 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lanthanide(III) complexes of aminoethyl-DO3A as PARACEST contrast agents based on decoordination of the weakly bound amino group†
Tereza Krchováa, Jan Kotek*a, Daniel Jirákb, Jana Havlíčkováa, Ivana Císařováa and Petr Hermanna
aDepartment of Inorganic Chemistry, Universita Karlova (Charles University), Hlavova 2030, 128 40 Prague 2, Czech Republic. E-mail: modrej@natur.cuni.cz; Fax: +420-22195-1253; Tel: +420-22195-1261
bDepartment of Radiodiagnostic and Interventional Radiology, Magnetic Resonance Unit, Institute of Clinical and Experimental Medicine, Vídeňská 1958/9, 140 21 Prague 4, Czech Republic
First published on 27th August 2013
Abstract
2-Aminoethyl DOTA analogues with unsubstituted (H3L1), monomethylated (H3L2) and dimethylated (H3L3) amino groups were prepared by improved synthetic procedures. Their solid-state structures exhibit an extensive system of intramolecular hydrogen bonds, which is probably present in solution and leads to the rather high value of the last dissociation constant. The protonation sequence of H3L1 in solution corresponds to that found in the solid state. The stability constants of the H3L1 complexes with La3+ and Gd3+ (20.02 and 22.23, respectively) are similar to those of DO3A and the reduction of the pKA value of the pendant amino group from 10.51 in the free ligand to 6.06 and 5.83 in the La3+ and Gd3+ complexes, respectively, points to coordination of the amino group. It was confirmed in the solid state structure of the [Yb(L1)] complex, where disorder between the SA′ and TSA′ isomers was found. A similar situation is expected in solution, where a fast equilibration among the isomers hampers the unambiguous determination of the isomer ratio in solution. The PARACEST effect was observed in Eu(III)–H3L1/H3L2 and Yb(III)–H3L1/H3L2 complexes, being dependent on pH in the region of 4.5–7.5 and pH-independent in more alkaline solutions. The decrease of the PARACEST effect parallels with the increasing abundance of the complex protonated species, where the pendant amino group is not coordinating. Surprisingly, a small PARACEST effect was also observed in solutions of Eu(III)/Yb(III)–H3L3 complexes, where the pendant amino group is dimethylated. The effect is detectable in a narrow pH region, where both protonated and deprotonated complex species are present in equilibrium. The data points to the new mechanism of the PARACEST effect, where the slow coordination–decoordination of the pendant amine is coupled with the fast proton exchange between the free amino group and bulk water mediates the magnetization transfer. The pH-dependence of the effect was proved to be measurable by MRI and, thus, the complexes extend the family of pH-sensitive probes.
Introduction
Contrast agents (CAs) that alternate the properties of bulk water have been used for a long time to improve the resolution and utilization of magnetic resonance imaging (MRI) in medicine and molecular biology. The classical molecular CAs are based on complexes of highly paramagnetic metal ions, mainly on trivalent gadolinium.1 The complexes change the relaxation times of the bulk water protons most frequently through the exchange of the whole coordinated water molecule. Another class of CAs is based on a completely different principle, a chemical exchange saturation transfer (CEST), was introduced some time ago.2 The method is based on irradiation (saturation) of protons exchangeable with bulk water protons leading, after the chemical exchange, to a decrease of the bulk water proton signal. As signals of the bulk water protons and the protons to be irradiated should be separated from each other as much as possible to reduce non-specific proton irradiation, paramagnetic complexes causing the extension of the NMR chemical shift scale were suggested (PARACEST).2,3 Most of these CAs are based on lanthanide(III) ions having suitable magnetic properties which are bound in macrocyclic ligands derived from DOTA (e.g., DOTA-tetraamides, Scheme 1), ensuring the high thermodynamic stability and kinetic inertness of the complexes. The exchangeable protons in the complexes should be endowed with suitable properties and the most important one is the rate of their exchange with bulk water. There are two proton pools exhibiting PARACEST effects in Ln(III)–DOTA-tetraamide complexes: (i) protons of a coordinated water molecule if it is in a slow-exchange region, typically in the microsecond range (e.g. [Yb(H2O)(DOTA-4AmCE)] complex (τM = 3 μs)4 or [Eu(H2O)(DOTAM)],5 [Eu(H2O)(DOTA-4AmCE)],6 and [Eu(H2O)(DOTA-4AmP)]4 complexes (τM = 55, 382 and 67 μs, respectively; for ligand structures see Scheme 1). The water molecule is bound directly to the metal ion and is close to the magnetic axis of the complexes and, thus, its protons exhibit a maximized difference in their chemical shift from the bulk water protons. (ii) Protons of O-coordinated amide group(s); however, these protons have their chemical shift much closer to the bulk water resonance. Nevertheless, their exchange depends on a number of external factors (e.g., τM for [Yb(DOTAM)]3+ lays, with a dependence on the pH, in the 400–5000 μs range7) and, therefore, the Ln(III) complexes of DOTA amide derivatives can be used as probes for various physiological parameters, such as pH, temperature, metabolite or metal ion concentrations, etc.2c,3 | ||
| Scheme 1 Ligands discussed in the paper. | ||
More recently, other PARACEST CAs based on different ligands (and, thus, on different pools of exchangeable protons) and/or metal ions have been suggested. Protons of the hydroxyl group are also labile and lanthanide(III) complexes of cyclen derivatives with alcohol pendant arms have been shown to exhibit a PARACEST effect.8 Some transition metal ions can also present suitable magnetic properties and complexes of high-spin iron(II) or nickel(II) with derivatives of cyclam, cyclen or 1,4,7-triazacyclononane having 2-hydroxypropyl, acetamide (e.g., DOTAM and NOTAM, see Scheme 1) or (5-aminopyridine-2-yl)methyl pendant arms have been shown to have a pronounced PARACEST effect.9–11 Another pool of exchangeable protons, a distant non-coordinated amino group, has also been explored in some complexes of lanthanide(III)12 as well as transition metal9 ions.
