 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A decatungstate-type polyoxoniobate with centered manganese: [H2MnIVNb10O32]8− as a soluble tetramethylammonium salt†
Jung-Ho
Son
*a and
William H.
Casey
*ab
aDepartment of Chemistry, University of California, Davis, One Shields Ave., Davis, CA 95616, USA. E-mail: junghoson@gmail.com; whcasey@ucdavis.edu; Fax: (+1) (530) 752 8995; Tel: (+1) (530) 752 3211
bDepartment of Geology, University of California, Davis, One Shields Ave., Davis, CA 95616, USA
First published on 1st August 2013
Abstract
A highly symmetric MnIV-centered polyoxoniobate [H2MnIVNb10O32]8− was synthesized via hydrothermal methods as a soluble tetramethylammonium salt. The structure is similar to decatungstate structure [W10O32]4−, except for the central heteroatom. The cluster is stable between 4 < pH < 10, as was characterized with ESI-MS and UV-Vis spectroscopy.
Polyoxometalates (POMs) comprise a class of metal–oxide ions that are widely used in catalysis and material science.1 The most familiar compounds are oxides of group 5 or 6 metals, such as vanadates, tungstates and molybdates. Unsubstituted POMs are usually diamagnetic in their most oxidized form and transition-metal-substituted POMs find use via their magnetic, electrochemical, and optical properties.2
Polyoxoniobates (PONb) are relatively less explored due to the synthetic challenge, although hexaniobate and decaniobate ions have been studied for many decades,3 in part from the pioneering work by Spinner4 and in spite of recent advances.5 Numbers of PONb substituted with main-group element have been synthesized by Nyman and coworkers.6 Transition-metal-substituted PONb have appeared in the literature more recently.7 As one of the earliest reported heteropolyoxoniobate compounds, Pope et al. and Stucky et al. reported two hexaniobate ions linked by MnIV or NiIV, i.e. Na12[MnNb12O38]·50H2O (MnNb12, Scheme 1A).8 Here, the hexaniobate ions cap and ligate the heteroatom in a sandwich-like geometry, but the heteroatom is not completely trapped in the PONb cage.9 This sandwich compound resembles the Weakley–Yamase type tungstate structure, ([Ln(W5O18)2]n−, Scheme 1B) where two W5O18 units are linked by a central lanthanide ion.10 Pope et al. synthesized a [Mn(CO)3]+-capped hexaniobate more recently.11 However, complete encapsulation of manganese within the PONb framework has not yet been achieved. Echoing the goals of the Pope et al.,11 manganese substitution in PONb might benefit attempts to sequester dangerous nuclides, such as 99Tc, in nuclear waste as niobates are generally stable at basic-pH conditions.
![Polyhedral drawing of MnNb12 (A), [Ln(W5O18)2]n− (B) and MnNb10 (C).](/image/article/2013/DT/c3dt51798e/c3dt51798e-s1.gif) | ||
| Scheme 1 Polyhedral drawing of MnNb12 (A), [Ln(W5O18)2]n− (B) and MnNb10 (C). | ||
Herein we report a new type of MnIV-substituted PONb cluster, [H2MnIVNb10O32]8− [MnNb10, Scheme 1C], as a tetramethylammonium (TMA) salt, TMA8[H2MnIVNb10O32]·22H2O (1). This cluster ion can be viewed as two MnNb5 Lindqvist motifs condensed by bridging oxygen atoms and sharing a MnIV site. We note that the structure of this cluster is similar to decatungstate ion,12 [W10O32]4−, which is used for photocatalyst in organic reactions.13 Structurally similar vanadates like [MV13O38]7− (M = MnIV, NiIV) are known,14 with additional vanadates surrounding the center site of the cluster shown in Scheme 1C. Decatungstate is unstable in aqueous solution and the vanadate derivative is only stable at 3 < pH < 5.
