Disentangling the impact of side chains and fluorine substituents of conjugated donor polymers on the performance of photovoltaic blends†
Liqiang
Yang
a,
John R.
Tumbleston
b,
Huaxing
Zhou
c,
Harald
Ade
b and
Wei
You
*ac
aCurriculum in Applied Sciences and Engineering, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3287, USA. E-mail: wyou@unc.edu
bDepartment of Physics, North Carolina State University, Raleigh, North Carolina 27695, USA
cDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA
First published on 18th October 2012
Abstract
Side chains and fluorine (F) substituents on conjugated polymers have shown significant impact on the photovoltaic properties of polymer-based bulk heterojunction (BHJ) solar cells, but their respective impact is largely studied independently. In order to disentangle the effect of side chains and F substituents, we comprehensively investigate a series of conjugated polymers with an identical backbone (PNDT–DTBT) but different combinations of side chains and F substituents. Surprisingly, these seemingly marginal changes to the polymer backbone strongly influence the morphology and structure in BHJ thin films (e.g., domain size/purity and the relative orientation of polymer crystallites), as manifested by resonant soft X-ray scattering (R-SoXS) and grazing-incidence wide-angle X-ray scattering (GI-WAXS), thereby exerting significant impact on the photovoltaic properties of these conjugated polymer-based BHJ cells. Devices based on the polymer with long bulky side chains and F substituents (C8,4-C6,2F) simultaneously exhibit large open circuit voltage (Voc), high short circuit current (Jsc) and good fill factor (FF), with an efficiency as high as 5.6% for this series of PNDT–DTBT polymers.
Broader contextPrevious studies showed that long branched side chains weaken intermolecular interactions, leading to an increased open circuit voltage (Voc) but a lower short circuit current (Jsc). Therefore, achieving both a high Voc and a high Jsc seemed irreconcilable via side chain optimization alone. However, we have shown in this study that the reduced Jsc introduced by long bulky side chains is significantly improved by fluorine (F) substitution on the conjugated backbone, which is partly due to an enhanced π–π stacking and optimized crystallite orientation relative to the electrodes. Further, the introduction of F substituents lowers the highest occupied molecular orbital (HOMO) level, yielding a higher Voc. Finally, introduction of F to the conjugated backbone noticeably increases the charge collection probability at all voltages as manifested by the observed improvements in the fill factor (FF). These observations imply that F substituents could mitigate the negative impact resulting from long and branched side chains, which indicates a possible new strategy to further improve the efficiency of conjugated polymer-based solar cells via optimal combination of side chains and F substituents. |
1. Introduction
Any conjugated polymer for photovoltaic applications contains three key constituent components: the conjugated backbone, the side chains and the substituents (both on the conjugated backbone).1 While the creative design and synthesis of conjugated backbones has received much attention and driven the efficiency of bulk heterojunction (BHJ) solar cells to record highs,2–11 the side chains and the substituents have largely been overlooked until recently.4,8,9,12–19Conjugated polymers require side chains to ensure their solubility in the processing solvent prior to device fabrication. Further, these side chains are critical to achieving high polymer molecular weights, which improve charge transport in the related BHJ solar cells and lead to high currents.16 However, recently we17 and others4,12–15 have shown that in addition to addressing the concerns on the solubility in the solvent and the molecular weight of related polymers, these seemingly “trivial” side chains can significantly affect the device characteristics of related BHJ solar cells (e.g., open circuit voltage, Voc, short circuit current, Jsc, and fill factor, FF). These chains influence intermolecular interactions (among polymers and between polymers and fullerenes) and related stacking/packing in the solid state, all of which have a large impact on the performance of the BHJ solar cell, a solid state device. Specifically, we have previously shown that long branched side chains weaken intermolecular interactions, leading to an increased Voc but a lower Jsc.17 On the other hand, short straight side chains promote intermolecular interactions, rendering an enhanced Jsc (though at the expense of Voc). Therefore, we concluded that side chain optimization of conjugated polymers requires a balance between Voc and Jsc to reach optimum efficiencies, since achieving both a high Voc and a high Jsc seemed irreconcilable.
While side chains do not significantly perturb the electronic and optical properties of related conjugated polymers (if anchored properly to minimize steric hindrance),16 substituents on the backbone such as fluorine (F) and oxygen (O) can fine-tune properties including the energy levels and band gaps.15,18 For example, we have recently shown that for two separate polymers, adding F atoms to the conjugated backbone leads to a higher Voc, a higher Jsc and a better FF for F-substituted polymer-based solar cells than those of their non-fluorinated analogs.8,9 Interestingly, in both cases, even with long and bulky side chains attached to the conjugated backbones (which would have led to a lower Jsc), very respectable currents were still obtained together with high Voc as well as better FF. These observations imply that F substituents could mitigate the negative impact on Jsc due to long and branched side chains – a very interesting observation that warrants further investigation.
In order to disentangle the intertwined influence on photovoltaic performance of side chains and F substituents, we carried out a systematic study on a series of polymers containing identical conjugated backbones but different side chains and either hydrogen or F substituents. The conjugated backbone of these polymers (PNDT–DTBT) is different from our previous two different F-containing polymers.8,9 Still, these PNDT–DTBT polymers are constructed following the “weak donor–strong acceptor strategy”,20,21 by alternating naphtho[2,1-b:3,4-b′]dithiophene (NDT) and 4,7-di(thiophen-2-yl)benzothiadiazole (DTBT) (Fig. 1). To minimize possible interference from molecular weight variations, all four polymers were synthesized with similar molecular weights as shown in Table 1. Interestingly, the C8,4-C6,2F polymer with long bulky side chains and F substituents exhibits the largest Voc and a very high Jsc as well as a high FF, resulting in the highest efficiency observed in its device among all four polymer-based devices. The observed differences in Voc, Jsc and FF, depending upon the side chains and F substituents, were thoroughly investigated via device characterization and morphological investigations with X-ray scattering. Our study clearly indicates that a proper combination of side chains and F substituents on the conjugated backbone is a viable approach to simultaneously obtain large Voc, high Jsc and good FF of the related BHJ devices.