In this work, we decided to investigate the PARACEST-related properties of complexes, where the CEST-causing amino group is directly coordinated to the central lanthanide(III) ions. To ensure complex stability, the ligands are based on a macrocyclic cyclen skeleton having three acetate and one 2-aminoethyl pendant arms, which contain primary (H3L1), partially methylated secondary (H3L2) and dimethylated tertiary (H3L3) amino groups, see Scheme 2. Some of these ligands have already been investigated as bifunctional ligands for gadolinium(III)-based targeted MRI CAs13 or radionuclides,14 and, during work on this project, as pH-sensitive gadolinium(III)-based positive MRI CAs.15
 | ||
| Scheme 2 Studied ligands. | ||
Results and discussion
Synthesis
The ligands were synthesized (Schemes 3 and 4) by reaction of t-Bu3DO3A with the appropriate amine-containing precursor. Alkylation of t-Bu3DO3A with N-(2-bromoethyl)-phthalimide followed by sequential deprotection by trifluoroacetic acid and hydrazine led, after chromatography on anion exchanger, to zwitterionic H3L1 in 59% overall yield (Scheme 3). The synthesis is more simple compared to the previously published procedure,14 and is easily scalable and avoids HPLC purification. | ||
Scheme 3 Synthesis of studied ligands H3L1 and H3L3: (i) DMF, K2CO3, TBAB, 40 °C; (ii) t-Bu3DO3A·HBr, MeCN, K2CO3, 60 °C; (iii) CF3COOH–CHCl3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux 24 h; (iv) aq. NH2NH2, 90 °C, 18 h; (v) (CH2O)n, HCOOH, reflux 24 h. :1), reflux 24 h; (iv) aq. NH2NH2, 90 °C, 18 h; (v) (CH2O)n, HCOOH, reflux 24 h. | ||
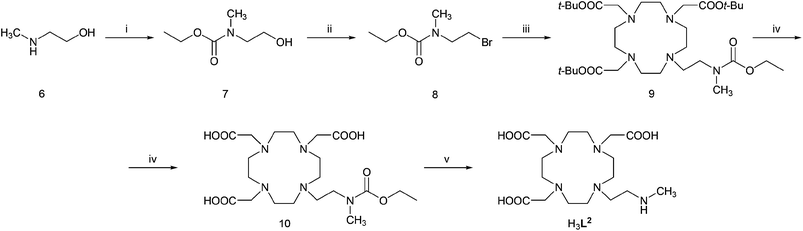 | ||
| Scheme 4 Synthesis of studied ligand H3L2: (i) CH3CH2OC(O)Cl, dioxan–H2O (1:1), RT, 2 h; (ii) CBr4, PPh3, THF, RT, 1 h; (iii) t-Bu3DO3A·HBr, K2CO3, MeCN, 60 °C; (iv) CF3COOH–CHCl3 (1:1), reflux 24 h; (v) 10% aq. NaOH, 90 °C, 24 h. | ||
To obtain ligand H3L2, 2-[N-(ethyloxycarbonyl)-N-methylamino]bromoethane was used as the alkylation agent reacting with t-Bu3DO3A and, after deprotection using trifluoroacetic acid and 10% aq. NaOH and chromatography on ion exchange resins, the ligand was isolated as a zwitterion in 30% overall yield (Scheme 4). Direct methylation of H3L1 with a formaldehyde–formic acid mixture easily produced H3L3 in high yield (Scheme 3); it is a significant improvement compared with the previous synthesis.15
All three studied ligands were structurally characterized in their zwitterionic forms by single-crystal X-ray diffraction analysis. Single-crystals of sufficient quality were obtained from solutions in water or aqueous ethanol. Title ligands were isolated in the form of hydrates (H3L1·5H2O, H3L2·6H2O and H3L3·3.5H2O, respectively). All three molecular structures are very similar, and therefore, only the molecular structure of H3L1 is shown here in Fig. 1; other structures are shown in the ESI (Fig. S1 and S2†). In all three cases, the protonation scheme is the same – two protons are bound to the macrocycle amino groups bearing acetate moieties and are located mutually trans, and the third one is bound to the pendant aminoethyl group. Such a protonation scheme is consistent with the form suggested on the basis of 1H NMR titration (see below, Scheme S1†). The conformation of the macrocyclic unit is (3,3,3,3)-B, as usually observed for double-protonated cyclen rings (Table S1†).16 Such a conformation is stabilized by intramolecular hydrogen bonds between the protonated and non-protonated macrocycle amino groups. Beside these interactions, the molecular structure is stabilized by further intramolecular hydrogen bonds, including protonated amines and deprotonated oxygen atoms of the carboxylate pendant arms, and the whole crystal structure is stabilized by extended hydrogen bond networks with water solvate molecules.
 | ||
| Fig. 1 Molecular structure of H3L1 found in the crystal structure of H3L1·5H2O showing an intramolecular hydrogen bond network. The carbon-bound hydrogen atoms were omitted for clarity. | ||
Stability of the complexes
The complexing properties of the most important ligand, H3L1, having a primary amino group, were investigated in detail. First, the conditions for the pendant amino group deprotonation and coordination were investigated by potentiometry. The ligand’s overall protonation constants βh10 were determined by potentiometry and are compiled together with corresponding pKA values in Table 1; the ligand distribution diagram is shown in Fig. S3.† As the highest protonation constant β110 is slightly behind the −log[H+] range suitable for potentiometric titrations, we also performed a 1H NMR titration in the pH range 1.2–13.5 to confirm such a high value of the first protonation constant and to assign the individual protonation sites. The values of the dissociation constants obtained by NMR (pKA = 13.3(3), 11.0(3), 9.3(2), 3.8(1) and 1.3(3)) are in a good agreement with those obtained by potentiometry (Table 1); the differences can be attributed to the different and non-controlled ionic strength during the NMR experiment and inaccurate (non-linear) potential–pH relationship in highly acidic/alkaline regions. However, the high value of the first protonation constant is confirmed and is clearly seen from the chemical shift dependence in the pH region 12.0–13.5 (Fig. S4†). In addition, it is possible, from pH-dependent changes of individual 1H NMR signals, to determine also the sites of consecutive protonations (Scheme S1†): the first proton is distributed over all ring nitrogen atoms; the rather high value of the corresponding protonation constant is a consequence of intramolecular hydrogen bonds involving the ring nitrogen atoms as well as the pendant arms. Two pKA values can be assigned to the partly simultaneous protonation of the pendant amino group and the second nitrogen atom of the macrocyclic ring, associated with re-location of the protons bound to the ring nitrogen atoms. The same proton distribution was also found in the zwitterionic forms of all studied ligands in the solid state (see above). The last two measured constants correspond to the protonation of the pendant carboxylate groups. Further protonations of the ring nitrogen atoms take place below pH 1. Such a protonation sequence is consistent with those commonly found for all DOTA-like ligands.The stability constants, log β011, determined for La(III) and Gd(III) ions were found to be 20.02 and 22.23, respectively (Table 1). Corresponding distribution diagrams are shown in Fig. S5 and S6.† The stability constants are lower than those reported for Ln(III)–DOTA (La(III) 22.9, Gd(III) 24.6) complexes and similar to those of Ln(III)–DO3A (Gd(III) 21.0) complexes.17 In both studied systems, protonated species with pKA 6.06 and 5.83 for the La(III) and Gd(III) complexes, respectively, are formed. The value for the protonation of the Gd(III)–H3L1 complex is in good agreement with the reported value (5.95) determined by relaxometric measurements.15 The distribution diagrams show that free lanthanide(III) ions are not present above pH 5.5. The protonation takes place on the pendant amino group as it is the most basic site of the in-cage complex molecule and a difference of ∼4–5 orders of magnitude between the constants, corresponding to its protonation in the complexes and analogous protonation in the free ligand, points to an easier deprotonation induced by coordination of the group to the metal ion. Thus, one can conclude, that studied ligands are coordinated in an octadentate fashion involving the pendant amino group in the [Ln(L1)] species, and that the amino group is decoordinated upon protonation in the weakly acidic region. The maximum abundance of the [Gd(HL1)]+ complex species in solution is observed at pH ∼5.
Structure of the complexes
To study the structure and properties of the lanthanide(III) complexes of H3L1, sample solutions were prepared by dissolving LnCl3 and the ligand in a Ln:L = 1:1.1 molar ratio, adjusting the pH with aq. LiOH to 6.0, heating the solution at 60 °C overnight, re-adjusting pH to 6.5 and heating at 60 °C overnight again.The complexes of the DOTA-like ligands are usually nona-coordinated (eight donor atoms coming from macrocyclic ligands and one site is occupied by a water molecule) and form two possible diastereomers – square-antiprismatic (SA) and twisted-square-antiprismatic (TSA) ones.1 These diastereoisomers differ in the relative screwing of the macrocyclic (δ/λ) and pendant (Δ/Λ) chelate rings, giving rise to Δλλλλ/Λδδδδ (SA) and Δδδδδ/Λλλλλ (TSA) combinations. In the 1H NMR spectra, the signals showing best the isomer ratio belongs to those of the “axial” protons of the macrocyclic chelate rings, which are the closest ones to the lanthanide(III) ion and to the magnetic axis of the complexes.18 In the Ln(III)–DOTA-like complexes, the TSA isomer with a larger coordination cage is strongly preferred at the beginning of the lanthanide series, with the SA one becoming dominant for heavier lanthanides.18 Although the situation is sometimes complicated by the possible presence of an octa-coordinated species (i.e., without a coordinated water molecule, usually denoted as SA′/TSA′), which have a similar chemical shift range in the 1H NMR spectra to the nona-coordinated SA/TSA species. Due to this fact, the abundance of twisted-square-antiprismatic isomers can slightly increase towards the end of the lanthanide series.18,19
The solution structures of the studied complexes were investigated by variable-temperature 1H NMR. To assure the coordination of the pendant amino group, the pH of the samples was adjusted to the weakly alkaline region. However, the spectrum of the Eu(III) complex at pH 9 (Fig. S7†) acquired at 25 °C was not resolved enough to observe the required signals and to determine the isomer ratio; only very broad signals in the 17–27 ppm region (after correction for bulk magnetic susceptibility shift) were observed. Such chemical shifts are just between the regions typical for SA and TSA species, although slightly closer to the TSA one.18 Both heating up to 90 °C or cooling to 0 °C led to a significant broadening and visual disappearance of these signals. It points to the fact that two relatively independent fluxional processes occur, affecting the position of the axial hydrogen atoms with respect to the lanthanide(III) ion. Therefore, we can only speculate that a mixture of the isomers is present, as it is common for most of Eu(III) complexes with DOTA-like ligands.18,19 A similar situation occurs for the Yb(III) complex. The 1H NMR spectrum acquired at 25 °C and pH 8.5 (Fig. S8†) shows signals of “axial” protons as broad peaks in the 86–112 ppm region. The chemical shifts,18,19 signal broadening and their virtual disappearance, observed either at lower and higher temperatures, point to the presence of a SA(′)/TSA(′) exchange process.