The compound 1 was synthesized by hydrothermal reaction of the mixture of hydrous niobium oxide, KMnO4, TMAOH at 110 °C for 3 days. We note that higher reaction temperatures lead to decomposition of the cluster over increased reaction times. The resulting brown solution after reaction was washed with isopropanol and extracted with ethanol. Purification to get analytically pure compound of 1 was challenging due to the similar solubility of 1 and dark colored impurity. The crystals of 1 were obtained as olive-gold colored crystals in the dark-brown viscous oily product. The dark, oily product was carefully washed with minimal amounts of ethanol to isolate crystals, since the dark colored impurity was only slightly more soluble in ethanol than 1. We note that the previously reported reaction condition to obtain MnNb12 sandwich complex8 involved lower temperature (∼90 °C) and shorter reaction times (less than an hour) compared to the conditions used to synthesize 1. We suspect that the MnIV encapsulated by the niobates results from a solution driven to a thermodynamically more stable end point by longer reaction times and higher temperatures.
The structure of 1 was determined by single crystal X-ray crystallography.‡ The cluster in 1 has manganese at the center surrounded by ten oxo-niobate frameworks, possessing idealized D4h symmetry [Scheme 1C, Fig. 1]. Eight TMA ions are found in the structure of 1, suggesting the charge of the cluster as −8. Bond valence sum (BVS) calculation (4.059) indicates the manganese oxidation state of MnIV. Two protons on the surface of the cluster are found in the electron-density map, thus the formula of the cluster can be expressed as [H2MnIVNb10O32]8−. These protons are bound to two μ2-O at the opposite sides of the cluster [Fig. 1]. Central site in the cluster shows regular MnO6 octahedron, with Mn–O lengths ranging between 1.875(3) and 1.916(3) Å and nearly orthogonal O–Mn–O angles from 89.46(12)° to 90.55(12)°. The Mn–μ2-O–Nb angles (103°–105°) are smaller than Nb–μ2-O–Nb angles (113°–120°), because of the significantly short Mn–μ6-O (1.876(3) Å) compared to Nb–μ6-O (2.475(3) and 2.484(3) Å) in trans position.
![Ball-and-stick model of [H2MnIVNb10O32]8− (yellow: Mn, large grey: Nb, red: O, small grey: H).](/image/article/2013/DT/c3dt51798e/c3dt51798e-f1.gif) | ||
| Fig. 1 Ball-and-stick model of [H2MnIVNb10O32]8− (yellow: Mn, large grey: Nb, red: O, small grey: H). | ||
We note that the cluster MnNb12 has similar, but more regular, Mn–O lengths of 1.863(26), 1.865(25) and 1.878(24) Å than MnNb10. However, the MnO6 in MnNb12 is elongated between the two hexaniobates, with two distinct O–Mn–O angles around 83° and 96°. This elongation is explained by Flynn and Stucky as being caused by the electrostatic repulsion of two hexaniobate units, which results in the axial stretching of the MnO6 octahedron.8d The coordination to MnIV in MnNb12 causes distortion within the hexaniobate ions, with the three oxygen atoms of the hexaniobates coordinating to MnIV being closer to each other, with O⋯O distances within 2.45–2.50 Å in the MnNb12 cluster, while the distances are normally 2.75–2.80 Å for uncoordinated μ2-O in the same hexaniobate units.
MnNb10 cluster is structurally similar to decatungstate except the existence of central MnIV. W–O bonds in decatungstate range 1.69–2.34 Å, while Nb–O bonds in 1 are slightly longer within a large 1.75–2.48 Å range due to the less positively charged NbV compared to WVI. PONb usually have higher negative charges than polyoxotungstates for the same reason. For example, a [Nb10O32] cluster without heteroatoms reducing the charge would have high charge of −14, compared to [W10O32]4−. Thus, incorporation of the central MnIV and two surface protons lowers the overall cluster charge to −8, thereby stabilizing it.
Electrospray-ionization mass spectrometry (ESI-MS) confirmed the identity of the cluster in the solution [Fig. 2]. The mass spectrum of 1 shows series of peaks for −4, −3 and −2 charged ions. The series of −4 ion peaks can be explained as due to fragmentation of the relatively highly charged H4[H2MnNb10O32]−4 ion during ionization in the ESI-MS. The mass spectrum of 1 also exhibits multiple peaks for −3 and −2 ions in higher m/z ranges, in addition to the TMAxHy[H2MnNb10O32]−(8−x−y) ions. These are commonly observed in the ESI-MS of the TMA salts of other PONb, but in much smaller intensities.7h–k These additional multiple peaks are interpreted as CH3+ adduct of the cluster ions, as the peak positions and shapes match well with calculations [Fig. S1†]. Thus the [H2MnNb10O32]8− cluster exhibits a somewhat unique behaviour in ESI-MS compared to other polyoxoniobates. Some methylated Anderson-type heteropolyoxomolybdates have been synthesized by ligand exchange with methanol.15 However, 1 exhibits the same ESI-MS spectra without contact with methanol. Thus actual existence of methylated cluster in our system is unlikely and we believe that the peaks corresponding to CH3+ adduct originated from the fragmentation of TMA during ionization.