 | ||
| Fig. 1 The chemical structures of four polymers based on the PNDT–DTBT backbone. | ||
| Polymer | M n (kg mol−1) | M w (kg mol−1) | PDI | E g (eV) | HOMOb (eV) | |
|---|---|---|---|---|---|---|
| 140 °C | r.t. | |||||
| a Calculated from the intersection of the tangents on the low energetic edge of the absorption spectrum (in dichlorobenzene) with the baseline. b HOMO levels are determined from cyclic voltammetry measurements (see ESI, Fig. S1†). | ||||||
| C6,2-C6,2 | 7.88 | 18.5 | 2.35 | 1.90 | 1.65 | −5.36 |
| C8,4-C6,2 | 7.62 | 16.2 | 2.13 | 1.90 | 1.79 | −5.37 |
| C6,2-C6,2F | 7.45 | 18.8 | 2.53 | 1.93 | 1.68 | −5.41 |
| C8,4-C6,2F | 10.5 | 28.3 | 2.70 | 1.93 | 1.72 | −5.43 |
2. Results and discussion
2.1 Optical and electrochemical properties
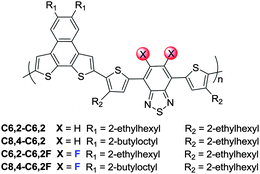
At high temperature, the effect of solubilizing chains on conjugated polymers has little impact on the optical properties of a polymer solution since the polymers are adequately solvated. Thus the absorption spectra of polymers with identical backbones collapse together, regardless of the side chain size and shape, as shown in Fig. 2a. Compared with those of non-fluorinated ones, the band edges of these F substituted polymers are slightly (∼0.03 eV) blue-shifted (1.93 eV vs. 1.90 eV), as observed in other similar systems.8,9,15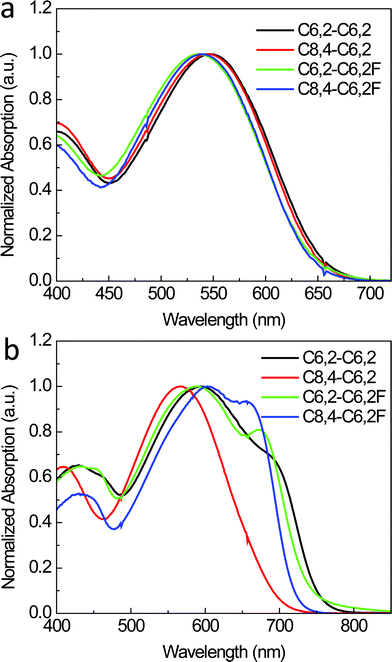 | ||
| Fig. 2 Normalized absorption spectra of four polymers in dichlorobenzene at (a) 140 °C and (b) room temperature. | ||
However, when these polymer solutions of identical concentration are cooled to room temperature, we observe noticeable differences in the optical properties of these polymer solutions (Fig. 2b). For example, the C6,2-C6,2 polymer with short side chains exhibits much stronger aggregation when compared with the C8,4-C6,2, as indicated by a pronounced absorption increase at longer wavelengths from about 690 nm to almost 750 nm. This red-shift in the absorption spectrum of the C6,2-C6,2 leads to a narrow band gap of 1.65 eV, roughly 0.14 eV smaller than that of the C8,4-C6,2 (1.79 eV). These results are consistent with our previous observation that introduction of short side chains to the polymer backbone strengthens π–π stacking ability and renders a strong aggregation in the relevant conjugated polymers.17 On the other hand, when F substituents are added to these two polymers, both C6,2-C6,2F and C8,4-C6,2F show pronounced absorption shoulders around 655 and 672 nm, respectively, indicating strong polymer aggregation even in solution at room temperature. Strong aggregation in these fluorinated PNDT–DTBTs is likely due to the induced intermolecular interactions via C–F⋯H, F⋯F and C–F⋯πF interactions.22,23 Because of the more pronounced aggregation introduced by F substituents, the C8,4-C6,2F demonstrates a red shift in its absorption spectrum when compared with that of the C8,4-C6,2, leading to a smaller band gap of 1.72 eV. Finally, introduction of F atoms to the C6,2-C6,2 does not further decrease its band gap, as observed from the similar absorption edge of the C6,2-C6,2F to that of the C6,2-C6,2. It seems that the short side chains on the C6,2-C6,2 already caused a strong red shift in absorption when compared with the C8,4-C6,2 rendering any additional red shift due to the incorporation of F substituents minimal. Additionally, it should be noted that the solubility of the C6,2-C6,2F was the lowest of the four polymers due to the shorter C6,2 side chains and the incorporation of F substituents, both of which contributed to enhanced aggregation in solution.
2.2 Morphology and structure of polymer:fullerene thin films
Given the differences in polymer aggregation in solution deduced from Fig. 2b, it is expected that blending these polymers with phenyl-C61-butyric acid methyl ester (PCBM) would lead to variations in film morphology and structure. As shown in Fig. 3, atomic force microscopy (AFM) phase images of the top surface of blend films (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w) based on the C8,4-C6,2 show less pronounced surface features than those based on the C6,2-C6,2 (see ESI, Fig. S2† for height images). This is possibly related to the resistance of the C8,4-C6,2 to aggregate in solution (Fig. 2b) as discussed in Section 2.1. While this is a noted difference due to changing the side chains, comparing the effect of F substitution reveals more striking variations: blends based on non-fluorinated polymers show much finer surface morphological features when compared to those with F substituted polymers.