The hypothesis of ongoing SA(′)/TSA(′) isomerism is supported by the solid-state structure of the [Yb(L1)]·5H2O complex (Fig. 2). The central ion is octacoordinated between mutually parallel macrocycle N4- and pendant-arm O3N-planes, but closer to the pendant-arm one. As a result of smaller values of the trans (“opening”) angles in the O3N-plane (120° and 125° for O311–Yb1–N52 and O211–Yb1–O411 angles, respectively), comparing to the limiting value (∼135°) for water coordination,20 no water molecule is directly bound to the metal centre. During the refining of the crystal structure, the set of difference maxima in the electron density map were located close to the nitrogen and carbon atoms of the macrocycle, and pointed to some disorder in the macrocyclic unit. It was successfully modelled, leading to a calculated ratio of the SA′:TSA′ isomers of 15:85% (Fig. 2 and S9†). The structural parameters of both coordination spheres fall into regions typical for the given isomers,20 with mean torsion angles between N4- and O3N-planes of 37.5 and 22.5°, and separation of the N4- and O3N-planes of 2.50 and 2.59 Å for the SA′ and TSA′ species, respectively. Further selected geometric parameters are compiled in Table S2.†
![Molecular structure of [Yb(L1)] found in the crystal structure of [Yb(L1)]·5H2O; TSA′ (Λλλλλ, 85%) species (A) and SA′ (Λδδδδ, 15%) species (B). Carbon-bound H-atoms are omitted for clarity.](/image/article/2013/DT/c3dt52031e/c3dt52031e-f2.gif) | ||
| Fig. 2 Molecular structure of [Yb(L1)] found in the crystal structure of [Yb(L1)]·5H2O; TSA′ (Λλλλλ, 85%) species (A) and SA′ (Λδδδδ, 15%) species (B). Carbon-bound H-atoms are omitted for clarity. | ||
Chemical exchange saturation transfer
PARACEST experiments (measurements of so-called Z-spectra) produced the clean saturation of bulk water after irradiation of the broad regions with maxima at +19.5 and +34 ppm for the Eu(III)–H3L1 complex (pH = 7.67, t = 25 °C), and +42 ppm and +89 ppm for the Yb(III)–H3L1 complex (pH = 7.40, t = 25 °C); chemical shifts are given with respect to the signal of the bulk water protons. In the 1H NMR spectra of the Eu(III) complex acquired in aqueous solution, a broad signal at 34 ppm is observable, and disappears on selective water pre-saturation or when acquiring the spectra in D2O (Fig. S10†). The other signal is overlapped with C–H proton signals and, therefore, cannot be distinguished in the 1H NMR spectra. Also in the case of the Yb(III) complex, there are C–H proton signals in the regions corresponding to the CEST effect and, thus, it prevents the direct observation of the signals.For both complexes, the two observed CEST signals have equal intensities. With respect to the chemical structure of the complexes, the observed effect can be theoretically caused by two exchangeable pools of protons, i.e., by protons of the amino group of the pendant arm and/or by protons of the coordinated water molecule. From relaxometric data, it is known that one water molecule is coordinated in the Gd(III) complex,15 and this fact is also undoubtedly valid for the complex of the slightly larger Eu(III) ion (but it cannot be easily confirmed by the normally used luminescence measurements due to the presence of two types of quenchers, O–H and N–H, which complicates the data evaluation). However, in the case of the complex with the significantly smaller Yb(III), an anhydrous species might be expected on the basis of the solid-state structure (see above). In such cases, three possible elucidations can be considered: (i) both signals come from the amino group, (ii) one signal belongs to the coordinated water molecule and other to the amine, and (iii) both signals belong to the water molecule. To distinguish between these possibilities, more detailed studies were carried out. The most interesting CEST behaviour was observed while changing the solution pH. The CEST effect started to be observable at pH ∼6 for both of the Eu(III) and Yb(III)–H3L1 complexes and reached a maximum at pH ∼8, and the saturation transfer efficiency was not changed with further pH increases up to ∼12; the mutual intensity ratio of the CEST peaks is not changed with pH. The results are shown in Fig. 3A and 3B and strongly support the first possibility, as when the CEST effect is caused by the coordinated water molecule (or by exchangeable protons of the amide7 or hydroxy8 moieties), the effect drops with increasing pH due to the faster prototropic (base-catalyzed) exchange of the corresponding protons (of the coordinated water molecule or the other mentioned pools). The behaviour reported in this work has not yet been reported in the literature. The magnitude of the CEST effect at different pH values follows an abundance of the fully deprotonated species – the CEST effect consistently disappears at lower pH with protonation and, thus, decoordination of the amino group (see potentiometric results, Table 1 and Fig. S5 and S6†). The observations mentioned above are fully consistent with the low “acidity” of the N–H bond in the studied amino derivative if compared to the generally higher acidity of the N–H proton in carboxylic amides; therefore, in the case of DOTAM complexes, base-catalysed proton exchange significantly decreases the CEST effect in alkaline solutions but, in the case of our complexes, the CEST effect remains unchanged even at very high pH.
 | ||
| Fig. 3 Z-spectra of a 80 mM aqueous solution (H2O–D2O 1:10) of Eu(III)–H3L1 complex (B0 = 7.05 T, satpwr = 29 dB ∼1000 Hz, satdly = 2 s) at T = 298 K (A) and pH = 7.40 (C). Z-spectra of a 100 mM aqueous solution (H2O–D2O 1:10) of Yb(III)–H3L1 complex (B0 = 7.05 T, satpwr = 29 dB ∼1000 Hz, satdly = 2 s) at T = 298 K (B) and pH = 7.40 (D). | ||
Thus, the observed CEST signals are assigned to hydrogen atoms of the coordinated pendant amino group. Two observed signals rise from the fact that two amine hydrogen atoms become magnetically non-equivalent after the group coordination. A similar situation has been observed for the amide hydrogen atoms of DOTAM (Scheme 1).7 The lanthanide-induced shift of the amine hydrogen atoms is higher than that of the amide hydrogen atoms in analogous complexes, as the coordinated amine group is much closer to the metal ion and to the magnetic axis of the complexes. The possibility that two signals arise from the presence of two (SA and TSA) isomers could be excluded as both signals have an equal intensity and it would imply that the SA and TSA isomers have the same abundance for both complexes, which is very improbable due to the general trends along the lanthanide series. In addition, as both isomers should have a very similar geometry of the N3O-plane, the NMR signals associated with the protons of the amino pendant arm should have similar chemical shifts in both the SA and TSA species; however, there is a significant difference in the chemical shifts between the signals (Δδ ∼15 and 50 ppm for Eu(III) and Yb(III) complexes, respectively). With increasing temperature, the saturation transfer becomes more effective and two amine hydrogen signals converge and, finally, coalescence around 55 °C and 65 °C for the Eu(III) and Yb(III) complexes, respectively (Fig. 3C and 3D). On further heating (up to 95 °C, Fig. S11†), the coalesced CEST signals approach slightly closer to signal of the bulk water but are still observable. Such a behaviour points to the dynamic averaging of the non-equivalent amine hydrogen signals; it can originate from a faster coordination–decoordination equilibrium or from a faster geometry change.