![ESI-MS of 1 with peak assignments. [1] = [H2MnIVNb10O32] (0.2 mM solution of 1, pH 7.7).](/image/article/2013/DT/c3dt51798e/c3dt51798e-f2.gif) | ||
| Fig. 2 ESI-MS of 1 with peak assignments. [1] = [H2MnIVNb10O32] (0.2 mM solution of 1, pH 7.7). | ||
The color of the aqueous solution of 1 is yellow and distinctly different from the reported orange color of MnNb12, probably due to the slightly different Mn–O environment. UV-Vis absorption spectrum of 1 is also different from that reported for the MnNb12, which has absorbances at 238, 300, 450 and 480 nm,8 while 1 shows absorption near 250, 300, 350 and 450 nm [Fig. S2–S4†].
The stability of the cluster was examined by using both UV-Vis and ESI-MS titration experiments. When dissolved into solution at 2 mM concentrations, the solution reaches a natural pH of 8.7. Adding acid or base to this solution leads to decomposition of the cluster, as is evident in the decreased abundance of peaks assignable to clusters in ESI-MS of 2 mM and 0.2 mM solutions [Fig. 3 and Fig. S5,† respectively]. When titrating with base, peaks assignable to the clusters significantly decrease in abundance at pH > 11 for a 2 mM solution of 1 and pH > 9 for 0.2 mM solution of 1. ESI-MS spectra taken during an acid titration of 1 indicates that the cluster is stable until pH ∼ 5, however, the initial golden yellow color of the solution started to fade at pH < 7 and changed to orange-pink color near pH ∼ 4 during the titration of a 2 mM solution of 1 with acid, while the yellow solution color did not change with base addition until pH ∼ 13. In addition to decomposition, we suspect that protonation of structural oxygens is affecting the color.
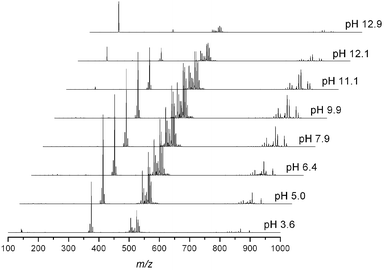 | ||
| Fig. 3 ESI-MS of 2 mM solution of 1 during titration in different pH. The solution concentration change for each pH was minimized by using 1 M acid or 2.75 M base in titration. Each spectrum is an averaged signal for acquisitions of 1 min duration. The peak at 196 m/z in basic condition corresponds to TMAOH. | ||
ESI-MS of the acidified solution indicated an increase in peaks assignable to H+ adducts of 1, with a decrease of the CH3+-adduct peak. The increase in CH3+ adduct peaks during base addition is expected and attributable to the increased TMA concentration, added to the solution as a TMAOH base and then fragmented in the ionization step of ESI-MS [Fig. 3 and Fig. S5†].
Among the UV-Vis absorption bands of 0.02 mM solution of 1, the strongest ligand-metal charge-transfer (LMCT) band at 250 nm was the most sensitive to decomposition of the cluster [Fig. S2†]. When 1 was titrated with base, the band at 250 nm decreased as pH increased, with two new bands appearing near 240 nm and 260 nm above pH 9 [Fig. S2†]. We suspect that these indicate decomposition of the cluster, in agreement with the ESI-MS results. The absorption spectra did not change significantly until acidified to pH 4.4. Below this pH, overall absorption gradually increased until the compound finally decomposed to form a colored precipitate that we assume is hydrous, manganese–niobium oxide. The behavior is similar in the higher concentration solutions, but the 2 mM solution begins to flocculate as early as pH 6.5, reflecting the higher sample concentration and existence of background salt (0.1 M TMACl), which was not used in ESI-MS titration experiment.
In summary, we report a new MnIV-substituted PONb, [H2MnIVNb10O32]8− as a soluble TMA salt. It has a relatively wide stability between 4 < pH < 10 in aqueous solutions, suggesting a robust use in applications, possibly complementing the decatungstate ion.
