DOI:
10.1039/C3DT52290C
(Paper)
Dalton Trans., 2013,
42, 16518-16526
Theoretical studies on concerted versus two steps hydrogen atom transfer reaction by non-heme MnIV/III![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O complexes: how important is the oxo ligand basicity in the C–H activation step?†‡
O complexes: how important is the oxo ligand basicity in the C–H activation step?†‡
Received
21st August 2013
, Accepted 4th September 2013
First published on 5th September 2013
Abstract
High-valent metal–oxo complexes have been extensively studied over the years due to their intriguing properties and their abundant catalytic potential. The majority of the catalytic reactions performed by these metal–oxo complexes involves a C–H activation step and extensive efforts over the years have been undertaken to understand the mechanistic aspects of this step. The C–H activation by metal–oxo complexes proceeds via a hydrogen atom transfer reaction and this could happen by multiple pathways, (i) via a proton-transfer followed by an electron transfer (PT-ET), (ii) via an electron-transfer followed by a proton transfer (ET-PT), (iii) via a concerted proton-coupled electron transfer (PCET) mechanism. Identifying the right mechanism is a surging topic in this area and here using [MnIIIH3buea(O)]2− (1) and [MnIVH3buea(O)]− (2) species (where H3buea = tris[(N′-tert-butylureaylato)-N-ethylene]aminato) and its C–H activation reaction with dihydroanthracene (DHA), we have explored the mechanism of hydrogen atom transfer reactions. The experimental kinetic data reported earlier (T. H. Parsell, M.-Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762) suggests that the mechanism between 1 and 2 is drastically different. By computing the transition states, reaction energies and by analyzing the wavefunction of the reactant and transitions states, we authenticate the proposal that the MnIII![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O undergoes a step wise PT-ET mechanism where as the MnIVO species undergo a concerted PCET mechanism. Both the species pass through a [MnIII–OH] intermediate and the stability of this species hold the key to the difference in the reactivity. The electronic origin for the difference in reactivity is routed back to the strength and basicity of the Mn–oxo bond and the computed results are in excellent agreement with the experimental results.
O undergoes a step wise PT-ET mechanism where as the MnIVO species undergo a concerted PCET mechanism. Both the species pass through a [MnIII–OH] intermediate and the stability of this species hold the key to the difference in the reactivity. The electronic origin for the difference in reactivity is routed back to the strength and basicity of the Mn–oxo bond and the computed results are in excellent agreement with the experimental results.
Introduction
Selective C–H bond oxidative cleavage of aromatic/aliphatic hydrocarbons is one of the important synthetic transformations in enzymatic and industrial processes. Naturally, mononuclear non-heme high-valent ferryl–oxo species serve as an astonishing paradigm for the activation of C–H bond in highly efficient and selective manner.1–4 Given the remarkable success of the oxidative potential of high-valent iron–oxo based model catalysts, many researchers were stimulated to target other metal based oxo complexes for achieving the C–H bond oxidative cleavage.5–9 Among the reported other metal based biomimetic model complexes, the manganese based oxo complexes have shown to possess prominent catalytic ability towards the C–H bond activation and O-atoms transfer reactions when compared to ferryl–oxo species and also these model complexes are capable of mimicking the catalytic properties of lipooxygenase, cytochrome P450 and ironoxido reductase enzyme.10–13
The catalytic reactivity of the metal–oxo species towards the C–H activation process is largely influenced by their redox properties.14–16 Besides some recent reports suggest that basicity of oxo ligands play a vital role in the rapid reactivity of manganese(IV/V)–oxo complexes involving C–H activation.17 The primacy of the basicity of oxo ligands was further legitimated by Borovik et al. through the C–H bond cleavage ability of monomeric Mn(III/IV)–oxo units with a tetradentate tripodal ligands ([MnIII/IVH3buea(O)]2−/−) (H3buea = tris[(N′-tert-butylureaylato)-N-ethylene]aminato) having trigonal bipyramidal geometry. Their study clearly shows that the basicity of oxo ligands affect the kinetic aspects of the C–H bond of activation.18 The KIE experiments reveal that kMn(O)H/kMn(O)D values are twice as much for 2 compared to 1 (2.6 vs. 6.8 for 1 and 2 respectively). Further a larger pKa measured for 1 compared to 2 lead to the suggestion of an anionic intermediate for 1 and a radical based intermediate for species 2. Based on the KIE experiments and estimated activation barrier for the two species, a different mechanistic route has been proposed for MnIIIO and MnIVO species. The C–H bond activation by [MnIIIH3buea(O)]2− (1) suggested to proceed through a two step mechanism, proton transfer followed by electron transfer whereas in [MnIVH3buea(O)]− (2) the reaction proceeds in a single step, i.e. via proton-coupled electron transfer (PCET) step. This PCET mechanism is also suggested as the possible mechanism for iron and manganese lipooxygenase and cytochrome P450 enzymes.19–24 Further the mechanistic studies suggest that 1 follows anionic mechanism attributed to the strong basic character of the oxo group (larger pKa) compared to species 2. Although the experimental study unfolds the differences in reactivity between 1 and 2, the precise reason for the preferred choice of the mechanism by these oxidants is unclear and this aspect is important to establish connection between observed reactivity of these models to that of enzymes. In this regard, we have undertaken a theoretical study based on density functional method to specifically address the following issues. (i) Between MnIIIO and MnIVO species, which one is a stronger oxidant and why? (ii) What are the exact mechanisms by which 1 and 2 activate the C–H bonds (PT-ET, ET-PT and PCET)? (iii) What are the electronic reasons behind the preferred choice of the mechanism?