Due to the insensitivity of the CEST effect to pH changes in the alkaline region, the process described above cannot be caused by neither hydroxide nor proton attack on the coordinated amino group. Therefore, the mechanism of the observed CEST effect is very probably associated with the (semi)labile coordination of the 2-aminoethyl pendant arm. Its coordination–decoordination is the rate-limiting step – when the pendant arm is coordinated, no exchange occurs but in the moment of the pendant arm decoordination, the exchange of protons of the free amino group with those of bulk water proceeds very quickly.
To support the hypothesis presented above, we prepared and studied complexes of mono- and dimethylated analogues, i.e., with ligands H3L2 and H3L3. The complexes of H3L2 showed significant CEST effects, observed as two close peaks with unequal intensity at 43 and 49.5 ppm for Eu(III) and one peak at 57 ppm for Yb(III), respectively (Fig. S12†). The two signals in the Z-spectra, observed in the case of the Eu(III) complex, can be attributed to two modes of coordination of the methylamino group, in which the methyl and hydrogen occupy equatorial/axial or reverse positions. In the case of the complex with the smaller Yb(III) ion, one of these possibilities is probably strongly preferred due to the larger steric strain around the methyl group, leading to only one observable signal in the Z-spectra. However, to our surprise, the Yb(III)–H3L3 complex also exhibits a CEST effect at 26 ppm, although the CEST peak has a small intensity and is observable only in a narrow pH range 6–8.5 (Fig. 4 and S13†). In this pH region, the protonation–deprotonation of the –NMe2 group is supposed (accordingly to the corresponding log KA ∼7.8 reported previously for its Gd(III) complex15). Thus, an equilibrium between the deprotonated (and mostly coordinated) and protonated (and uncoordinated) amino group in the complex is present. Therefore, the observation of the CEST effect can be explained by the protonation–deprotonation of the uncoordinated dimethylamino group, which is still located sufficiently close to the highly paramagnetic centre to mediate the saturation transfer to the bulk water through this process. Such an explanation is consistent with the fact that the observed CEST effect disappears at a relatively low temperature ∼65 °C, which is probably due to the much faster chemical exchange process (Fig. S13†), contrary to the complexes of H3L1 where the analogous signal disappearance cannot be reached even at 95 °C (see above). In the case of the Eu(III)–H3L3 complex, a slight CEST effect was also observed, causing some asymmetric broadening of the water signal in the Z-spectra (Fig. S13†). This feature can be explained in a similar way to the above.
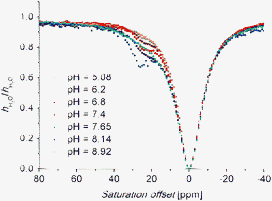 | ||
| Fig. 4 Z-spectra of a 50 mM aqueous solution (H2O–D2O 1:10) of the Yb(III)–H3L3 complex (B0 = 7.05 T, T = 298 K, satpwr = 29 dB ∼1000 Hz, satdly = 2 s). | ||
To test the applicability of the complexes as MRI pH probes, the CEST effect was measured in phantoms containing Eu(III)–H3L1 and Yb(III)–H3L1 complex solutions having different pH and concentrations. The final CEST images were obtained after irradiation at the CEST and symmetrical negative frequencies and subtracting the images (Fig. 5 and S14†). Normalized signal intensities are shown in Fig. S15 and S16.† The results clearly show that the prepared complexes can be successfully employed as CEST probes in the pH region relevant to living systems.
![MRI-CEST images of phantoms consisting of one vial containing 80 mM aq. solution of [La(H2O)(L1)] as a diamagnetic standard and eight vials containing 80 mM aq. solutions of [Eu(H2O)(L1)] and [Yb(L1)] with different pH values; pH is shown in figures A. Experimental conditions: MSME pulse sequence, B0 = 4.7 T, B1 = 20 μT, T = 293 K, satdly = 2 s, TR = 5 s, TE = 8.9 ms, scan time = 8 min. I.A: T1-weighted image, satfrq = 34 ppm from the bulk water signal. I.B: T1-weighted image, satfrq = −34 ppm from the bulk water signal. I.C: The difference between images I.A and I.B in false colours. II.A: T1-weighted image, satfrq = 89 ppm from the bulk water signal. II.B: T1-weighted image, satfrq = −89 ppm from the bulk water signal. II.C: The difference between images II.A and II.B in false colours.](/image/article/2013/DT/c3dt52031e/c3dt52031e-f5.gif) | ||
| Fig. 5 MRI-CEST images of phantoms consisting of one vial containing 80 mM aq. solution of [La(H2O)(L1)] as a diamagnetic standard and eight vials containing 80 mM aq. solutions of [Eu(H2O)(L1)] and [Yb(L1)] with different pH values; pH is shown in figures A. Experimental conditions: MSME pulse sequence, B0 = 4.7 T, B1 = 20 μT, T = 293 K, satdly = 2 s, TR = 5 s, TE = 8.9 ms, scan time = 8 min. I.A: T1-weighted image, satfrq = 34 ppm from the bulk water signal. I.B: T1-weighted image, satfrq = −34 ppm from the bulk water signal. I.C: The difference between images I.A and I.B in false colours. II.A: T1-weighted image, satfrq = 89 ppm from the bulk water signal. II.B: T1-weighted image, satfrq = −89 ppm from the bulk water signal. II.C: The difference between images II.A and II.B in false colours. | ||
Experimental
Materials and methods
Commercially available chemicals had synthetic purity and were used as received. Deionized water, used for the synthesis of the ligands and their complexes, was prepared using a deionization water system ROWAPUR 200/100. Water used for potentiometric titrations was prepared by a Milli-Q (Millipore).t-Bu3DO3A·HBr [1,4,7-tris(t-butylcarboxymethyl)-1,4,7,10-tetraazacyclododecane hydrobromide] was prepared according to the published procedure.21 Dry solvents were prepared by the standard purification procedures22 and stored over molecular sieves under an argon atmosphere: dry MeCN was obtained by distillation from P2O5, THF was dried by refluxing with potassium and distilled under an argon atmosphere.
The ESI-MS spectra were acquired on a Bruker ESQUIRE 3000 spectrometer equipped with an electrospray ion source and ion-trap detection. Measurements were carried out in the positive and negative modes. NMR characterization data (1D: 1H, 13C; 2D: HSQC, HMBC, 1H–1H COSY) were recorded on VNMRS300, VarianUNITYINOVA 400 or Bruker Avance III 600, using 5-mm sample tubes. Chemical shifts δ are given in ppm and coupling constants J are reported in Hz. For the 1H and 13C measurements in D2O, t-BuOH was used as the internal standard (δH = 1.25, δC = 30.29). For the measurements in CDCl3, TMS was used as the internal standard (δH = 0.00, δC = 0.00). Abbreviations s (singlet), t (triplet), q (quartet), m (multiplet) and br (broad) are used in order to express the signal multiplicities. Elemental analysis was performed at the Institute of Macromolecular Chemistry of the Academy of Science of the Czech Republic (Prague).
Synthesis
1H NMR (299.9 MHz, CDCl3): δ 3.59 (2H, t, 3JHH = 6.6, NCH2CH2Br); 4.09 (2H, t, 3JHH = 6.6, NCH2CH2Br); 7.70–7.73 (2H, m, arom.); 7.84–7.86 (2H, m, arom.).