:1 w/w) based on the C8,4-C6,2 show less pronounced surface features than those based on the C6,2-C6,2 (see ESI, Fig. S2† for height images). This is possibly related to the resistance of the C8,4-C6,2 to aggregate in solution (Fig. 2b) as discussed in Section 2.1. While this is a noted difference due to changing the side chains, comparing the effect of F substitution reveals more striking variations: blends based on non-fluorinated polymers show much finer surface morphological features when compared to those with F substituted polymers.
 | ||
| Fig. 3 AFM phase images (2 × 2 μm) of (a) C6,2-C6,2, (b) C8,4-C6,2, (c) C6,2-C6,2F and (d) C8,4-C6,2F-based BHJ devices processed with dichlorobenzene. | ||
While AFM provides insights into the top surface morphology, it is not necessarily representative of the bulk morphology of the thin films.24 To gain a more complete picture of film morphology, the distributions of domain spacing along with relative domain purities are assessed with resonant soft X-ray scattering (R-SoXS).25,26Fig. 4 shows circular averages of 2D scattering data for a photon energy of 284.2 eV, with domain spacing and relative domain purity values listed in Table 2. At 284.2 eV, the materials contrast between polymer and fullerene is nearly maximized but below the absorption edge of the materials to minimize X-ray fluorescence (see ESI, Fig. S3†). The scattering peaks in Fig. 4 are caused by the contrast between polymer and fullerene and are therefore indicative of domain spacing between polymer-rich and PCBM-rich domains. Scattering peaks observed for the C6,2-C6,2F and C8,4-C6,2F-based blends are at relatively small q and for the C6,2-C6,2 and C8,4-C6,2-based blends at larger q. This indicates that the mode of the distribution corresponding to the dominant domain spacing for fluorinated polymer-based blends (2π/q = 60–80 nm) is larger than those based on their non-fluorinated analogs (30–50 nm). The increase in domain spacing is representative of an increase in domain size, which has also been observed when adding fluorine substituent atoms in other systems.27 This increase in domain size is also consistent with the AFM measurements. Interestingly, an additional scattering peak at 0.01 nm−1 is observed in the C6,2-C6,2F-based blend, which indicates a second domain distribution at ∼600 nm. Such a large domain size may result from strong aggregations of the C6,2-C6,2F in solution, which may explain this feature in the thin film. This is consistent with the observed limited solubility resulting from the short side chain and the strong intermolecular interaction introduced by F substituents, both of which facilitate the formation of aggregations of the C6,2-C6,2F in solution.
 | ||
| Fig. 4 R-SoXS scattering profiles for the photon energy of 284.2 eV, which are representative of the distributions of domain spacings between polymer-rich and PCBM-rich regions in the bulk of the thin films. Please note that the 1-D intensity I multiplied by q2, i.e., Iq2, corresponds to the azimuthal integration of our 2-D data. | ||
| Polymer | (100) d-spacing (Å) [±0.1] | (010) d-spacing (Å) [±0.1] | (010) OOP/IP intensity ratio [±10%] | Mode of domain spacing (nm) [±5] | Relative domain purity (%) [±10] |
|---|---|---|---|---|---|
| a Relative domain purities are determined by taking the square root of the total scattering intensity (TSI) and normalizing to the blend with the highest TSI (blend based on C8,4-C6,2F in this case). Uncertainties for d-spacings and intensity ratios from GI-WAXS are from largest uncertainties of multi-peak fits, while uncertainties for domain spacings and purity from R-SoXS are estimated from multiple measurements of identical samples. Errors in domain purity due to truncation of the integral of TSI and uncertainty in thickness measurements are also considered. “OOP” and “IP” stand for “out-of-plane” and “in-plane”, respectively. | |||||
| C6,2-C6,2 | 19.6 | 3.6 | 5.8 | 33 | 58 |
| C8,4-C6,2 | 21.0 | 3.7 | <0.13 | 48 | 79 |
| C6,2-C6,2F | 19.6 | 3.6 | 31 | 58 | 55 |
| C8,4-C6,2F | 20.8 | 3.7 | 2.0 | 83 | 100 |
Along with the identification of the domain size, R-SoXS can also be used to determine relative domain purity differences between films (Table 2). Since the polymer/fullerene blend ratios are identical, and the scattering has been corrected for differences in active layer thickness (i.e., illuminated volume), differences in the total scattering intensity are related to differences in domain purity.28 Both C8,4-based blends exhibit higher total scattering intensity (corresponding to integration of profiles in Fig. 4) than the total scattering intensity of blends based on the C6,2-C6,2 and C6,2-C6,2F. This indicates that the domains in the C8,4-based blends are purer than their C6,2-based counterparts. This could be explained by the long bulky C8,4 side chains requiring large space volumes next to the polymer backbone and hence repelling fullerenes to form larger and more pure domains. Alternatively, the more highly solvent-miscible C8,4-based polymers stay in solution longer during casting, which gives the forming thin film more time to phase separate and be less quenched in a state far from equilibrium. In summary, the R-SoXS measurements reveal that both the side chains and F substituents play an important role in determining the domain size and purity of the polymer/fullerene thin films.