Computational details
All the calculations were carried out using the Gaussian 09 suite of programs.25 The geometry optimizations have been performed with the B3LYP functional.26,27 The B3LYP has a proven track record of predicting the structures and the energetics accurately for such metal mediated catalytic reactions28–30 (see ESI‡ for discussion on chosen methodology). The LACVP basis set comprising the LanL2DZ–Los Alamos effective core potential for the Mn31–34 and a 6-31G*35 basis set for the other atoms have been employed for geometry optimization and the optimized geometries were then used to perform single point energy calculations using a TZVP basis set on all atoms.36,37 Solvation energies have been computed using PCM solvation model employing acetonitrile as the solvent. Frequency calculations were performed on the optimized structures at the B3LYP level to verify that they are minima on the potential-energy surface (PES) and also to obtain free energy corrections. The quoted DFT energies are the B3LYP/TZVP solvation energies incorporating free energies correction at the B3LYP/LACVP level, unless otherwise mentioned. The transition states were characterized by single imaginary frequency corresponding to the reaction coordinate and are verified by animating the frequency using visualization software such as Molden.38,39 The broken-symmetry approach available in Gaussian 09 is employed to aid smooth convergence in case of radical intermediates.40 The energy decomposition analysis (EDA)41–43 was performed at the same level of theory using AOMIX software.44–46
Result and discussion
Mechanism of C–H activation by MnIII![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O and MnIVO species
O and MnIVO species
Here we have investigated the mechanistic aspects of the C–H bond cleavage of dihydroanthracene (DHA) reaction by monomeric MnIII/IV–oxo complexes of tetradentate tripodal ligands with anionic nitrogen donors (H3buea) ligand using density functional methods. The proposed reaction mechanism involves two different pathways (anionic and radical) which are classified based on the type of intermediate formation (Scheme 1). In the anionic pathway, the monomeric oxomanganese species (1) [MnIII(H3buea)O]2− abstract a proton from DHA leading to the formation of anionic monohydroanthracene (MHA) intermediate (1intA) and [MnIII(H3buea)–OH]− species which further undergoes rapid electron transfer leading to the formation of [MnII(H3buea)–OH]2− and MHA radical intermediate (1intR). Further a second hydrogen abstraction (ts2) from the MHA leads to the formation of anthracene and [MnII(H3buea)–OH]2− species. In the radical pathway, species 2 ([MnIV(H3buea)O]−) undergoes PCET mechanism leading to the formation of MHA radical intermediate (2intR) and [MnIII(H3buea)–OH]1− species. Subsequent hydrogen abstraction from the MHA radical intermediate lead to the formation of anthracene and [MnIII(H3buea)–OH]− species. This adapted mechanism is consistent with the experimental observation.18
 |
| | Scheme 1 Mechanistic scheme adopted for C–H activation reaction involving monomeric oxomanganese(III/IV) complexes. | |
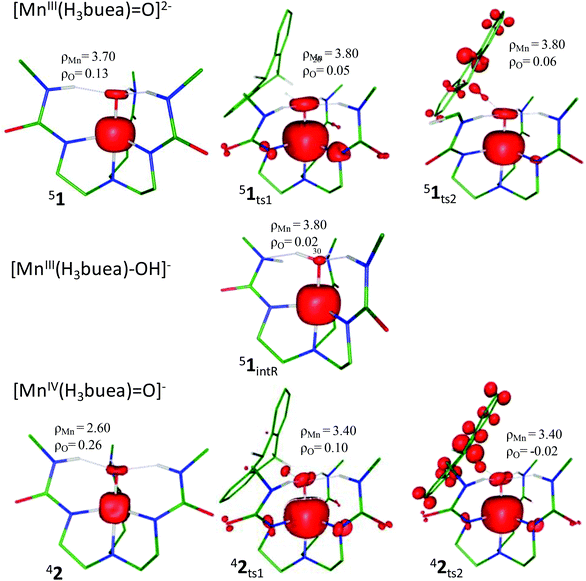