13C{1H} NMR (75.4 MHz, CDCl3): δ 28.55 (1C, NCH2CH2Br); 39.70 (1C, NCH2CH2Br); 123.93 (2C, arom.); 132.23 (2C, arom. quaternary); 134.63 (2C, arom.); 168.22 (2C, CO).
Elemental analysis: found (calcd for C10H8BrNO2, Mr = 254.1) C: 47.68 (47.27), H: 3.07 (3.17), N: 5.32 (5.51), Br: 31.15 (31.45).
A portion (3.35 g) of this mixture containing compound 4 was dissolved in CF3COOH and CHCl3 (30 ml, 1:1); the resulting solution was refluxed for 24 h. After evaporation to dryness the oily residue was dissolved in a small amount of distilled water and then loaded onto a strong cation exchange column (Dowex 50, 50–100 mesh, H+-form, 2 × 9 cm). Acidic impurities were removed by elution with water and the product 5 was eluted with 5% aq. NH3. Fractions containing the product (TLC check) were combined and evaporated to give 2.82 g of yellow oil containing compound 5. The crude product 5 was used in the next step without purification.
The portion (2.80 g) of the crude product 5 was dissolved in 80% aq. NH2NH2·H2O (15 ml); the resulting mixture was then heated for 18 h at 90 °C and concentrated in vacuo. The residue was dissolved in a small amount of distilled water. Then the solution was filtered and loaded onto a strong anion exchange column (Dowex 1, OH−-form, 1.25 × 8 cm). Impurities were removed by elution with water and the product H3L1 was eluted with 5% aq. CH3COOH. Fractions containing the pure product (TLC and 1H NMR check) were combined and evaporated to give H3L1 (1.90 g) as an yellow oil. The crude product was purified by crystallization from hot EtOH to give H3L1·3H2O (1.15 g, 59%) as a white powder.
Compound 4. HPLC: Solvent: A = CH3CN, B = 0.1% of CF3COOH in H2O, C = H2O. Gradient: A: 20–50%, 0–24 min; 50%, 24–40 min; 50–20%, 40–41 min; 20%, 41–61 min. B: 20%, 0–61 min. tR = 17.0 min.
1H NMR (299.9 MHz, CDCl3): δ 1.45–1.48 (27H, m, (C(CH3)3); 3.15–3.43 (18H, m, CH2); 3.53 (4H, s, CH2CO2); 3.86 (2H, s, CH2CO2); 4.02 (2H, br, CH2); 7.73–7.76 (2H, m, arom.); 7.84–7.87 (2H, m, arom.).
13C{1H} NMR (150.9 MHz, CDCl3): δ 27.92 (9C, C(CH3)3); 32.58, 48.97, 49.92, 50.13, 51.17, 51.86, 54.77 (13C, CH2); 82.96 (2C, C(CH3)3); 84.28 (1C, C(CH3)3); 123.58 (2C, arom.); 131.65 (2C, arom. quaternary); 134.37 (2C, arom.); 167.85 (3C, CH2CO2); 169.22 (2C, NCO).
MS-ESI: (+): 688.5 ([M + H]+, calcd 688.4); 710.4 ([M + Na]+; calcd 710.4).
Compound 5. MS-ESI: (−): 518.0 ([M − H]−, calcd 518.2).
H3L1·3H2O. 1H NMR (600.2 MHz, D2O, pD = 5.4, Fig. S17†): 2.85–2.89 (4H, m, CH2NCH2CO); 2.94 (2H, br, CH2CH2NH2); 3.02–3.04 (4H, m, CH2N(CH2)2NH2); 3.06 (2H, t, 3JHH = 5.1, CH2NH2); 3.13 (2H, s, CH2CO); 3.33–3.37 (2H, m, CH2NCH2CO); 3.40–3.45 (2H, m, CH2NCH2CO); 3.54–3.58 (2H, m, CH2NCH2CO); 3.71–3.77 (2H, m, CH2NCH2CO); 3.90 (4H, AB-multiplet, CH2CO).
13C{1H} NMR (150.9 MHz, D2O, pD = 5.4, Fig. S18†): δ 37.01 (1C, CH2NH2); 48.90 (2C, CH2N(CH2)2NH2); 48.99 (2C, CH2NCH2CO); 50.82 (1C, CH2CH2NH2); 51.00 (2C, CH2NCH2CO); 53.06 (2C, CH2NCH2CO); 55.85 (1C, CH2CO); 58.07 (2C, CH2CO); 171.02 (2C, CH2CO); 178.77 (1C, CH2CO).
MS-ESI: (+): 412.0 ([M + Na]+, calcd 412.2); 428.0 ([M + K]+, calcd 428.2). (−): 425.8 ([M + K − 2H]−, calcd 426.2).
Elemental analysis: found (calcd for C16H37N5O9, Mr = 443.5) C: 43.17 (43.33), H: 8.40 (8.41), N: 15.39 (15.79).
:1), ethyloxycarbonylchloride (0.96 g, 8.85 mmol) was added dropwise. The solution was stirred at room temperature for 2 h and concentrated in vacuo. The oily residue was dissolved in CH2Cl2 (30 ml) and extracted with H2O (3 × 15 ml) and 3% aq. HCl (1 × 15 ml). The organic portion was dried over Na2SO4 and concentrated in vacuo to give 1.10 g of 7 as a colourless oil.1H NMR (299.9 MHz, CDCl3): δ 1.24 (3H, t, 3JHH = 7.2, CH2CH3); 2.94 (3H, s, NCH3); 3.41 (2H, br, NCH2); 3.73 (2H, br, CH2OH); 4.11 (2H, q, 3JHH = 7.2, CH2CH3).
13C{1H} NMR (75.4 MHz, CDCl3): δ 14.85 (1C, CH2CH3); 35.42 (1C, NCH3); 51.92 (1C, CH2CH2OH); 61.40 (1C, CH2OH); 61.74 (1C, CH2CH3); 158.17 (1C, CO).
MS-ESI: (+): 148.5 ([M + H]+, calcd 148.1); 170.4 ([M + Na]+, calcd 170.1).
:9). Fractions containing the product 8 (TLC check) were combined and evaporated to give 8 (1.32 g) as a colourless oil.1H NMR (299.9 MHz, CDCl3): δ 1.26 (3H, t, 3JHH = 6.9, CH2CH3); 2.97 (3H, s, NCH3); 3.45 (2H, br, NCH2); 3.62 (2H, br, CH2Br); 4.14 (2H, q, 3JHH = 6.9, CH2CH3).
13C{1H} NMR (75.4 MHz, CDCl3): δ 14.89 (1C, CH2CH3); 29.50 (1C, CH2Br); 35.60 (1C, NCH3); 51.30 (1C, CH2CH2Br); 61.76 (1C, CH2CH3); 156.31 (1C, CO).
A portion (3.80 g) of the mixture containing compound 9 was dissolved in a mixture of CF3COOH and CHCl3 (30 ml, 1:1); the resulting solution was refluxed for 24 h and evaporated on a rotary evaporator. The oily residue was dissolved in a small amount of distilled water and evaporated (this procedure was then repeated three more times) to give 5.00 g of yellow oil containing compound 10 (it was used in the next step without purification).
The crude product 10 (5.00 g) was dissolved in 10% aq. NaOH (50 ml) and stirred for 24 h at 90 °C. Then the solution was loaded onto a strong anion exchange column (Dowex 1, OH−-form, 1.25 × 20 cm). Impurities were removed by elution with water and the product H3L2 was eluted with 5% aq. CH3COOH. Fractions containing the product (TLC and 1H NMR check) were combined and evaporated to give H3L2 (1.97 g) as a brownish oil. The crude product was dissolved in small amount of distilled water and anhydrous EtOH was added dropwise. Crystallization started after a few minutes, and the suspension was left overnight. The white crystalline solid was filtered off and dried under a vacuum to give H3L2·5.5H2O (0.66 g, 30%) as a white powder.
Compound 9. MS-ESI: (+): 644.5 ([M + H]+, calcd 644.5); 666.4 ([M + Na]+; calcd 666.5).
