 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the double exchange interaction in a mixed valence {VIII–VII} pair: a theoretical perspective†
Soumen Ghosh,
Saurabh Kumar Singh,
Subrata Tewary and
Gopalan Rajaraman*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: rajaraman@chem.iitb.ac.in
First published on 8th August 2013
Abstract
Combined DFT–TD-DFT methodology has been employed to fully characterise a mixed valence {VII–VIII} complex of molecular formula [(PY5Me2)2V2(m-5,6-dimethylbenzimidazolate)]4+. These calculations offer viable ways to enhance double-exchange parameters – a key ingredient in the synthesis of SMMs.
In the area of single molecule magnets (SMMs),1 apart from large spin ground state (S) and negative anisotropy (D), a very large exchange interaction (J) between the metal ions is another important requirement to obtain an isolated ground state.2 This means that one should have a very large isotropic exchange (J), however Js are often small and very large values are obtained only in a few occasions.3 On the other hand in mixed-valence compounds the double exchange parameter (B), which stabilises the high-spin ground state, is generally large compared to J and is often in the order of thousands of wave number.4 The requirement of an isolated ground state can thus be easily achieved if B is considered as a key ingredient in the synthesis of SMMs. This has an advantage of maximizing the S value and also yielding an isolated ground state. Despite these facts, only a few mixed valence complexes have been studied in this direction.5 The properties of these complexes are controlled by the amount of localization of unpaired spins in these complexes and this demands a thorough characterization, which is often non-trivial. Depending on the amount of localization and delocalization of spins in these complexes, they have been categorised in three classes (class-I, II and III).6
In recent years theoretical methods have emerged as a powerful tool in the area of molecular magnetism.7 Bencini et al. in their seminal work proposed a protocol using DFT to compute the parameters associated with mixed valence compounds.4a As this method is generally applied to molecules possessing relatively high symmetry, only a few systems have been subjected to such study4 and this severely restricts the general understanding of the key parameters involved in mixed valence complexes.
Recently Long et al.8 have reported a group of imidazole bridged divanadium VII–VII compounds of molecular formula [(PY5Me2)2V2(Lbr)]4+ (where Lbr = im−, 2-mim−, 4,5-dpim−, bzim−, 5,6-dmbzim− (1)). Magnetic studies reveal an antiferromagnetic coupling between the two metal centres leading to an S = 0 ground state and the fit to the magnetic susceptibility yield J value of −6 cm−1. Complex 1 (Fig. 1) upon one electron oxidation yields a mixed valence VIII–VII compound (1ox), which has an S = 5/2 ground state. Magnetic and spectral analysis suggests that the 1ox is a class-II type compound and yields an estimate of the delocalization parameters as J = −6 cm−1, B = 220 cm−1, and the inter-valence charge transfer (IVCT) band is observed at 4190 cm−1 (solid state spectrum).8 Further, Coronado et al. have studied the spin states of 1ox and suggested that the electric field can be used to manipulate ground state spin.9 By modelling the properties using semi-classical theory and incorporating vibronic effects using quantum mechanical approach they have illustrated that S = 5/2 or S = 3/2 or S = 1/2 can be the ground state for 1ox depending on the vibronic coupling parameter (λ).9 However, the estimate of the delocalization parameters are seemingly different from the original one (B of 220 cm−1 of Long et al.8 vs. 666 cm−1 of Coronado et al.9). These differences essentially highlight the difficulties in extracting these parameters using experimental and analytical methods. This necessitates a generic theoretical method for computation of these parameters.