We have computed all the possible mechanistic pathways for the C–H bond activation reaction for both the oxidants 1 and 2 with DHA. Our B3LYP/TZVP calculations clearly show that both the oxidants possess a high spin (51 and 42) ground state with excited S = 1 and S = 1/2 states lying at 127.8 and 81.3 kJ mol−1 higher in energy for 1 and 2 respectively (Fig. 1 and 2). The MnIII/IVO bond in 1 and 2 is stabilized predominately by the presence of the intramolecular H-bonds between the terminal oxo ligand with H-bond donors of the ureate nitrogen atoms. The Mn–O bond length in species 2 (1.68 Å) is found to be shorter than that of species 1 (1.73 Å) (shown in Fig. 3 and Table S1 of ESI‡), this shorter and stronger MnO bond observed in 2 is also reflected in the computed bond order (Wiberg bond index of 1.16 vs. 1.32 for 1 and 2 respectively). The computed Mn–O bond lengths and other structural parameters are in agreement with the reported X-ray47–52 and previous theoretical studies.53–55 In addition to this, the Mulliken spin density on the oxo-oxygen in species 2 (ρMn = 2.6, ρO = 0.26, Fig. 4 and Table S2 of ESI‡) is substantially delocalized more than that of the oxo-oxygen in species 1 (ρMn = 3.7, ρO = 0.13, Fig. 4 and Table S2 of ESI‡). This implies that species 2 is having larger Mn–O covalent bond character and that leads to a shorter Mn–O bond. Further we would like to note here that the spin density on the oxygen atoms for both the species is significantly less compared to the corresponding iron–oxo species5 and thus can very well be denoted as metal–oxo complexes rather than oxy-radical type species. Moreover, species 1 is found to have shorter Mn–Neq bonds (average Mn–Neq bond length = 2.13 Å) and longer Mn–Nax bonds (2.21 Å). In 2 although the same trend is visible, the Mn–Nax bonds are much longer and Mn–Neq bond are much shorter than that of 1. A shorter and stronger Mn–O bond attributes to longer Mn–Nax bonds in the high-valent MnIVO species.56 The larger basicity of species 1 compared to species 2 is also reflected in the computed NPA charges (−0.82 vs. −0.63 for 1 and 2 respectively). Due to this variation in the basicity, the strength of three weak N–H⋯O(Mn) hydrogen bonding interactions also varies significantly between 1 and 2. The three weak N–H⋯O(Mn) hydrogen bond lengths are equal (1.79 Å) in 1 while they differ drastically in 2 (see Fig. 3). On comparing with species 1, the weak N–H⋯O(Mn) bonds are elongated by 0.025–0.035 Å in 2 while the N–H distances are proportionately shortened. The longer and weaker N–H⋯O(Mn) distances in 2 are also reflected in the OLP→BD*N–H donor–acceptor interactions in the NBO analysis. For 1 the stabilization energies are found to be in the range of 32.7–22.1 kJ mol−1 while for 2 they are in the range of 28.5–8.7 kJ mol−1. These electronic and structural differences between 1 and 2 hold the key to the difference in the reactivity between these two species.57
 |
| | Fig. 1 Computed reaction profile of C–H activation reaction involving DHA and species 1. | |
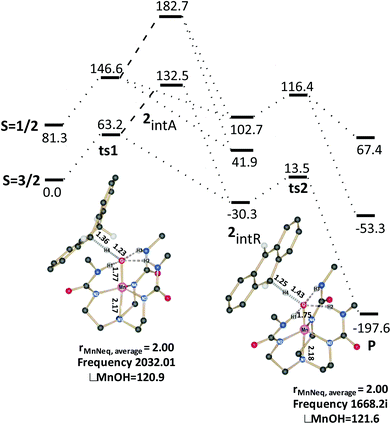 |
| | Fig. 2 Computed reaction profile of C–H activation reaction involving DHA and species 2. | |
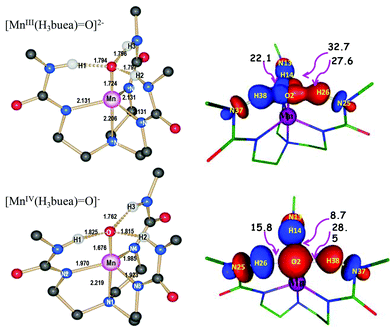 |
| | Fig. 3 (a) Optimized geometries of species 1 and 2 along with geometrical parameters and (b) weak H-bond stabilization interaction between O(LP)→BD*(NH) bonds. | |
 |
| | Fig. 4 Computed spin density plots for ts1, ts2 and 51intR of species 1 and 2. | |
Based on the computed reaction profile diagram (Fig. 1 and 2), it is clear that the C–H activation of DHA by these two oxidants occurs on the high spin surface. The high spin S = 2 surface in 1 is well separated from other spin states which suggests that the excited spin states are unlikely to participate in the reaction mechanism. The barrier for the proton transfer (51ts1) from DHA is estimated to be 54.2 kJ mol−1 and the proton transfer leads to the formation of an anionic intermediate (51intA). This step is estimated to be endothermic (+52.4 kJ mol−1) in nature. In