Compound 10. MS-ESI: (−): 474.1 ([M − H]−, calcd 474.3); 512.0 ([M + K − 2H]−, calcd 512.0).
H3L2·5.5H2O. 1H NMR (299.9 MHz, D2O, pD = 1.15): δ 2.63 (3H, s, NCH3); 2.80–3.15 (12H, m, CH2); 3.20–3.63 (10H, m, CH2); 3.97 (4H, br, CH2CO2).
1H NMR (299.9 MHz, D2O, pD = 5.3, Fig. S19†): δ 2.77 (3H, s, NCH3); 2.80–2.91 (4H, m, CH2); 2.94–3.08 (8H, m, CH2); 3.13 (2H, s, CH2CO2); 3.31–3.46 (4H, m, CH2); 3.52–3.60 (2H, m, CH2); 3.70–3.76 (2H, m, CH2); 3.89 (4H, AB-multiplet, CH2CO2).
13C{1H} NMR (150.9 MHz, D2O, pD = 5.6, Fig. S20†): δ 34.93 (1C, NCH3); 46.89 (1C, CH2NCH3); 48.89 (2C, CH2); 49.03 (2C, CH2); 50.42 (1C, CH2CH2NCH3); 51.04 (2C, CH2); 53.10 (2C, CH2); 55.68 (1C, CH2CO2); 58.16 (2C, CH2CO2); 171.08 (2C, CH2CO2); 178.63 (1C, CH2CO2).
MS-ESI: (+): 404.3 ([M + H]+, calcd 404.2); 426.9 ([M + Na]+, calcd 426.2); 442.8 ([M + K]+, calcd 442.2).
Elemental analysis: found (calcd for C17H44N5O11.5, Mr = 502.6) C: 40.84 (40.63), H: 9.27 (9.22), N: 13.80 (13.84).
1H NMR (600.2 MHz, D2O, pD = 4.8, Fig. S21†): δ 2.87–2.90 (2H, m, CH2); 2.93 (6H, s, N(CH3)2); 2.96 (2H, t, 3JHH = 6.0 Hz, NCH2CH2N(CH3)2); 3.05–3.08 (4H, m, CH2); 3.13–3.17 (2H, m, CH2); 3.27 (2H, t, 3JHH = 6.0 Hz, CH2N(CH3)2); 3.36 (2H, s, CH2CO2); 3.38–3.40 (2H, m, CH2); 3.47–3.56 (6H, m, CH2); 3.88 (4H, AB-multiplet, CH2CO2).
13C{1H} NMR (150.9 MHz, D2O, pD = 4.8, Fig. S22†): δ 44.32 (2C, N(CH3)2); 48.08 (1C, CH2, NCH2CH2N(CH3)2); 48.70 (2C, CH2); 49.14 (2C, CH2); 51.32 (2C, CH2); 52.68 (2C, CH2); 55.63 (1C, CH2CO2); 56.08 (1C, CH2N(CH3)2); 57.49 (2C, CH2CO2); 170.51 (2C, CH2CO2); 177.96 (1C, CH2CO2).
MS-ESI: (+): 456.9 ([M + K]+, calcd 456.2).
Elemental analysis: found (calcd for C18H43N5O10, Mr = 489.6) C: 44.29 (44.16), H: 8.86 (8.85), N: 14.14 (14.31).
Eu3+–H3L1. MS-ESI: (+): 540.1 ([M + H]+, calcd 540.2); 562.1 ([M + Na]+; calcd 562.1). (−): 573.9 ([M + Cl]−, calcd 574.1).
Yb3+–H3L1. MS-ESI: (+): 561.1 ([M + H]+, calcd 561.2); 683.1 ([M + Na]+; calcd 683.2). (−): 594.9 ([M + Cl]−, calcd 595.1).
Eu3+–H3L2. MS-ESI: (+): 553.6 ([M + H]+, calcd 554.2). (−): 587.7 ([M + Cl]−, calcd 588.1).
Yb3+–H3L2. MS-ESI: (+): 581.8 ([M + Li]+, calcd 581.2). (−): 609.6 ([M + Cl]−, calcd 609.1).
Eu3+–H3L3. MS-ESI: (+): 574.7 ([M + Li]+, calcd 574.2). (−): 602.6 ([M + Cl]−, calcd 602.1).
Yb3+–H3L3. MS-ESI: (+): 595.8 ([M + Li]+, calcd 595.2). (−): 623.6 ([M + Cl]−, calcd 623.1).
X-Ray diffraction
Single-crystals of H3L1·5H2O were prepared by slow evaporation of the aqueous solution. The crystals of H3L2·6H2O were prepared by evaporation of the ethanolic solution, and the crystals of H3L3·3.5H2O were prepared by the slow cooling of a saturated hot ethanolic solution. The Yb3+–H3L1 complex for crystallization was prepared by mixing the ligand with 1.1 equiv. of the YbCl3 in a small amount of distilled water. The pH was adjusted to ∼6 with 1 M NaOH, and the mixture was stirred overnight at 60 °C. Then the pH was re-adjusted to 6.5 and the solution was stirred overnight at 60 °C. The complex was purified on a Al2O3 column by chromatography. The pure product was eluted using a mixture of EtOH–H2O–conc. aq. NH3 (10:8:1). Single crystals of [Yb(L1)]·5H2O were prepared from a concentrated aqueous solution of the complex by slow diffusion of the THF vapours.The diffraction data were collected by employing a ApexII CCD diffractometer using Mo-Kα radiation (λ = 0.71073 Å) at 150(1) K and analyzed using the SAINT V8.27B (Bruker AXS Inc., 2012) program package. The structures were solved by direct methods (SHELXS9724) and refined by full-matrix least-squares techniques (SHELXL9725). The relevant data for the structures have been provided.†
Crystal data
H3L1·5H2O: C16H41N5O11, M = 479.54, monoclinic, a = 7.80380(10), b = 16.7236(3), c = 17.8081(3) Å, β = 92.1431(12)°, U = 2322.47(6) Å3, space group P21/c, Z = 4, 5358 total reflections, 4408 intense reflections, R1[I > 2σ(I)] = 0.0344, wR2(all data) = 0.0873. CCDC 952386.† All non-hydrogen atoms were refined anisotropically. Although all hydrogen atoms could be found in the electron difference map, those bound to carbon atom were fixed in theoretical positions using a riding model with Ueq(H) = 1.2Ueq(C) to keep the number of refined parameters low. Hydrogen atoms bound to nitrogen or oxygen atoms were fully refined.H3L2·6H2O: C17H45N5O12, M = 511.58, monoclinic, a = 9.4479(7), b = 17.7199(14), c = 15.5379(9) Å, β = 106.573(2)°, U = 2493.2(3) Å3, space group P21/n, Z = 4, 5723 total reflections, 4765 intense reflections, R1[I > 2σ(I)] = 0.0415, wR2(all data) = 0.1129. CCDC 952384.† The strategy of refinement was the same as that described in the previous case. Beside this, several solvate water molecules were refined as twisted in two positions with some disordered hydrogen atoms in two positions with half-occupancy.
H3L3·3.5H2O: C18H42N5O9.50, M = 480.57, monoclinic, a = 7.4798(2), b = 15.9561(4), c = 20.0951(6) Å, β = 91.5670(10)°, U = 2397.42(11) Å3, space group P21/n, Z = 4, 5503 total reflections, 4686 intense reflections, R1[I > 2σ(I)] = 0.0370, wR2(all data) = 0.0952. CCDC 952385.† The refining strategy was the same as described above.