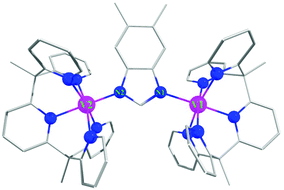 | ||
| Fig. 1 Crystal structure of the complex 1. Color code: V in pink, N in blue, for clarity hydrogens are omitted. | ||
To overcome these issues, here we have outlined a combined DFT–TD-DFT approach for complete characterization of mixed valence transition metal dimers. We have studied complex 1ox using this procedure and have illustrated the applicability of this method to large systems and have developed magneto-structural correlation for the double-exchange parameter (B) for the first time. To begin with, we have computed the J value of complex 1 using the B3LYP functional and TZV basis set using the established procedure to compute the J values7 (see ESI† for Hamiltonian employed). The DFT calculations yield an antiferromagnetic J of −7 cm−1 compared to −6 cm−1 determined by experiment.8 Antiferromagnetic interactions result from the dxz|π*|dxz overlap as revealed by overlap integral analysis (see ESI Table S1†). Complex 1 and 1ox essentially differs in two structural aspects, the V–N(1) and V–N(2) distances and V–N(1)–C(1) and V–N(2)–C(1) angles. Since a switch in antiferro- to ferromagnetic coupling occurs as we go from 1 to 1ox, we have also developed a magneto-structural correlation in 1 to see if such a switch is possible with these structural parameters (see Fig. 2). These correlations were developed by performing single point calculations by varying one parameter at a time. Increasing the V–N distances or V–N–C angles symmetrically led to a less antiferromagnetic interaction; however at larger distances/angles the curve tends to saturate and no switching from antiferro-to-ferro has been observed. This saturation although occurs at all V–N distances, for angles at large values J tends to decrease further; this suggests that at large angle the interaction can in fact be ferromagnetic as one can expect due to orbital orthogonality. Orbital analysis suggests that the dxz–dxz overlap prevails even at larger distances/angles leading to an antiferromagnetic interaction, due to efficient π-type overlap with the dxz orbitals on both ends. This analysis clearly illustrates that these structural parameters are unlikely to switch the magnetic interaction.
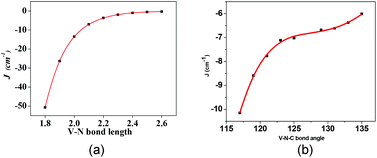 | ||
| Fig. 2 Magneto-structural correlations for complex 1. (a) J vs. V–N bond distance, (b) J vs. V–N–C bond angle (see ESI† for fitting details, also see Tables S2 and S3†). | ||
To obtain insight into the electron delocalization and double exchange interaction, calculations have been performed for the S = 5/2, 3/2 and 1/2 spin states on the crystal structure of 1ox. Calculations reveal that S = 5/2 is the ground state for 1ox, and this is consistent with experimental observations.8 Structural optimization also led to the same conclusion where S = 5/2 is predicted to be the ground state with S = 1/2 and 3/2 lying at 1239 cm−1 and 5280 cm−1 higher in energy, respectively. In the S = 5/2 state (the ideal configuration expected for the VIII–VII scenario is (dxy)1(dyz)1(dxz)1–(dxy)1(dyz)1(dxz)0); the extra electron is found to be completely delocalized as revealed by the spin density distribution (2.54 vs. 2.55 on V(1) and V(2) respectively, see Fig. 3a). The HOMO and LUMO of 1ox also have equal dxz orbital contributions on both the vanadium atoms (see Fig. 3b and 3c), suggesting that the electron is completely delocalized. In this spin state, the inter-valence charge transfer (IVCT) band occurs between symmetric and antisymmetric combinations of dxz|π*|dxz orbitals (see Fig. 3b and c). Our TD-DFT calculations estimate this band to be at 4037 cm−1, in excellent agreement with the experimental observation of 4190 cm−1. In contrast, in the S = 3/2 state the electron is trapped in one of the vanadium atoms, as reflected in the computed spin density where spin densities of 2.81 on V(1) and 0.1 on V(2) are computed (see Fig. 3d). Similar to the S = 3/2 state, in S = 1/2 the electron is also trapped, where V(1) is found to have spin density of 2.94 while V(2) has a spin density value of −2.13 (see Fig. 3e and Table S6†). Due to the difference in sign between the two atoms, spin delocalization due to unpaired spins does not occur in this electronic state. The optimized structures essentially reflect the electronic symmetry of these spin states (see Fig. S1 and Table S5†). To validate the nature of the DFT-computed S = 3/2 state, we have also attempted to compute this spin state using state-average CASSCF calculations (see Table S8 and Fig. S3†). These calculations suggest that the electron distribution obtained by DFT in S = 3/2 is the lowest lying with 99% weight and very little mixing with other states, and this adds confidence to the DFT computed parameters.