the next step this intermediate undergoes a rapid electron transfer from the anionic DHA to the metal complex and this leads to a radical intermediate (51intR). This radical intermediate is stabilized by 41.3 kJ mol−1 compared to the 51intA intermediate suggesting that the electron transfer is feasible and the reaction proceeds further from the 51intR species. For species 2 the initial hydrogen abstraction (42ts1) from DHA requires a barrier height of 63.2 kJ mol−1 which is slightly higher than that computed for species 1. In contrast to the mechanistic steps described for 1, in 2 the formation of the radical intermediate 42intR is thermodynamically more favored compared to the anionic intermediate 42intA (energy margin of 162.8 kJ mol−1). Also the nature of the transition state advocates the direction of the course of reaction that the species go forth. Comparing just the Mn–O bond distance, it is clear that species 51ts1 is reactant like while the 42ts1 is product like (see Table S1 of ESI‡). In case of 42ts1, significant spin densities are detected at the DHA moiety (group spin densities on the DHA ρDHA = −0.25) suggesting a development of a partial radical character at the transition state (see Fig. 4). The computed energetics and the electronic structure is consistent with earlier theoretical reports on a MnIVO complexes.58 A similar radicaloid character for the transition state has also been reported for the non-heme FeIVO species.59 In 51ts1 the DHA moiety is found to have negligible spin densities (ρDHA = 0.01) but a large accumulated negative charge and this suggests a preference for an anionic type intermediate in this case. For species 1, although the 51intA and 51intA species are not separated by an energy barrier, these two species are related by one electron being in DHA or in the MnIIIO complex. The nature of the transition state clearly reveals that it is converging to an anionic intermediate and does not possess any radical character. Although the anionic and radical intermediate in the potential energy surface is shown to have no barrier, the electron transfer from the MHA− to MH˙ might have some barrier associated with the deformation of MHA molecule, besides the potential energy surface for such reactions are likely to be multidimensional in nature and thus warrant a valence-bond theory based approach60 to further probe the nature of the transition state. As far as other spin states of the metal–oxo complexes are concerned, those states are very high in energy compared to the ground state. Although their structure and bonding are not described here, a similar scenario to the ground state is also witnessed for other excited spin states for both 1 and 2 (see ESI Table S1 and Fig. S1‡ for the optimized structures of the transition states). Put together, all these data clearly indicates that 1 and 2 proceeds via different routes with the former favoring an anionic intermediate while for the latler the reaction is routed through a radical intermediate.
Electronic structure origin of difference in the reactivity
The energy decomposition analysis (EDA) were performed for 1 and 2 by treating H3buea, Mn and O as separate fragments (H3buea + Mn + O, see Table 1). The EDA results for 1 and 2 show that ΔEint for MnIVO is 1.6 times larger than that of the MnIIIO suggesting that species 2 is very stable compared to species 1 and accounts for the fact that 1 is more reactive than 2. A significant contribution to the interaction energy for 2 arise from orbital stabilization and this indicates a stronger Mn–L bond, particularly the Mn–oxo bond for 2 compared to 1.