This work was supported by an NSF CCI grant through the Center for Sustainable Materials Chemistry, number CHE-1102637.
Notes and references
- (a) M. T. Pope, Heteropoly and Isopolyoxometalates, Springer-Verlag, Berlin, 1983 Search PubMed; (b) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef; (c) Polyoxometalates. From Platonic Solids to Anti-Retroviral Activity, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1994 Search PubMed; (d) C. L. Hill, ed. Special issue on “Polyoxometalates”, Chem. Rev, 1998, 98, 1–390 Search PubMed; (e) C. Ritchie, A. Ferguson, H. Nojiri, H. N. Miras, Y.-F. Song, D.-L. Long, E. Burkholder, M. Murrie, P. Kögerler, E. K. Brechin and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 5609–5612 CrossRef CAS; (f) J.-D. Compain, P. Mialane, A. Dolbecq, I. Mbomekallé, J. Marrot, F. Sécheresse, E. Rivière, G. Rogez and W. Wernsdorfer, Angew. Chem., Int. Ed., 2009, 121, 3123–3127 CrossRef.
- (a) Polyoxometalate Molecular Science, ed. J. J. Borrás-Almenar, E. Coronado, A. Müller and M. T. Pope, Springer, 2003 Search PubMed; (b) Polyoxometalate Chemistry from Topology via Self-Assembly to Applications, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2001 Search PubMed; (c) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS; (d) D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS; (e) L. Cronin and A. Müller, eds. Special issue on “Polyoxometalate cluster science”, Chem. Soc. Rev, 2012, 4122, 7325–7648 Search PubMed; (f) D.-L. Long and L. Cronin, eds. Special issue on “Polyoxometalates”, Dalton Trans, 2012, 4133, 9799–10106 Search PubMed.
- (a) I. Lindqvist, Ark. Kemi, 1953, 5, 247 CAS; (b) E. J. Graeber and B. Morosin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2137 CrossRef.
- (a) S. Si Larbi, D. Bodiot and B. Spinner, Rev. Chim. Miner., 1976, 13, 497–507 CAS; (b) A. Goiffon and B. Spinner, Talanta, 1977, 24, 130–132 CrossRef CAS.
- M. Nyman, Dalton Trans., 2011, 40, 8049–8058 RSC.
- (a) M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansl, T. M. Nenoff and A. M. Sattler, Science, 2002, 297, 996–998 CrossRef CAS; (b) M. Nyman, F. Bonhomme, T. M. Alam, J. B. Parise and G. M. B. Vaughan, Angew. Chem., Int. Ed., 2004, 43, 2787–2792 CrossRef CAS; (c) T. M. Anderson, S. G. Thoma, F. Bonhomme, M. A. Rodriguez, H. Park, J. B. Parise, T. M. Alam, J. P. Larentzos and M. Nyman, Cryst. Growth Des., 2007, 7, 719–723 CrossRef CAS; (d) F. Bonhomme, J. P. Larentzos, T. M. Alam, E. J. Maginn and M. Nyman, Inorg. Chem., 2005, 44, 1774–1785 CrossRef CAS; (e) M. Nyman, A. J. Celestian, J. B. Parise, G. P. Holland and T. M. Alam, Inorg. Chem., 2006, 45, 1043–1052 CrossRef CAS; (f) M. Nyman, J. P. Larentzos, E. J. Maginn, M. E. Welk, D. Ingersoll, H. Park, J. B. Parise, I. Bull and F. Bonhomme, Inorg. Chem., 2007, 46, 2067–2079 CrossRef CAS; (g) Y. Hou, M. Nyman and M. Rodriguez, Angew. Chem., Int. Ed., 2011, 50, 12514–12517 CrossRef CAS.