[Yb(L1)]·5H2O: C16H38N5O11Yb, M = 649.55, monoclinic, a = 16.8708(7), b = 7.5238(4), c = 18.4880(9) Å, β = 90.4230(10)°, U = 2346.7(2) Å3, space group P21/c, Z = 4, 5374 total reflections, 5160 intense reflections, R1[I > 2σ(I)] = 0.0137, wR2(all data) = 0.0306. CCDC 933967.† Preliminary refinement revealed a number of different maxima of the electronic density, which were located close to the carbon and nitrogen atoms of the macrocyclic part of the molecule. It pointed to a disorder of the macrocycle in a similar way as already reported in the literature (see, e.g., ref. 20c, Fig. S9†). It was successfully modelled using the anisotropic refinement of atoms occupying positions with a higher occupancy, whilst the less occupied part was refined in isotropic mode. All other non-hydrogen atoms were refined anisotropically. Although hydrogen atoms could be found in the electron difference map, they were fixed in theoretical (C–H) or original (N–H, O–H) positions using the riding model with Ueq(H) = 1.2Ueq(X) to keep the number of refined parameters low.
HPLC
The analytical HPLC system consisted of a gradient pump Beta 10 (ECOM) equipped with a mixer Knauer A0285 and dual UV-detector (ECOM). Analysis was performed on a LunaPHC8 column (150 × 4.6 mm, Phenomenex, the flow rate 1 ml min−1). The mobile phase was continuously vacuum degassed in a DG 3014 degasser (ECOM) and it was mixed in the gradient pump from the stock solutions. The detector wavelengths were set to 210 and 254 nm. Injection volumes were 20 μl (concentrations of the samples were 1 mg ml−1).The preparative HPLC system was composed of a gradient pump LCD 50 K (ECOM) and UV-Vis detector LCD 2083 (ECOM). Preparation was performed on a LunaPHC8 (250 × 21.1 mm, Phenomenex, the flow rate was maintained at 20 ml min−1). The detector wavelengths were set to 210 nm. The mobile phase was prepared separately and degassed using an ultrasound probe (Cole-Parmer 750-Watt Ultrasonic Homogenizer). Injection volumes were 1 ml. The collection and processing of the data was performed using Clarity (DataApex) software.
Potentiometry
Potentiometric titrations26 were carried out in a vessel thermostatted at 25.0 ± 0.1 °C, at a constant ionic strength (I = 0.1 M (Me4N)Cl) using a PHM 240 pH-meter, a GK 2401B combined glass electrode and a 2 ml ABU 900 automatic piston burette (all Radiometer). The initial volume was ca. 5 ml and the ligand concentration in the titration vessel was ca. 0.004 M. An inert atmosphere was provided by the constant passage of argon saturated with the vapour of the solvent [0.1 M (Me4N)Cl]. Extra HCl was added to the starting solution to run titrations in the −log[H+] range 2.0–12.0 (for protonation constants) and 1.8–6.0 for the Gd3+ and La3+ stability constants. Titrations were performed with a (Me4N)OH solution. Titrations with metal ions were performed at metal-to-ligand molar ratios of 1:1. Ligand titrations consisted of about 40 points per titration and were run in triplicate. As the complexation was too slow for conventional titration, the “out-of-cell” method was used;26 about 25 points per titration, two parallel titrations, three weeks at room temperature for equilibration. To calculate the protonation constants of the ligand and the stability constants of the complexes, the OPIUM27 software package was used.The overall protonation constants βh10 are concentration constants defined as βh10 = [HhL]/([H]h·[L]) (they can be transferred into stepwise dissociation constants as pKA(HhL) = log βh − log βh−1). The concentration stability constants βhlm are generally defined by βhlm = [HhLlMm]/([H]h·[L]l·[M]m). The value of pKw used was 13.81.
1H NMR titrations
The 1H NMR titration over the whole pH region (31 points), for determination of the protonation constants, was performed at 25 °C (ligand concentration: 0.05 M; no ionic strength control) in H2O on a Brucker Avance (III) 600 (B0 = 14.1 T), using 5 mm sample tubes. 1H NMR spectra were measured without presaturation of the water signal. A coaxial capillary with D2O (for the lock) and t-BuOH (as external standard; δH = 1.25 ppm) was used. The solution pH (0.5–13.5) was adjusted with aqueous HCl or NMe4OH solutions and measured with a combined glass electrode (Mettler Toledo) and pH-meter (3505 pH Meter, JENWAY) calibrated with standard buffers. Protonation constants were calculated with the OPIUM software package.27CEST experiments
All Z-spectra were recorded on a VNMRS300 operating at 299.9 MHz (B0 = 7.05 T), using 5 mm sample tubes and a coaxial capillary with D2O and t-BuOH as the external standard. Samples were of 25–100 mM concentration in the mixture of H2O and D2O (1:10) or in H2O; pH was adjusted with aqueous HCl or LiOH solutions. Standard pulse sequences for presaturation experiments were used. Saturation offsets were set using the array-function (increment 200–250 Hz). Other measurement parameters are listed below each figure.MRI CEST images were measured with phantoms consisting of one vial containing an aqueous solution of the La3+-complex as a control and eight (resp. five) vials containing aqueous solution of Eu3+ or Yb3+-complexes with different pH values or concentrations. All MRI CEST images were acquired on a 4.7 T scanner (Bruker BioSpec, Germany) using RARE (Rapid Acquisition with Refocused Echoes) or MSME (Multi Slice Multi Echo) pulse sequences with presaturation pulse. Experimental conditions: TR = 5000 ms, TE = 8.9 ms, resolution 0.35 × 0.35 × 2 mm, turbo factor = 4 (in RARE sequence). Other measurement parameters are listed below each figure. Parameters of presaturation pulse: B1 = 20 μT, satdly = 2 s. For all MR experiments, the resonator coil was used.
All MR images were processed using an in-house program, written in Matlab (The Mathworks Inc, USA), where the signal intensity was normalized to unit slope and receiver gain. Then the difference image between the images acquired with negative and positive frequency offset of the saturation frequency was calculated. Difference images were analysed using ImageJ software (NIH, USA), regions of interest were outlined manually.
Conclusions
We prepared 2-aminoethyl derivatives of DO3A and showed that the coordinated amino group mediates NMR saturation transfer to the bulk water. We suggest a new mechanism for the PARACEST effect, consisting of the coordination–decoordination of the amino group having an optimal rate, combined with the very fast protonation–deprotonation of the group while the group is non-bound to the paramagnetic metal ion. The pH-dependence of the saturation transfer parallels with the abundance of the species with the coordinated amino group. This novel type of PARACEST agent broadens the arsenal of pH-sensitive probes for possible in vivo pH measurements by MRI.Acknowledgements
We thank J. Plutnar and Z. Tošner for their help with the setting of the CEST pulse sequences. The financial support of the Ministry of Education of the Czech Republic (no. MSM0021620857), the Grant Agency of the Czech Republic (no. 207/11/1437) and the Grant Agency of the Charles University (no. 110213) is acknowledged.Notes and references
- (a) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed; (b) P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027–3047 RSC; (c) C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- (a) J. Zhou and P. C. M. van Zijl, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 109–136 CrossRef CAS; (b) Y. Wu, M. Evbuomwan, M. Melendez, A. Opina and A. D. Sherry, Future Med. Chem., 2010, 2, 351–366 CrossRef CAS PubMed; (c) E. Terreno, D. D. Castelli and S. Aime, Contrast Media Mol. Imaging, 2010, 5, 78–98 CAS; (d) P. C. M. van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed; (e) G. Liu, X. Song, K. W. Y. Chan and M. T. McMahon, NMR Biomed., 2013, 26, 810–828 CrossRef CAS PubMed.
- (a) S. Zhang, M. Merritt, D. E. Woessner, R. E. Lenkinski and A. D. Sherry, Acc. Chem. Res., 2003, 36, 783–790 CrossRef CAS PubMed; (b) M. Woods, D. E. Woessner and A. D. Sherry, Chem. Soc. Rev., 2006, 35, 500–511 RSC; (c) S. Viswanathan, Z. Kovács, K. N. Green, S. J. Ratnakar and A. D. Sherry, Chem. Rev., 2010, 110, 2960–3018 CrossRef CAS PubMed; (d) T. C. Soesbe, Y. Wu and A. D. Sherry, NMR Biomed., 2013, 26, 829–838 CrossRef CAS PubMed.