Table 1 Summary of EDA performed for species 1 and 2. The ΔE are given in kJ mol−1
| Species |
|
{H3buea + Mn + O} |
|
1
|
ΔEint |
−8609.7 |
| ΔEsteric |
−6305.2 |
| ΔEorb |
−2304.5 |
|
2
|
ΔEint |
−14096.2 |
| ΔEsteric |
−9339.0 |
| ΔEorb |
−4757.2 |
The trigonal bipyramidal structure observed in 1 and 2 and its related frontier orbitals are entirely different from that of an octahedral complexes of high-valent manganese or iron–oxo species.61Fig. 5 and 6 show the FMO of the 51 and 42 and their respective first transition states. The occupation of four electrons in 51 is found to be in 
 δxy1
δxy1
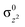 in which the two π* orbitals (π*xz and π*yz) are degenerate, similarly the two δ orbitals (δxy and δx2−y2) are also degenerate and lie 0.35 eV higher in energy with respect to the π* orbitals. The empty σz2 orbital is found to lie much higher at 4.19 eV. In the transition state 51ts1, the degeneracy of the π* and δ orbitals are slightly perturbed and the δ-type orbitals are further destabilized. In the case of 52, the three unpaired electrons are found to be in
in which the two π* orbitals (π*xz and π*yz) are degenerate, similarly the two δ orbitals (δxy and δx2−y2) are also degenerate and lie 0.35 eV higher in energy with respect to the π* orbitals. The empty σz2 orbital is found to lie much higher at 4.19 eV. In the transition state 51ts1, the degeneracy of the π* and δ orbitals are slightly perturbed and the δ-type orbitals are further destabilized. In the case of 52, the three unpaired electrons are found to be in 
 δxy1
δxy1
 orbitals. Unlike in 1, here the degeneracy of the two π* orbitals (π*xz and π*yz) are lifted and the σz2 orbital is significantly destabilized. In the first transition state 42ts1, one of the C–H bond electron is found to be transferred to δx2−y2 orbitals (vide infra) and this reduces the energy gap between the π*xz and π*yz orbitals (see Scheme 2). We would like to note here that earlier report on an octahedral MnIVO species suggest such a transfer to π*xz orbital while here our calculations indicate the electron being transferred to δ type δx2−y2 orbital.59 This orbital has significant density on the oxygen and also the orbital is polarized to accept the electron (see Fig. 6). Such electron transfer does not occur in 51ts1 for two reasons: (i) electron transfer to
orbitals. Unlike in 1, here the degeneracy of the two π* orbitals (π*xz and π*yz) are lifted and the σz2 orbital is significantly destabilized. In the first transition state 42ts1, one of the C–H bond electron is found to be transferred to δx2−y2 orbitals (vide infra) and this reduces the energy gap between the π*xz and π*yz orbitals (see Scheme 2). We would like to note here that earlier report on an octahedral MnIVO species suggest such a transfer to π*xz orbital while here our calculations indicate the electron being transferred to δ type δx2−y2 orbital.59 This orbital has significant density on the oxygen and also the orbital is polarized to accept the electron (see Fig. 6). Such electron transfer does not occur in 51ts1 for two reasons: (i) electron transfer to  demands a very high energy as this orbital is significantly destabilized, (ii) the other option of transferring the down-ward spin electron (β-electron) of the σC–H bond to the singly occupied orbitals will lift the degeneracy and this again will demand a significant energy (see Scheme 2). The above two factors in fact favour electron transfer in 2 where the δx2−y2 orbital is not too high lying (see Scheme 2) and other singly occupied orbitals are non-degenerate and thus the MnIVO readily accepts an electron from the DHA leading to a transition state with a MnIII–OH like character.
demands a very high energy as this orbital is significantly destabilized, (ii) the other option of transferring the down-ward spin electron (β-electron) of the σC–H bond to the singly occupied orbitals will lift the degeneracy and this again will demand a significant energy (see Scheme 2). The above two factors in fact favour electron transfer in 2 where the δx2−y2 orbital is not too high lying (see Scheme 2) and other singly occupied orbitals are non-degenerate and thus the MnIVO readily accepts an electron from the DHA leading to a transition state with a MnIII–OH like character.