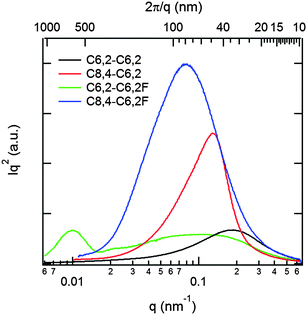
We further conducted grazing-incidence wide-angle X-ray scattering (GI-WAXS) to gain insight into the structural differences of the blend films. This measurement provides detailed information on the relative crystallite orientation along with a relative measure of the strengths of intermolecular interactions between polymer chains as reflected in changes in the crystal d-spacing.13,17,29Fig. 5 presents 2D GI-WAXS data of these four polymer/PCBM films measured on PEDOT:PSS-coated Si substrates. Representative d-spacing values and peak intensities are listed in Table 2, corresponding to multi-peak fitting in Fig. S4.† It should be noted that the isotropic and broad ring around q = 1.4 Å−1 arises from amorphous PCBM where sharp crystal reflections are not observed. In terms of polymer crystallites, the strong (100) peak and higher order peaks visible for some films represent lamellar polymer spacing between backbones. On the other hand, the π–π stacking between coplanar π-conjugated polymers is represented by the (010) peak. Polymers with short C6,2 side chains (C6,2-C6,2 and C6,2-C6,2F) exhibit both smaller (100) and (010) spacing than their corresponding polymers with C8,4 side chains. The smaller (100) spacing by ∼1 Å for the C6,2-based polymers in their blends is consistent with the shorter C6,2 side chains when compared with the blends with C8,4-based polymers. The same argument could be used to explain the slight reductions in (010) spacing for the C6,2-based polymer blends where these side chains require less space next to the polymer backbone. This reduction in spacing indicates stronger intermolecular interactions between polymer chains as argued previously to correlate with device performance.17
 | ||
| Fig. 5 GI-WAXS data of (a) C6,2-C6,2, (b) C8,4-C6,2, (c) C6,2-C6,2F and (d) C8,4-C6,2F-based polymer/PCBM BHJ films where the color scale corresponds to the ranges of 2.3 to 3.6 for (a and c) and 2.15 to 3.45 for (b and d) to highlight the important features of each sample. (e) Corresponding 20° sector averages in-plane (qx−y) and out-of-plane (qz), where in-plane and out-of-plane traces for each sample have been scaled by a common factor for ease of comparison. (f) Zoom-in of the (010) peaks where the intensities are normalized to the PCBM peak intensities near 1.4 Å−1 for clarity. Please note that the scattering vector components (i.e., qz and qx−y) are approximate as the 2D data have not been corrected for the “missing wedge” of data along the out-of-plane direction.30 | ||
By comparing the 2-D GI-WAXS data and intensity ratios of out-of-plane (OOP) to in-plane (IP) diffraction in the 〈010〉 direction, relative comparisons of the orientation of π–π stacking planes can also be made. It should be noted that 2-D angular data cannot capture a certain range of qz and qx−y intensities along the OOP direction, resulting in a “missing wedge”.30 Hence, the presented analysis assumes that the true OOP intensities are representative of the data measured here. This assumption is not expected to significantly change the conclusions drawn from the intensity ratios, since they each differ by an order of magnitude, and the peak widths in the 〈010〉 direction are similar for all samples. With addition of fluorine or when shortening the side chains, the polymer orientation becomes increasingly “face-on” with the π–π stacking direction perpendicular to the substrate. This can also be noted in the intensity distribution of the (100) peaks for the IP and OOP directions where the (100) and (200) peaks have larger IP components for polymers that are more “face-on”. From the ratio of (010) intensities, blends with the C6,2-C6,2F exhibit the most “face-on” polymer configuration while those with the C8,4-C6,2 exhibit the least and are preferentially “edge-on” with the side chains perpendicular to the substrate. This is supported by this blend having the highest IP (010) intensity and largest anisotropy between IP and OOP (100) intensity. Interestingly, shortening (010) polymer d-spacing via shortening the side chain and/or adding F substituents promotes increasingly “face-on” polymer orientations. It is possible that strong π–π stacking ability helps the coplanar π-conjugated backbone interact strongly with the substrate, thereby facilitating a “face-on” structure. Finally, compared to polymer crystallite orientation, less significant changes occur for the polymer crystallite size calculated from the inverse full width at half maximum of the (010) peak width (see ESI, Fig. S4†).
2.3 Photovoltaic properties of BHJ devices
Quite surprisingly, the minor changes in the optical and electrochemical properties resulted in significant changes in the morphological and structural properties of the BHJ blend films as discussed in Section 2.2. Correspondingly, significant differences were noted in the photovoltaic properties of BHJ devices where the efficiency varies as much as three fold (from 1.9% to 5.6%). The current–voltage characteristics of solar cells based on these four polymers are shown in Fig. 6a with representative performance parameters listed in Table 3. We note that for fair comparison and accurate interpretation of structure–property relationships, we maintained identical processing conditions for all polymers for their representative devices (e.g., weight ratio of polymer to PCBM was 1:1 in o-dichlorobenzene (DCB)). Detailed analysis of the BHJ device characteristics further discloses the impact on related photovoltaic properties introduced by the subtle changes in alkyl chains and substituents on these polymers. In the following, we will individually discuss the impact of F substituents and side chains on Jsc, FF, and Voc of related BHJ devices based on this series of four polymers.
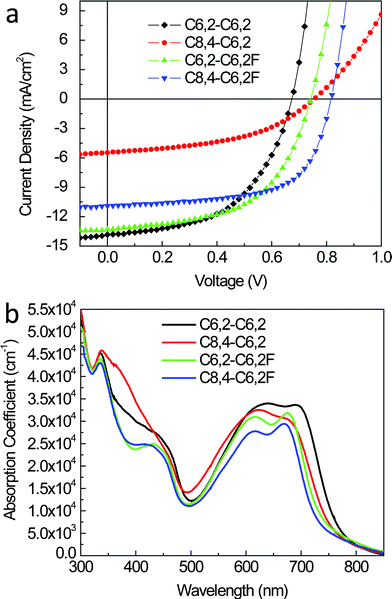 | ||
| Fig. 6 (a) Current density vs. voltage characteristics of optimized BHJ solar cells under 1 Sun illumination (100 mW cm−2). (b) Absorption coefficient of polymer/PCBM thin films. | ||
| Polymer | Polymer:PCBM | Processing solvent | Thickness (nm) | V oc (V) | J sc (mA cm−2) | FF (%) | η (%) |
|---|---|---|---|---|---|---|---|
| C6,2-C6,2 | 1:1 |
DCB | 85 ± 5 | 0.67 ± 0.01 | 13.8 ± 0.7 | 53.1 ± 2.1 | 4.9 ± 0.1 |
| C8,4-C6,2 | 1:1 |
DCB | 83 ± 5 | 0.75 ± 0.01 | 5.5 ± 0.1 | 46.5 ± 0.4 | 1.9 ± 0.1 |
| C6,2-C6,2F | 1:1 |
DCB | 116 ± 5 | 0.75 ± 0.01 | 13.3 ± 0.4 | 54.4 ± 0.9 | 5.4 ± 0.2 |
| C8,4-C6,2F | 1:1 |
DCB | 118 ± 5 | 0.81 ± 0.01 | 10.9 ± 0.3 | 63.6 ± 1.2 | 5.6 ± 0.2 |
We further applied optical modeling of complete devices to quantify these optical effects. Though the weaker intrinsic absorption of the fluorinated polymer-based blends is compensated by thicker active layers in their optimized devices, this thickness change amounts to a minor modification (∼4%) in absorption as deduced from optical modeling (see ESI, Fig. S6†). On the other hand, differences in absorption strength from the absorption coefficients (Fig. 6b) play a major role and suggests 11% higher absorption for the C6,2-C6,2-based blend when compared with its fluorinated analogue. However, the Jsc improvement amounts to only 4%, signifying a non-optical enhancement to the Jsc for the blend with the C6,2-C6,2F. The same holds true when comparing the two C8,4-based blends where the absorption is 16% higher for the non-fluorinated polymer C8,4-C6,2-based active layer, but the Jsc is actually 2 times lower for its corresponding devices. Both comparisons indicate that addition of fluorine creates an electrical enhancement that compensates for the weaker intrinsic absorption of fluorinated polymers. This yields nearly equivalent Jsc when comparing devices with C6,2-based polymers (C6,2-C6,2 vs. C6,2-C6,2F) or significantly higher Jsc when comparing those with C8,4 side chains (C8,4-C6,2 vs. C8,4-C6,2F). As postulated above, changes in Jsc not related to absorption differences could be related to modifications in the structure introduced by F substituents, such as polymer crystal orientation or morphology (e.g., domain size and domain purity), leading to improved charge generation and/or transport. These will be considered below.