- S. Zhang and A. D. Sherry, J. Solid State Chem., 2003, 171, 38–43 CrossRef CAS.
- M. Woods, A. Pasha, P. Zhao, G. Tireso, S. Chowdhury, G. Kiefer, D. E. Woessner and A. D. Sherry, Dalton Trans., 2011, 40, 6759–6764 RSC.
- S. Zhang, K. Wu, M. C. Biewer and A. D. Sherry, Inorg. Chem., 2001, 40, 4284–4290 CrossRef CAS PubMed.
- S. Zhang, L. Michaudet, S. Burgess and A. D. Sherry, Angew. Chem., Int. Ed., 2002, 41, 1919–1921 CrossRef CAS.
- (a) C.-H. Huang and J. R. Morrow, Inorg. Chem., 2009, 48, 7237–7243 CrossRef CAS PubMed; (b) C.-H. Huang and J. R. Morrow, J. Am. Chem. Soc., 2009, 131, 4206–4207 CrossRef CAS PubMed; (c) C.-H. Huang, J. Hammell, S. J. Ratnakar, A. D. Sherry and J. R. Morrow, Inorg. Chem., 2010, 49, 5963–5970 CrossRef CAS PubMed; (d) J. Hammell, L. Buttarazzi, C.-H. Huang and J. R. Morrow, Inorg. Chem., 2011, 50, 4857–4867 CrossRef CAS PubMed; (e) D. D. Castelli, E. Terreno and S. Aime, Angew. Chem., Int. Ed., 2011, 50, 1798–1800 CrossRef PubMed.
- S. J. Dorazio, P. B. Tsitovich, K. E. Siters, J. A. Spernyak and J. R. Morrow, J. Am. Chem. Soc., 2011, 133, 14154–14156 CrossRef CAS PubMed.
- (a) S. J. Dorazio and J. R. Morrow, Inorg. Chem., 2012, 51, 7448–7450 CrossRef CAS PubMed; (b) S. J. Dorazio, P. B. Tsitovich, S. A. Gardina and J. R. Morrow, J. Inorg. Biochem., 2012, 117, 212–219 CrossRef CAS PubMed; (c) P. B. Tsitovich and J. R. Morrow, Inorg. Chim. Acta, 2012, 393, 3–11 CrossRef CAS.
- A. O. Olatunde, S. J. Dorazio, J. A. Spernyak and J. R. Morrow, J. Am. Chem. Soc., 2012, 134, 18503–18505 CrossRef CAS PubMed.
- (a) B. Yoo and M. D. Pagel, J. Am. Chem. Soc., 2006, 128, 14032–14033 CrossRef CAS PubMed; (b) G. Liu, Y. Li and M. D. Pagel, Magn. Reson. Med., 2007, 58, 1249–1256 CrossRef CAS PubMed; (c) T. Chauvin, P. Durand, M. Bernier, H. Meudal, B.-T. Doan, F. Noury, B. Badet, J.-C. Beloeil and É. Tóth, Angew. Chem., Int. Ed., 2008, 47, 4370–4372 CrossRef CAS PubMed; (d) T. Chauvin, S. Torres, R. Rosseto, J. Kotek, B. Badet, P. Durand and É. Tóth, Chem.–Eur. J., 2012, 18, 1408–1418 CrossRef CAS PubMed; (e) V. R. Sheth, G. Liu, Y. Li and M. D. Pagel, Contrast Media Mol. Imaging, 2012, 7, 26–34 CrossRef CAS PubMed; (f) V. R. Sheth, Y. Li, L. Q. Chen, C. M. Howison, C. A. Flask and M. D. Pagel, Magn. Reson. Med., 2012, 67, 760–768 CrossRef PubMed.
- (a) J. A. Duimstra, F. J. Femia and T. J. Meade, J. Am. Chem. Soc., 2005, 127, 12847–12855 CrossRef CAS PubMed; (b) A. Mishra, J. Pfeuffer, R. Mishra, J. Engelmann, A. K. Mishra, K. Ugurbil and N. K. Logothetis, Bioconjugate Chem., 2006, 17, 773–780 CrossRef CAS PubMed.
- A. K. Mishra and J.-F. Chatal, New J. Chem., 2001, 25, 336–339 RSC.
- G. B. Giovenzana, R. Negri, G. A. Rolla and L. Tei, Eur. J. Inorg. Chem., 2012, 2035–2039 CrossRef CAS.
- M. Meyer, V. Dahaoui-Gindrey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 178–180, 1313–1405 CrossRef CAS.
- K. Kumar, C. A. Chang, L. C. Francesconi, D. D. Dischino, M. F. Malley, J. Z. Gougoutas and M. F. Tweedle, Inorg. Chem., 1994, 33, 3567–3575 CrossRef CAS.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- (a) J. Rudovský, P. Cígler, J. Kotek, P. Hermann, P. Vojtíšek, I. Lukeš, J. A. Peters, L. Vander Elst and R. N. Muller, Chem.–Eur. J., 2005, 11, 2373–2384 CrossRef PubMed; (b) P. Lebdušková, P. Hermann, L. Helm, É. Tóth, J. Kotek, K. Binnemans, J. Rudovský, I. Lukeš and A. E. Merbach, Dalton Trans., 2007, 493–501 RSC; (c) T. Vitha, V. Kubíček, J. Kotek, P. Hermann, L. Vander Elst, R. N. Muller, I. Lukeš and J. A. Peters, Dalton Trans., 2009, 3201–3214 Search PubMed; (d) D. D. Castelli, M. C. Caligara, M. Botta, E. Terreno and S. Aime, Inorg. Chem., 2013, 52, 7130–7138 CrossRef PubMed.
- (a) I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef; (b) P. Vojtíšek, P. Cígler, J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2005, 44, 5591–5599 CrossRef PubMed; (c) J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2006, 45, 3097–3102 CrossRef CAS PubMed.
- (a) A. Dadabhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2002, 348–357 RSC; (b) D. A. Moore, Org. Synth., 2008, 85, 10–14 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
- N. Cesari, C. Biancalani, C. Vergelli, V. Dal Piaz, A. Graziano, P. Biagini, C. Ghelardini, N. Galeotti and M. P. Giovannoni, J. Med. Chem., 2006, 49, 7826–7835 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS97, Program for Crystal Structure Solution from Diffraction Data, University of Göottingen, Göottingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement from Diffraction Data, University of Göottingen, Göottingen, 1997 Search PubMed.
- (a) P. Táborský, P. Lubal, J. Havel, J. Kotek, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2005, 70, 1909–1942 CrossRef; (b) M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- M. Kývala and I. Lukeš, International Conference Chemometrics 95, p. 63, Pardubice, Czech Republic, 1995. Full version of “OPIUM” available (free of charge) on: http://web.natur.cuni.cz/~kyvala/opium.html.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of H3L2·6H2O and H3L3·3.5H2O, and selected geometric parameters. The distribution diagram of H3L1. Chemical shift dependence of H3L1 on pH, and its protonation scheme. Distribution diagrams of the Ln3+:H3L1 systems. Temperature dependence of the 1H NMR spectra of [Eu(H2O)(L1)] and [Yb(L1)]. Disorder of the [Yb(L1)] molecule found in the solid-state structure of [Yb(L1)]·5H2O, and its selected geometric parameters. Set of Z-spectra of the [Eu(H2O)(L1,2,3)] and [Yb(L1,2,3)] complexes at different temperatures and pH. MRI-CEST images of phantoms of [Eu(H2O)(L1)] and [Yb(L1)], and the normalized intensity of the CEST effect. 1H and 13C{1H}NMR spectra of H3L1, H3L2 and H3L3. CCDC 933967, 952384–952386. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52031e |
| This journal is © The Royal Society of Chemistry 2013 |