 |
| | Fig. 5 Schematic MO diagrams of species 1 and its corresponding first transition state 51ts1. The energy differences given here are in eV. | |
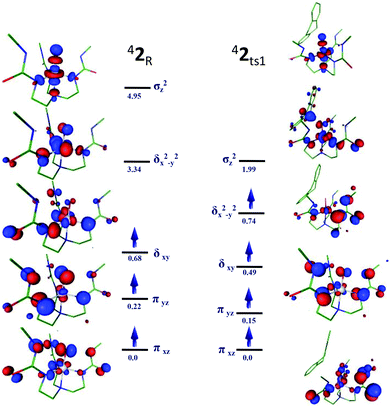 |
| | Fig. 6 Schematic MO diagrams of species 2 and its corresponding first transition state 42ts1. The energy differences given here are in eV. | |
 |
| | Scheme 2 The feasible electron shifts between the σC–H bond and Mn–O d-based orbitals for MnIIIO and MnIVO species. | |
Thermodynamic rational for the difference in the reactivity
To understand the origin of the difference in the mechanism and to probe various possible pathways by which the hydrogen atom transfer can takes place (PT-ET, ET-PT and PCET), a thermodynamic formation energy cycle for the MnIIIO and MnIVO has been constructed62 and is shown in Fig. 7. A quick look at the thermodynamic energies suggests a favourable PCET for both the species, however if the kinetic results discussed above are assimilated, one can easily differentiate the subtle mechanistic change between the two species. The first transition state for the MnIIIO species is clearly a PT transition state as discussed above and thermodynamics suggest that this step is endothermic by 43.1 kJ mol−1 and the subsequent ET step more than compensate the energy loss, leading to the final [MnII–OH] product. Given the barrier height of 52.5 kJ mol−1 for the first transition state, a meta-stable [MnIII–OH]+ species is certainly feasible (note the characterized transition state has a strong MnIII–OH character, see below) and thus one can suggest a PT-ET mechanism is operational for this species. On the other hand, the ET-PT mechanism is unlikely as the ET step is exceedingly endothermic compared to the transition state barrier, thus the transition state is unlikely to converge to a [MnIIO] meta-stable species. The spin density plots of MnIIIO, 51ts1 and high-spin state of [MnIII–OH] are shown in Fig. 4. As one can see the transition state clearly resembles [MnIII–OH] type species (3.776 vs. 3.783) and this affirmatively suggests that the reaction proceed through a PT-ET mechanism here and not a through a PCET or ET-PT mechanism.
 |
| | Fig. 7 Relative thermodynamic free energies between MnIII/IVO and its hydroxo complexes. | |
Unlike 1, for species 2 both the initial PT (for PT-ET mechanism) and the alternative initial ET (for ET-PT mechanism, see Fig. 7) steps are exceedingly endothermic and are much higher than the barrier height computed for 42ts1 (63.2 kJ mol−1). The spin density plots for the MnIVO, 42ts1 and MnIII–OH are shown in Fig. 4 clearly suggest that the transition state here also resembles the [MnIII–OH] type species (spin density values 2.60 vs. 3.40 in 2 and 42ts1 respectively) and not [MnIV–OH]+ and this clearly directs the discussion towards a concerted PCET mechanism for species 2. This step is an energetically favourable process (−41.7 kJ mol−1). The reason for the difference between 1 and 2 is related to the stability of the electronically preferred [MnIII–OH] type species, independent of the starting oxidation state of the oxo species. As the Mn–O bonds are stronger and less basic in MnIVO compared to MnIIIO, a protonated meta-stable intermediate for the MnIVO is extremely unstable and this leads to a switch in the mechanism. Thus the thermodynamic cycle constructed essentially verifies the mechanistic insights discussed earlier and provide confidence to the established mechanism for species 1 and 2.
Second HAT reaction
The second hydrogen abstraction takes place (51ts2) with the barrier of 21.3 kJ mol−1 and leads to the formation of anthracene in 1. For 2, the barrier height for second hydrogen abstraction (42ts2) is computed to be 43.8 kJ mol−1. In accord to the first step the second hydrogen atom transfer reaction is also found to proceed via the similar mechanism, i.e. species 1 is found to follow a PT-ET mechanism while 2 is found to follow a PCET path. This is again easily visible from the spin density plots computed for the 51ts2 and 42ts2 species (see Fig. 4) where again both the species resemble a [MnIII–OH] complex as discussed for the first hydrogen atom transfer reaction.
Correlation to experiments
Comparing both the transition states ts1 and ts2 barrier heights for both the species, it is clear that for both the species the first transition state is the rate-limiting. The computed barrier height of 54.2 (for 1) and 63.2 kJ mol−1 for the rate-determining step correlates well with the experimental kinetic data (75.3 and 79.5 kJ mol−1 for species 1 and 2 respectively), although the absolute values are slightly underestimated.18 Besides the computed imaginary frequencies corresponding to the transition states 51ts1 and 51ts2 are 1280.9i and 1191.7i respectively while for 42ts1 and 42ts2 are 2032i and 1668.2i respectively. Larger imaginary frequencies are obtained for species 2 compared to species 1 and this suggest a large tunneling contribution for 2 and a narrow and sharper reaction barrier than that of 1. This is clearly envisaged from the larger kinetic isotopic effect (KIE) values (6.8) obtained for species 2.