As discussed earlier, polymer crystallites in BHJ films exhibit more “face-on” or “edge-on” structure depending on the side chain and substituent atoms (Fig. 5, Section 2.2). This could influence carrier recombination since a preferential “face-on” orientation would provide more efficient hole transport and improve charge collection efficiency.31–34 To test this hypothesis, the hole mobilities of BHJ films were measured with space charge limited current (SCLC) diodes (Table 4) (see ESI, Fig. S7† for current–voltage curves). The hole mobility correlates quite well with the polymer crystallite orientations (Table 2). For example, the BHJ blend consisting of the C8,4-C6,2 has the lowest hole mobility together with the least “face-on” crystallite orientation, whereas the C6,2-C6,2F-based BHJ blend has the highest mobility and the most “face-on” orientation. When coupled with an enhanced optical absorption, the higher hole mobility of the C6,2 (C6,2-C6,2 or C6,2-C6,2F) based blend than that of its corresponding C8,4 polymer (C8,4-C6,2 or C8,4-C6,2F) based device helps explain the larger Jsc for the C6,2-based devices. Similarly, the lowest Jsc of the C8,4-C6,2-based device (among all four devices) can (at least partially) be ascribed to the least “face-on” orientation of polymers in its BHJ blend which, not surprisingly, has the lowest hole mobility.
In order to gain more insights into the observed difference in device performance (especially the Jsc), we further investigated the charge generation by comparing the saturated photocurrent (Jsatph) from these four devices. In practice, we first calculated the photocurrent (Jph) by subtracting the current density in the dark from the current density under illumination (see ESI, Fig. S8† for light and dark currents).35,36 From the resulting Jph–V characteristics, we further determined the compensation voltage (V0) at which Jph = 0. Fig. 7a plots Jph against the effective voltage across the device (given by V0 − V). For large reverse voltages (V0 − V > 1 V), all free carriers are extracted by the applied field and collected by electrodes with minimal recombination, leading to the saturated photocurrent that is field-independent (Jsatph).35 The saturated photocurrent is therefore decided by the optical absorption and any other electric-field-independent losses, such as diffusion of charge neutral excitons to donor–acceptor interfaces. Fig. 7a shows that Jsatph is noticeably different for all the devices. In particular, the Jsatph for the C8,4-C6,2 based device is significantly lower than those for the other three, which essentially accounts for the lowest Jsc for the C8,4-C6,2-based device. However, we cannot ascribe the lowest Jsatph for the C8,4-C6,2 based device to a poor exciton diffusion in its BHJ blend, because the C8,4-C6,2-based BHJ blend has smaller and less pure domains when compared with the C8,4-C6,2F-based blend. Generally, domains that are smaller and relatively impure should have optimal exciton diffusion efficiency as predicted by modeling.37,38 Therefore, the low Jsatph in the C8,4-C6,2-based device does not result from poor exciton diffusion efficiency. It is possible that the “edge-on” orientation and the weak π–π stacking of these C8,4-C6,2 conjugated backbones facilitate the formation of exciton traps and restrict free carrier generation in the C8,4-C6,2/PCBM thin films.
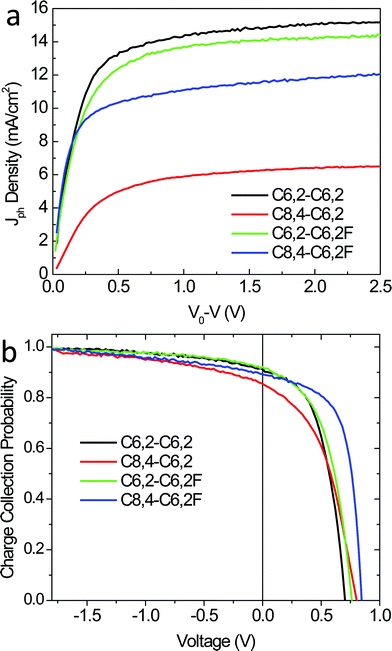 | ||
| Fig. 7 (a) Photocurrent density vs. effective voltage and (b) charge collection probability vs. applied voltage of optimized BHJ solar cells under 1 Sun conditions (100 mW cm−2). | ||
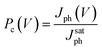 | (1) |
This practice equalizes the photocurrent at high electric fields thereby removing differences in optical absorption and other voltage-independent losses that do not depend on the internal field. As shown in Fig. 7b, Pc(V) becomes independent of the applied field at large applied reverse voltages (V < −1 V) and all field-dependent losses become negligible. For smaller applied reverse voltages (V > −1 V), the Pc(V) varies significantly due to differences in field-dependent recombination losses. These could include dissociation of bound electron–hole (e–h) pairs into free carriers by the applied field35 and bimolecular recombination.39 The dominant loss process in BHJ solar cells is currently under debate as it has recently been argued that dissociation of bound e–h pairs is independent of applied voltage.40 It has also been outlined that the photocurrent dependence on cell voltage could arise from geminate pair recombination depending on the system,41 where it has been shown that bimolecular recombination only dominates in the range of voltages close to Voc.36 Furthermore, losses related to the impact and possibility of the recently observed molecular ordering at the donor–acceptor interface also need to be assessed.42 Instead of identifying the exact recombination loss process, we simply use the charge collection probability to quantify differences in field-dependent losses of the different blends under weak applied fields where the FF is determined.