- (a) M. Nyman, L. J. Criscenti, F. Bonhomme, M. A. Rodriguez and R. T. Cygan, J. Solid State Chem., 2003, 176, 111–119 CrossRef CAS; (b) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 5634–5636 CrossRef CAS; (c) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Dalton Trans., 2009, 2677–2678 RSC; (d) J.-Y. Niu, G. Chen, J.-W. Zhao, P.-T. Ma, S.-Z. Li, J.-P. Wang, M.-X. Li, Y. Bai and B.-S. Ji, Chem.–Eur. J., 2010, 16, 7082–7086 CAS; (e) G. Guo, Y. Xu, J. Cao and C. Hu, Chem. Commun., 2011, 47, 9411–9413 RSC; (f) G. Guo, Y. Xu, J. Cao and C. Hu, Chem.–Eur. J., 2012, 18, 3493–3497 CrossRef CAS; (g) P. Huang, C. Qin, X.-L. Wang, C.-Y. Sun, G.-S. Yang, K.-Z. Shao, Y.-Q. Jiao, K. Zhou and Z.-M. Su, Chem. Commun., 2012, 48, 103–105 RSC; (h) J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2012, 41, 12674–12677 RSC; (i) J.-H. Son, C. A. Ohlin, E. C. Larson, P. Yu and W. H. Casey, Eur. J. Inorg. Chem., 2013, 1748–1753 CrossRef CAS; (j) J.-H. Son, C. A. Ohlin, R. L. Johnson, P. Yu and W. H. Casey, Chem.–Eur. J., 2013, 19, 5191–5197 CrossRef CAS; (k) J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2013, 42, 7529–7533 RSC.
- (a) B. W. Dale and M. T. Pope, Chem. Commun., 1967, 792 RSC; (b) B. W. Dale, J. M. Buckley and M. T. Pope, J. Chem. Soc. A, 1969, 301–304 RSC; (c) C. M. Flynn Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 332–334 CrossRef; (d) C. M. Flynn Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 335–344 CrossRef.
- D. Atencio, J. M. V. Coutinho, A. C. Doriguetto, Y. P. Mascarenhas, J. Ellena and V. C. Ferrari, Am. Mineral., 2008, 93, 81–87 CrossRef CAS.
- (a) R. D. Peacock and T. J. R. Weakley, J. Chem. Soc. A, 1971, 1836–1839 RSC; (b) J. Iball, J. N. Low and T. J. R. Weakley, J. Chem. Soc., Dalton Trans., 1974, 2021–2024 RSC; (c) T. Ozeki, M. Takahashi and T. Yamase, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1370–1374 CrossRef; (d) M. Sugeta and T. Yamase, Bull. Chem. Soc. Jpn., 1993, 66, 444–449 CrossRef CAS.
- A. V. Besserguenev, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40, 2582–2586 CrossRef CAS.
- J. Fuchs, H. Hartl and W. Schiller, Angew. Chem., Int. Ed. Engl., 1973, 12, 420 CrossRef.
- (a) C. Tanielian, Coord. Chem. Rev., 1998, 1165–1181 CrossRef CAS; (b) I. N. Lykakis, C. Tanielian, R. Seghrouchni and M. Orfanopoulos, J. Mol. Catal. A: Chem., 2007, 262, 176–184 CrossRef CAS; (c) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC; (d) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS.
- (a) C. M. Flynn Jr. and M. T. Pope, J. Am. Chem. Soc., 1970, 92, 85–90 CrossRef; (b) S. Liu, D. Li, L. Xie, H. Cheng, X. Zhao and Z. Su, Inorg. Chem., 2006, 45, 8036–8040 CrossRef CAS.
- (a) D. Honda, S. Ikegami, T. Inoue, T. Ozeki and A. Yagasaki, Inorg. Chem., 2007, 46, 1464–1470 CrossRef CAS; (b) S. Ikegami, K. Kani, T. Ozeki and A. Yagasaki, Chem. Commun., 2010, 46, 785–787 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, ESI-MS peak assignment with calculated spectrum, UV-Vis titration data of 0.02 mM, 0.2 mM and 2 mM solution of 1, ESI-MS of 0.2 mM solution of 1 during titration and FT-IR spectrum. CCDC 943073. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51798e |
‡ Crystal data. (1) CCDC 943073. C32H101N8O54MnNb10, M = 2446.25, monoclinic, a = 13.9537(6), b = 34.0884(14), c = 18.0134(7) Å, β = 92.907(1)°, U = 8557.2(6) Å3, T = 88 K, space group P21/n (no. 14), Z = 4, 125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 508 reflections measured, 17489 unique (Rint = 0.0391) which were used in all calculations. The final wR(F2) was 0.0937 (all data). 508 reflections measured, 17489 unique (Rint = 0.0391) which were used in all calculations. The final wR(F2) was 0.0937 (all data). |
| This journal is © The Royal Society of Chemistry 2013 |