Conclusions
DFT calculation have been used to investigate the kinetic aspects of the C–H bond activation reactions of monomeric MnIII/IVoxo units with tetradentate tripodal ligands. The initial H-abstraction on the high spin surface is found to be the rate determining step for both the species 1 and 2. From a detailed electronic and structural analysis of the transition states along with the computation of reaction energies of various species in the hydrogen atom transfer reaction, our calculations unequivocally suggest that the highly reactive 1 prefers to undergo a step-wise proton transfer followed by electron transfer (PT-ET) mechanism while relatively less reactive 2 prefers a concerted proton-coupled electron transfer (PCET) path (see Scheme 3). This drastic difference in reactivity between 1 and 2 is mainly attributed to the strength of the MnO bond, its basicity and the nature of electron delocalization during the C–H bond activation. Interestingly the computed transition state for the hydrogen atom transfer reactions for the MnIIIO and MnIVO species reveal the course of the reaction where structurally and electronically resembling [MnIII–OH] like transition state has been detected for both the species. This essentially en route to a PT-ET type mechanism for 1 and a PCET mechanism for species 2. All our computed results are in excellent agreement to the experimental reports.
 |
| | Scheme 3 A schematic DFT computed mechanism for HAT reaction by MnIIIO and MnIVO complexes. | |
Although the presented results are likely to be general for MnIII/IVO species with different set of ligand architecture, factors which are expected to directly affect the Mn–O bond covalency (such as H-bonding interaction etc.) can lead to some difference in the predicted reactivity pattern.
To this end, here for the first time using DFT methods we have demonstrated that a subtle mechanistic difference in hydrogen atom transfer reactions for such large metal–oxo complexes can be apprehended if an apt bunch of computational tools are employed (PES modelling, reaction energy computation, MO/NBO analysis). This procedure will be expanded to other examples in future.
Acknowledgements
GR would like to acknowledge financial support from the Government of India through the Department of Science and Technology (SR/S1/IC-41/2010; SR/NM/NS-1119/2011) and Indian Institute of Technology, Bombay to access the high performance computing facility. MJ would like to thank IITB for postdoctoral fellowship, AA to CSIR New Delhi for JRF fellowship and BP would like to thank UGC New Delhi for JRF fellowship.
Notes and references
-
(a)
O. Hayaishi, Molecular Mechanism of Oxygen Activation, Academic Press, New York, 1974 Search PubMed;
(b) M.-H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385–2419 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
-
(a) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed;
(b) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- L. Que Jr. and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607–2624 CrossRef.
- A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC.
-
(a)
Cytochrome P450: Structure, Mechanism and Biochemistry, ed. P. R. Ortiz de Montellano, Plenum, New York, 1995 Search PubMed;
(b) J. M. Bollinger Jr. and C. Krebs, J. Inorg. Biochem., 2006, 100, 586–605 CrossRef PubMed.
- L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger Jr. and C. Krebs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738–14743 CrossRef CAS PubMed.
- D. P. Galonic, E. W. Barr, C. T. Walsh, J. M. Bollinger Jr. and C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116 CrossRef CAS PubMed.
- C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger Jr., Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que Jr., S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed.
- D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119–5125 CrossRef CAS PubMed.
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS PubMed.
- K. A. Prokop, S. P. de Visser and D. P. Goldberg, Angew. Chem., Int. Ed., 2010, 49, 5091–5095 CrossRef CAS PubMed.
- T. H. Parsell, M.-Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762–2763 CrossRef CAS PubMed.
- J. P. Roth and J. M. Mayer, Inorg. Chem., 1999, 38, 2760–2761 CrossRef CAS PubMed.
- Y. Wang, H. Hirao, H. Chen, H. Onaka, S. Nagano and S. Shaik, J. Am. Chem. Soc., 2008, 130, 7170–7171 CrossRef CAS PubMed.
- Y. Wang, H. Chen, M. Makino, Y. Shiro, S. Nagano, S. Asamizu, H. Onaka and S. Shaik, J. Am. Chem. Soc., 2009, 131, 6748–6762 CrossRef CAS PubMed.
- J. J. Warren, T.A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- O. Tishchenko, D. G. Truhlar, A. Ceulemans and M. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 7000–7010 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. V. Soudackov, J. Phys. Chem. B, 2008, 112, 14108–14123 CrossRef CAS PubMed.