Under weak applied fields where the charge collection probability varies significantly, the device based on the C8,4-C6,2F achieved the highest charge collection probability while its non-fluorinated counterpart achieved the least (for example, V = 0.5 V). The two C6,2-based blends have charge collection probability that is in-between the blends consisting of the two C8,4-based polymers. When comparing the two C8,4-based blends, the optimal crystallite orientation and improved mobilities in the C8,4-C6,2F blend retard the recombination of free charge carriers during charge transport, leading to an improved charge collection probability and hence a higher FF. However, the mobilities and “face-on” crystallite orientation are highest for the two C6,2-based blends, which do not correlate with the charge collection probability at low fields or the FF.
The inability to globally correlate the FF with crystallite orientation is partially resolved by considering the measured domain purity from R-SoXS (Table 2). Both C8,4-based blends have domain purities that are significantly higher than the two blends consisting of C6,2-based polymers. Recent modeling has shown that low domain purity restricts charge collection37 and could explain the lower FF for the two C6,2-based blends than that of the C8,4-C6,2F-based blend even though the hole mobilities of the former two blends are higher (Table 2). Furthermore, the lower domain purity in the case of the C8,4-C6,2-based blend when compared with that of the C8,4-C6,2F may further hamper the charge collection probability of the device consisting of the C8,4-C6,2, which would be exacerbated by its lower hole mobility. Furthermore, a potentially important effect not probed with these techniques is that fluorine (F) is the most electronegative element in the periodic table, with a Pauling electronegativity of 4.0, much larger than that of hydrogen (2.2).43 It is plausible that introduction of the most electronegative element (F) creates strong internal dipole moments which lower the Coulombic potential between e–h pairs.44 This would lead to weaker Coulombic attractions between e–h pairs after exciton splitting, leading to low recombination rates with the introduction of fluorine substituents. Ultimately, the slight modification of the side chain and the substituent atom yields dramatic differences in morphology and structure, resulting in significant differences in Jsc and FF.
While this argument, the lower HOMO level leading to a larger Voc, holds for atomic substitution, it does not account for differences in Voc depending on the side chain since the side chain does not significantly impact the HOMO level of conjugated polymers in this study. Still, using bulky C8,4 side chains instead of C6,2 leads to an improvement in Voc of ∼0.07 V (Table 3). Improvements in Voc when using longer and bulkier side chains have been observed previously, owing to a weakening of intermolecular interactions between donor and acceptor materials.17,45 This is supported by the structural and morphological measurements of these blends as discussed above. The greater and closer π–π crystallite stacking of the two C6,2-based blends implies a stronger intermolecular interaction between polymer chains in polymer crystallites and potentially between polymer/PCBM, while the lower domain purities of these two blends support greater molecular mixing of polymer and PCBM potentially due to stronger polymer/PCBM interactions.46 Each of these would support a morphological explanation for lower Voc for the blends based on the polymers of shorter C6,2 side chains. Thus the synergistic effects of incorporating both long bulky side chains and F substituents in the case of the C8,4-C6,2F lead to the largest Voc in its BHJ device in this series of polymers (0.81 V). When combined with the highest FF and a reasonable Jsc, a high overall efficiency exceeding 5.6% was obtained for this device, the highest of the blends studied in this work.
3. Conclusions
This detailed study of PNDT–DTBT polymers with an identical conjugated backbone but different side chains and F substituents complements our previous independent discoveries of the effects of the side chains and F substitutions on photovoltaic properties of polymer-based solar cells. It becomes clear that side chains and F substituents strongly influence the morphology and structure in BHJ thin films, thereby resulting in significant impacts on the photovoltaic properties of conjugated polymer-based BHJ cells. For the C8,4-C6,2 polymer with long chains of C8,4, introduction of the most electronegative element, F, to the conjugated backbone noticeably increases the saturated photocurrent and charge collection probability as manifested in improvements in the Jsc and FF, respectively. Furthermore, the C8,4-C6,2F polymers tend to adopt an increasingly “face on” orientation to the substrate. This preferred orientation is correlated with a higher hole mobility which, in conjunction with domain purification for the fluorinated C8,4-based BHJ devices, helps explain a significantly higher Jsc of the C8,4-C6,2F-based devices than that of its non-fluorinated counterpart. Additionally, long, branched side chains such as C8,4 help weaken the polymer/PCBM intermolecular interaction, which, together with a lower HOMO level by the electronegative F substituents, leads to the highest Voc for the C8,4-C6,2F-based BHJ device. These factors contribute to the observed highest efficiency of the C8,4-C6,2F-based solar cells in the studied series of polymers. On the other hand, polymers with shorter side chains (e.g., C6,2-C6,2) have a strong tendency to aggregate, which indeed helps optical absorption and hole mobility yielding a higher Jsc, when compared with the C8,4-C6,2-based devices. In spite of the high mobilities, the FF is lower than the champion C8,4-C6,2F device for the two C6,2-C6,2-based devices regardless of fluorination. This is explained due to lower domain purity which yields slightly lower overall performance of two C6,2-based BHJ blends. Overall, our results indicate that an educated choice of side chains and atomic substituents can maximize the energy harvesting potential of a given conjugated backbone when used in a BHJ organic solar cell.4. Experimental
4.1 Reagents and instrumentation