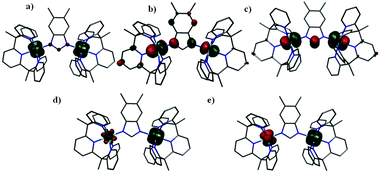 | ||
| Fig. 3 DFT computed spin density plots for complex 1ox (a) spin density, (b) HOMO and (c) LUMO of high spin state, (d) spin density of S = 3/2 state, (e) spin density of S = 1/2 state. The iso-density surface represented corresponds to a value of 0.03 bohr3 per e−. The green and red colour indicate positive and negative densities. | ||
The double exchange parameter (B) and isotropic exchange (J) are determined by both dynamic and static mechanisms of that particular spin state.4 The asymmetric stretching vibration of the V–N bond is likely to be responsible for localization10 and this is computed in the present case to be 122 cm−1. The PES (potential energy surface) has been constructed by fixing the V–V distance and symmetrically varying the V–N distance on both sides. The computed PESs for S = 5/2, 3/2 and 1/2 are shown in Fig. 4. Double minima of those PESs clearly indicate the localization in this mixed valence complex for all the spin states. For all spin states a symmetric structure is obtained at Q = 0. For S = 5/2, the Q = 0 is only 0.8 cm−1 higher in energy while the same in S = 3/2 and 1/2 are much higher (358 cm−1 and 819 cm−1, respectively). This barrier height represents the nature of localization/delocalization, i.e. a larger barrier height leads to a localized structure.
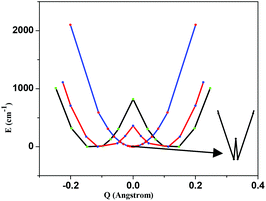 | ||
| Fig. 4 DFT computed potential energy surfaces for different spin states of complex 1ox (see also Fig. S2 of ESI†). | ||
The parameters involved in the characterization of 1ox are described by the following equations,10–12
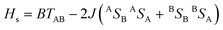 | (1) |
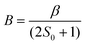 | (2) |
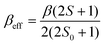 | (3) |
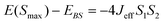 | (4) |
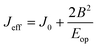 | (5) |
To calculate the double exchange parameter B, β has to be calculated. Previously it was calculated by transferring the electron from the symmetric orbital to anti-symmetric orbital using Slater transition state formalism for the symmetric structure.4 Although this method yields a good estimate of β, it lacks generality as it demands higher symmetry structures. An alternative and more general approach would be to employ the TD-DFT method13 to estimate this transition energy (see Table S7†). Here we have employed this protocol and computed β for the S = 5/2 state as 1999 cm−1 at the Q = 0 point. Earlier analytical modelling9 also renders the β value as 2000 cm−1 and this strikingly correlates to our estimate, and offers confidence on the computed parameter. Using the β parameter, the double exchange has been estimated using eqn (2) and this yields B as 666 cm−1. To probe the extent of delocalization in 1ox further, we have computed the localization parameter (λ) using the transition energy in the optimized structure (Eop) (where Eop = 2λ).11 This yields the λ value as 1997 cm−1.4b Interestingly, the β/λ value estimated here is ∼1.0 and this indicates that complex 1ox is not a class-II complex as nas previously been suggested8, but is in the regime of class-II/III.14–16 This observation is again supported by recent analytical modelling.9 This is also nicely illustrated in the computed PES of the S = 5/2 state, where only a very small barrier for complete delocalization was observed.