-
M. J. Frisch, et al., GAUSSIAN 09 (Revision 02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- H. Hirao, D. Kumar, L. Que and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590–8606 CrossRef CAS PubMed.
- C. M. Bathelt, J. Zurek, A. J. Mulholland and J. N. Harvey, J. Am. Chem. Soc., 2005, 127, 12900–12908 CrossRef CAS PubMed.
- P. E. M. Siegbahn and T. Borowski, Acc. Chem. Res., 2006, 39, 729–738 CrossRef CAS PubMed.
-
T. H. J. Dunning and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976, vol. 3, p. 1 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- C. Schaefer, C. Huber and R. Ahlrichs, Chem. Phys., 1994, 100, 5829–5835 Search PubMed.
-
P. Fluckiger, H. P. Luthi, S. Portmann and J. Weber, Molekel 4.3, Swiss Center for Scientific Computing, Manno, Switzerland, 2000 Search PubMed.
- S. Portmann and H. P. Luthi, Chimia, 2000, 54, 766–770 CAS.
- L. Noodleman and W. G. Han, JBIC, J. Biol. Inorg. Chem., 2006, 11, 674–694 CrossRef CAS PubMed.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- H. M. von and G. Frenking, Comput. Mol. Sci., 2012, 2, 43–62 CrossRef.
- T. Ziegler and A. Rauk, Theor. Chem. Acc., 1977, 46, 1–10 CrossRef CAS.
- S. I. Gorelsky, S. Ghosh and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 278–290 CrossRef CAS PubMed.
-
S. I. Gorelsky, AOMIX, Department of Chemistry, York University, Toronto, ON, http://www.sg-chem.net, 2011 Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young, G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 2556–2567 CrossRef CAS PubMed.
- R. Gupta, C. E. MacBeth, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2002, 124, 1136–1137 CrossRef CAS PubMed.
- D. C. Lacy, J. Mukherjee, R. L. Lucas, V. W. Day and A. S. Borovik, Polyhedron, 2013, 52, 261–267 CrossRef CAS PubMed.
- Z. Shirin, V. G. Young Jr. and A. S. Borovik, Chem. Commun., 1997, 1967–1968 RSC.
- Z. Shirin, B. S. Hammes, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2000, 122, 1836–1837 CrossRef CAS.
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 5810–5817 CrossRef CAS PubMed.
- R. Latifi, M. A. Sainna, E. V. Rybak-Akimova and S. P. de Visser, Chem.–Eur. J., 2013, 19, 4058–4068 CrossRef CAS PubMed.
- A. S. Borovik, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed.
- T. H. Parsell, R. K. Behan, M. T. Green, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2006, 128, 8728–8729 CrossRef CAS PubMed.
- C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young Jr., G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 2556–2567 CrossRef CAS PubMed.
- A. Dey, R. K. Hocking, P. Larsen, A. S. Borovik, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 9825–9833 CrossRef CAS PubMed.
- J. F. Hull, D. Balcells, E. L. O. Sauer, C. Raynaud, G. W. Brudvig, R. H. Crabtree and O. Eisenstein, J. Am. Chem. Soc., 2010, 132, 7605–7616 CrossRef CAS PubMed.
- K.-B. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2012, 3, 2851–2856 CrossRef CAS.
- C. Li, D. Danovich and S. Shaik, Chem. Sci., 2012, 3, 1903–1918 RSC.
-
(a) D. Janardanan, Y. Wang, P. Schyman, L. Que Jr. and S. Shaik, Angew. Chem., Int. Ed., 2010, 49, 3342–3345 CrossRef CAS PubMed;
(b) R. Latifi, L. Tahsini, B. Karamzadeh, N. Safari, W. Nam and S. P. de Visser, Arch. Biochem. Biophys., 2011, 507, 4–13 CrossRef CAS PubMed;
(c) A. Ansari, A. Kaushik and G. Rajaraman, J. Am. Chem. Soc., 2013, 135, 4235–4249 CrossRef CAS PubMed;
(d) M. Jaccob and G. Rajaraman, Dalton Trans., 2012, 41, 10430–10439 RSC.
-
(a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed;
(b) T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed;
(c) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Peter Comba, University of Heidelberg, Germany, on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: DFT computed structural parameters, spin density values and optimized structures of higher spin states. See DOI: 10.1039/c3dt52290c |
| § Present address: Department of Chemistry, Loyola College, Chennai-600034, Tamilnadu, India. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a