All reagents and chemicals were purchased from commercial sources (Aldrich, Acros, Strem, and Fluka) and used without further purification unless stated otherwise. Reagent grade solvents were dried when necessary and purified by distillation. Microwave assisted polymerizations were conducted in a CEM Discover Benchmate microwave reactor. Gel permeation chromatography (GPC) measurements were performed with a Polymer Laboratories PL-GPC 220 instrument using 1,2,4-trichlorobenzene solvent (stabilized with 125 ppm BHT) at 150 °C. The obtained molecular weight is relative to the polystyrene standard. UV-Visible absorption spectra were obtained using a Shimadzu UV-2401PC spectrophotometer. For the measurements of thin films, polymers were spun coated onto pre-cleaned glass slides from 10 mg mL−1 polymer solutions in chlorobenzene. The thicknesses of films were recorded using a profilometer (Alpha-Step D-100, Tencor Instruments). AFM images were collected using an Asylum Atomic Force Microscope (Asylum MFP-3D, Asylum Research). The microscope was operated in AC mode under ambient conditions (T = 21 °C, RH = 45%), using silicon cantilevers (Tap300Al, BudgetSensors) with resonance frequencies of approximately 300 kHz.4.2 Electrochemistry
Cyclic voltammetry measurements were carried out using a Bioanalytical Systems (BAS) Epsilon potentiostat equipped with a standard three-electrode configuration. Typically, a three electrode cell equipped with a glass carbon working electrode, a Ag/AgNO3 (0.01 M in anhydrous acetonitrile) reference electrode, and a Pt wire counter electrode was employed. The measurements were carried out in anhydrous acetonitrile with tetrabutyl ammonium hexafluorophosphate (0.1 M) as the supporting electrolyte under an argon atmosphere at a scan rate of 100 mV s−1. Polymer films were drop cast onto the glassy carbon working electrode from a 2.5 mg mL−1 chloroform solution and dried under house nitrogen stream prior to measurements. The electrochemical onsets were determined at the position where the current starts to differ from the baseline. The potential of the Ag/AgNO3 reference electrode was internally calibrated by using the ferrocene/ferrocenium redox couple (Fc/Fc+), which has a known reduction potential of −4.8 eV. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of copolymers were calculated from the onset oxidation potentials (Eoxonset) and onset reductive potentials (Eredonset), respectively, according to eqn (2) and (3).| HOMO= −(Eoxonset + 4.8) (eV) | (2) |
| LUMO= −(Eredonset + 4.8) (eV) | (3) |
4.3 Grazing-incidence wide-angle X-ray scattering (GI-WAXS)
Samples for GI-WAXS were prepared on PEDOT:PSS-coated Si substrates using the same preparation conditions as for devices. Measurements were carried out at Beamline 7.3.3 of the Advanced Light Source using a Pilatus 1M detector.47 A grazing incident angle of 0.12° was used where the air scatter was minimized by purging the air between the X-ray source, sample, and detector with helium gas. The photon energy was 10 keV, the sample detector distance was ∼366 mm, and the CCD pixel size was 0.172 mm. The exposure time for all images was 15 s.4.4 Resonant soft X-ray scattering (R-SoXS)
Samples for R-SoXS were prepared by floating sections of the films used for GI-WAXS onto silicon nitride windows 1.5 mm × 1.5 mm in size. Measurements were carried out at Beamline 11.0.1.2 at the Advanced Light Source in transmission mode using a cooled Princeton Instrument PI-MTE CCD camera in a vacuum48 following a procedure similar to those used previously.25,28 2-D scattering data were averaged along the azimuth of 180° sector averages and scaled by q2. The contrast between the polymer and fullerene was calculated from polymer and fullerene optical constants via near edge X-ray absorption fine structure (NEXAFS) measurements. NEXAFS spectra of neat films were obtained by scanning transmission X-ray microscopy (STXM) measurements at beamline 5.3.2.2 at the Advanced Light Source49 (see ESI, Fig. S3† for details).4.5 Polymer solar cell fabrication and testing
Glass substrates coated with patterned indium-doped tin oxide (ITO) were purchased from Thin Film Devices, Inc. The 150 nm sputtered ITO pattern had a resistivity of 15 Ω □−1. Prior to use, the substrates were ultrasonicated for 20 minutes in acetone followed by deionized water and then 2-propanol. The substrates were dried under a stream of nitrogen and subjected to the treatment of UV–ozone over 30 minutes. A filtered dispersion of PEDOT:PSS in water (Baytron PH500) was then spun cast onto clean ITO substrates at 4000 rpm for 60 seconds and then baked at 140 °C for 10 minutes to give a thin film with a thickness of 40 nm. Blends of the polymer and phenyl-C61-butyric acid methyl ester (PCBM) (1:1 w/w, 10 mg mL−1 for polymers) were dissolved in DCB with heating at 140 °C for 6 hours. All the solutions were spun cast at optimized rpm (vary from 400 to 600) for 60 seconds onto the PEDOT:PSS layer. The substrates were then dried at room temperature in the glovebox under nitrogen atmosphere for 12 hours. The devices were finished for measurement after thermal deposition of a 30 nm film of calcium and a 70 nm aluminum film as the cathode at a pressure of ∼2 × 10−6 mbar. There are 8 devices per substrate, with an active area of 12 mm2 per device. Device characterization was carried out under AM 1.5G irradiation with an intensity of 100 mW cm−2 (Oriel 91160, 300 W) calibrated by a NREL certified standard silicon cell. Current versus voltage (I–V) curves were recorded with a Keithley 2400 digital source meter. EQE was detected under monochromatic illumination (Oriel Cornerstone 260 ¼ m monochromator equipped with an Oriel 70613NS QTH lamp) and the calibration of the incident light was performed with a monocrystalline silicon diode. All fabrication steps after adding the PEDOT:PSS layer onto ITO substrate and characterizations were performed in gloveboxes under nitrogen atmosphere. For mobility measurements, the hole-only devices in a configuration of ITO/PEDOT:PSS (40 nm)/copolymer-PCBM/Pd (50 nm) were fabricated. The experimental dark current densities J of polymer/PCBM blends were measured when applied with voltage from 0 to 6 V. The applied voltage V was corrected from the built-in voltage Vbi which was taken as a compensation voltage Vbi = Voc + 0.05 V and the voltage drop Vrs across the indium tin oxide/poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonic acid) (ITO/PEDOT:PSS) series resistance and contact resistance, which is found to be around 35 Ω from a reference device without the polymer layer. From the plots of J0.5vs. V (see ESI, Fig. S7†), hole mobilities of copolymers can be deduced from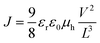 | (4) |
Acknowledgements
This work was supported by Office of Naval Research (N000141110235). WY is a Camille Dreyfus Teacher-Scholar. GI-WAXS and R-SoXS characterization and optical modeling contributed by JT and HA, supported by DOE, OS, BES, MSE (DE-FG02-98ER45737). Data were acquired at beamlines 7.3.3 (GI-WAXS), 5.3.2.2. (STXM/NEXAFS), and 11.0.1.2 (R-SoXS) of the ALS, which is supported by DOE (DE-AC02-05CH1123). We thank Alexander Hexemer and Steven Alvarez at beamline 7.3.3, Cheng Wang and Anthony Young at beamline 11.0.1.2, and David Kilcoyne at beamline 5.3.2.2 for assistance with data acquisition.References
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297 CrossRef CAS.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS.
