistry 2013 |