The PESs developed for S = 3/2 and S = 1/2 reveal a greater amount of localization compared to the ground state. Employing eqn (3),11 effective transfer integrals have been calculated for both the surfaces and the values are 802 cm−1 and 1375 cm−1 for S = 1/2 and 3/2 respectively. This is consistent with the fact that β should decrease as S decreases.17 Besides, the activation barrier has been calculated to be 96 cm−1 and 357 cm−1 for S = 3/2 and S = 1/2 (see ESI† for equation). In both cases, λ > βeff, suggesting that these two spin states belong to the class-II type. Again, this is reflected in the computed PES where a larger barrier height for localization was observed.
In the situation where the electrons are trapped, the isotropic exchange can be calculated using eqn (4) and (5).11,12 From eqn (4), Jeff is calculated as 206 cm−1.18 In eqn (5)10,11 the contribution due to the first term can be negative or positive depending on whether the interaction is ferromagnetic or antiferromagnetic, while the second term is always positive because it is the contribution of electron delocalization. Using this we have computed J0, i.e. the isotropic coupling constant, as −15.4 cm−1. J0 in the experimental paper is assumed to be equal to that of 1, while our calculations suggest that it is more than double that of 1 while retaining the sign. A large J0 is also consistent with the computed large overlap integral between the magnetic orbitals (see Table S4†). This invariably illustrates the complexity involved in deducing different parameters in mixed valence systems.
Since our protocol has been successfully employed to thoroughly characterize this mixed valence system, we have taken a leap forward towards prediction where a possibility to maximize the B parameter has been explored using magneto-structural correlation. Two parameters, V–N(1) and V–N(2) bond distances and V–N(1)–C and V–N(2)–C angles, were varied and the developed correlations are shown in Fig. 5.
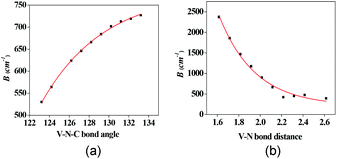 | ||
| Fig. 5 Magneto-structural correlation developed for the double exchange parameter. (a) B vs. V–N–C bond angle, (b) B vs. V–N bond length. | ||
As the V–N distance decreases, the B parameter increases exponentially, while larger distances decrease the value marginally and at much longer distances the B value essentially saturates. This also correlates with the computed overlap integral, where larger overlap values were detected at shorter distances. Similarly for angles as well, an exponential dependence is detected with larger angles leading to larger B values. However, the magnitude of variation is much smaller compared to the bond distance. This suggests that one can obtain large a B value by synthesising mixed valence complexes having relatively shorter M–L distances.
To this end, we have developed a combined DFT–TD-DFT protocol to estimate different parameters involved in mixed-valance complexes such as J, B, β and λ, and our study offers new insight into the way by which double exchange can be maximized.
Acknowledgements
GR would like to acknowledge financial support from DST India (SR/S1/IC-41/2010; SR/NM/NS-1119/2011) and IIT Bombay for access to the high performance computing facility. SKS and ST acknowledge CSIR and UGC for fellowship.References
- (a) R. Sessoli, H.-L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1 RSC.
- K. Hegetschweiler, B. Morgenstern, J. Zubieta, P. J. Hagrman, N. Lima, R. Sessoli and F. Totti, Angew. Chem., Int. Ed., 2004, 43, 3436 CrossRef CAS PubMed.