- H. Zhou, L. Yang, S. C. Price, K. J. Knight and W. You, Angew. Chem., Int. Ed., 2010, 49, 7992 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595 CrossRef CAS.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995 CrossRef CAS.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. m. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250 CrossRef CAS.
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS.
- D. A. M. Egbe, S. Türk, S. Rathgeber, F. Kühnlenz, R. Jadhav, A. Wild, E. Birckner, G. Adam, A. Pivrikas, V. Cimrova, G. n. Knör, N. S. Sariciftci and H. Hoppe, Macromolecules, 2010, 43, 1261 CrossRef CAS.
- J. M. Szarko, J. Guo, Y. Liang, B. Lee, B. S. Rolczynski, J. Strzalka, T. Xu, S. Loser, T. J. Marks, L. Yu and L. X. Chen, Adv. Mater., 2010, 22, 5468 CrossRef CAS.
- S. Ko, E. Verploegen, S. Hong, R. Mondal, E. T. Hoke, M. F. Toney, M. D. McGehee and Z. Bao, J. Am. Chem. Soc., 2011, 133, 16722 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS.
- H. Zhou, L. Yang, S. Xiao, S. Liu and W. You, Macromolecules, 2010, 43, 811 CrossRef CAS.
- L. Yang, H. Zhou and W. You, J. Phys. Chem. C, 2010, 114, 16793 CAS.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586 CrossRef CAS.
- H. J. Son, W. Wang, T. Xu, Y. Liang, Y. Wu, G. Li and L. Yu, J. Am. Chem. Soc., 2011, 133, 1885 CrossRef CAS.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377 CAS.
- S. C. Price, A. C. Stuart and W. You, Macromolecules, 2010, 43, 797 CrossRef CAS.
- K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- T. Okamoto, K. Nakahara, A. Saeki, S. Seki, J. H. Oh, H. B. Akkerman, Z. Bao and Y. Matsuo, Chem. Mater., 2011, 23, 1646 CrossRef CAS.
- M. A. Ruderer, S. Guo, R. Meier, H.-Y. Chiang, V. Körstgens, J. Wiedersich, J. Perlich, S. V. Roth and P. Müller-Buschbaum, Adv. Funct. Mater., 2011, 21, 3382 CrossRef CAS.
- H. Yan, B. A. Collins, E. Gann, C. Wang, H. Ade and C. R. McNeill, ACS Nano, 2011, 6, 677 CrossRef.
- S. Swaraj, C. Wang, H. Yan, B. Watts, J. Lüning, C. R. McNeill and H. Ade, Nano Lett., 2010, 10, 2863 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2012 DOI:10.1002/aenm.201200377.
- D. M. DeLongchamp, R. J. Kline and A. Herzing, Energy Environ. Sci., 2012, 5, 5980 CAS.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488 CrossRef CAS.
- P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514 CrossRef CAS.
- Z. M. Beiley, E. T. Hoke, R. Noriega, J. Dacuña, G. F. Burkhard, J. A. Bartelt, A. Salleo, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2011, 1, 954 CrossRef CAS.
- J. Guo, Y. Liang, J. Szarko, B. Lee, H. J. Son, B. S. Rolczynski, L. Yu and L. X. Chen, J. Phys. Chem. B, 2010, 114, 742 CrossRef CAS.
- A. T.-H. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Frechet, J. Am. Chem. Soc., 2012, 134, 2180 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- B. P. Lyons, N. Clarke and C. Groves, Energy Environ. Sci., 2012, 5, 7657 CAS.
- C. R. McNeill, S. Westenhoff, C. Groves, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 19153 CAS.
- C. G. Shuttle, R. Hamilton, B. C. O'Regan, J. Nelson and J. R. Durrant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16448 CrossRef CAS.
- F. C. Jamieson, T. Agostinelli, H. Azimi, J. Nelson and J. R. Durrant, J. Phys. Chem. Lett., 2010, 1, 3306 CrossRef CAS.
- J. D. Servaites, M. A. Ratner and T. J. Marks, Energy Environ. Sci., 2011, 4, 4410 CAS.
- B. A. Collins, J. E. Cochran, H. Yan, E. Gann, C. Hub, R. Fink, C. Wang, T. Schuettfort, C. R. McNeill, M. L. Chabinyc and H. Ade, Nat. Mater., 2012, 11, 536 CrossRef CAS.
- K. Peer, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2011, 133, 20468 CrossRef CAS.
- M. D. Perez, C. Borek, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9281 CrossRef CAS.
- B. A. Collins, E. Gann, L. Guignard, X. He, C. R. McNeill and H. Ade, J. Phys. Chem. Lett., 2010, 1, 3160 CrossRef CAS.
- H. Alexander, B. Wim, G. James, S. Eric, G. Eliot, K. Rick, M. Alastair, C. Matthew, R. Bruce and P. Howard, J. Phys.: Conf. Ser., 2010, 247, 012007 CrossRef.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS.
- A. L. D. Kilcoyne, T. Tyliszczak, W. F. Steele, S. Fakra, P. Hitchcock, K. Franck, E. Anderson, B. Harteneck, E. G. Rightor, G. E. Mitchell, A. P. Hitchcock, L. Yang, T. Warwick and H. Ade, J. Synchrotron Radiat., 2003, 10, 125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CV curves of all polymers, AFM images of thin films of blends, GI-WAXS multi-peak Gaussian fitting, UV-vis absorption spectra of pure polymer films, EQE spectra and SCLC curve of each device, optical modeling, and R-SoXS details. See DOI: 10.1039/c2ee23235a |
| This journal is © The Royal Society of Chemistry 2013 |