- (a) V. Barone, A. Bencini, I. Ciofini, C. A. Daul and F. Totti, J. Am. Chem. Soc., 1998, 120, 8357 CrossRef CAS; (b) V. Barone, A. Bencini, D. Gatteschi and F. Totti, Chem.–Eur. J., 2002, 8, 5019 CrossRef CAS PubMed; (c) A. Bencini, E. Berti, A. Caneschi, D. Gatteschi, E. Giannasi and I. Invernizzi, Chem.–Eur. J., 2002, 8, 3660 CrossRef CAS PubMed; (d) M. A. Castro and A. E. Roitberg, J. Chem. Theory Comput., 2013, 9, 2609 CrossRef CAS; (e) M. Shoji, K. Koizumi, T. Taniguchi, Y. Kitigawa, S. Yamanaka, M. Okumura and K. Yamaguchi, Int. J. Quantum Chem., 2007, 107, 116 CrossRef CAS.
- (a) T. C. Stamatatos and G. Christou, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 113 CrossRef CAS PubMed; (b) Z.-M. Zhang, S. Yao, Y.-G. Li, H.-H. Wu, Y.-H. Wang, M. Rouzieres, R. Clerac, Z.-M. Su and E.-B. Wang, Chem. Commun., 2013, 49, 2515 RSC; (c) M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2002, 124, 3512 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247 CrossRef CAS.
- (a) T. Rajeshkumar and G. Rajaraman, Chem. Commun., 2012, 48, 7856 RSC; (b) S. K. Singh, N. K. Tibrewal and G. Rajaraman, Dalton Trans., 2011, 40, 10897 RSC; (c) S. K. Singh and G. Rajaraman, Dalton Trans., 2013, 42, 3623 RSC; (d) E. Ruiz, S. Alvarez, A. Rodriguez-Fortea, P. Alemany, Y. Pouillon and C. Massobrio, in Magnetism: Molecules to Materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001, vol. II, p. 227 Search PubMed; (e) A. Bencini and F. Totti, Int. J. Quantum Chem., 2005, 101, 819 CrossRef CAS.
- B. Bechlars, D. M. D'Alessandro, D. M. Jenkins, A. T. Iavarone, S. D. Glover, C. P. Kubiak and J. R. Long, Nat. Chem., 2010, 2, 362 CrossRef CAS PubMed.
- (a) C. Bosch-Serrano, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariço, A. Palii and B. Tsukerblat, ChemPhysChem, 2012, 13, 2662 CrossRef CAS PubMed; (b) C. Bosch-Serrano, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Palii and B. Tsukerblat, Phys. Rev. B: Condens. Matter, 2012, 86, 024432 CrossRef.
- G. Blondin and J.-J. Girerd, Chem. Rev., 1990, 90, 1359 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed.
- (a) L. Noodleman and E. J. Baerends, J. Am. Chem. Soc., 1984, 106, 2316 CrossRef CAS; (b) L. Noodleman, C. Y. Peng, D. A. Case and L.-M. Mouesca, Coord. Chem. Rev., 1995, 144, 199 CrossRef CAS.
- C. Lambert, C. Risko, V. Coropceanu, J. Schelter, S. Amthor, N. E. Gruhn, J. C. Durivage and J. Bredas, J. Am. Chem. Soc., 2005, 127, 8508 CrossRef CAS PubMed.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655 CrossRef CAS PubMed.
- S. D. Glover, B. J. Lear, J. C. Salsman, C. H. Londergan and C. P. Kubiak, Philos. Trans. R. Soc. London, Ser. A, 2008, 366, 177 CrossRef CAS PubMed.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168 RSC.
- Here we would like to note that significant spin contamination, particularly of the S = 1/2 state, has been detected and values are not corrected for this contamination (see ESI Table S7† for details).
- Note here that a class-II type been assumed for S = 5/2 although this is not strictly valid. Thus Jeff should be only being treated as a guide to understand electron delocalization.
Footnote |
| † Electronic supplementary information (ESI) available: Computed overlap integrals, geometry of the optimized structures, computed spin densities, individual developed PESs, TD-DFT estimated transition energies. See DOI: 10.1039/c3dt51935j |
| This journal is © The Royal Society of Chemistry 2013 |