DOI:
10.1039/C3DT51791H
(Paper)
Dalton Trans., 2013,
42, 14826-14835
Co-ordinative properties of a tripodal trisamide ligand with a capped octahedral preference†
Received
4th July 2013
, Accepted 31st July 2013
First published on 1st August 2013
Abstract
An investigation into the co-ordination chemistry of tris(6-pivaloylamino-2-pyridylmethyl)amine (TPPA) shows a preference for capped octahedral geometry when no additional donors are present. This tripodal ligand yields capped octahedral complexes upon co-ordination to a series of first row transition metals when the counter-ion is a perchlorate, bromide or iodide ion. The exact geometry has been confirmed by shape mapping calculations. The largest variation from the capped octahedral geometry is observed in the case of nickel(II), indicating a greater octahedral preference of this metal ion.
Introduction
The tripodal tetraamine ligand, tris(2-pyridylmethyl)amine (TPA), was first synthesised in 1967 by Anderegg and Wenk,1 and its co-ordination chemistry has since been extensively studied.2–6 The TPA framework (Fig. 1) is sterically suited to co-ordinating via a trigonal pyramidal arrangement of donor atoms, and typically an additional ligand will approach axially to give a trigonal bipyramidal co-ordination geometry around the metal cation. Although there are several exceptions to this, such as the co-ordination to Pt(II) and Pd(II), which prefer square planar geometries, causing the ligand to bind in a hypodentate manner in which one pyridine does not co-ordinate.7,8 Similar tridentate co-ordination modes have also been observed upon co-ordination to ruthenium(II)9 and iron(II).10
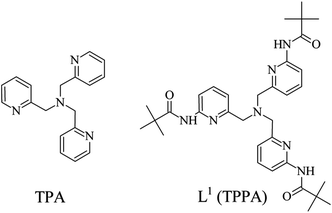 |
| | Fig. 1 The ligand under investigation, L1 (right), alongside TPA (left). | |
While seven- and eight-coordinate complexes of TPA have been reported, for example, [Fe(TPA)2](BPh4)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and [Mn(TPA)2](ClO4)211 (each contains two tetradentate TPA groups per metal ion), it is interesting that derivatives of TPA (such as TPPA), where the pyridyl groups are substituted with additional donor atoms,5,12,13 generally result in the isolation of five co-ordinate species even though they are potentially heptadentate. Presumably, this is due to poor metal–ligand interactions in this geometry meaning that the five co-ordinate geometry is favourable. However, the nature of these substituents and the position on the pyridyl rings have been shown to have significant effects on the structural and physical properties of their metal complexes.14–16
10 and [Mn(TPA)2](ClO4)211 (each contains two tetradentate TPA groups per metal ion), it is interesting that derivatives of TPA (such as TPPA), where the pyridyl groups are substituted with additional donor atoms,5,12,13 generally result in the isolation of five co-ordinate species even though they are potentially heptadentate. Presumably, this is due to poor metal–ligand interactions in this geometry meaning that the five co-ordinate geometry is favourable. However, the nature of these substituents and the position on the pyridyl rings have been shown to have significant effects on the structural and physical properties of their metal complexes.14–16
A common set of derivatives of TPA is generated by alkyl substitution at the 6-position of the pyridyl groups. This simple modification can produce unfavourable steric interactions which influence the ligand's ability to bind to the metal. In a few reported examples of TPA, substituents at the 6-position on each of the pyridyl groups have been shown to block further co-ordination entirely; for example, some TPA derivatives containing methyl15,16 and phenyl17 functionalities have been reported which, upon complexation to Cu(I) cations, lead to four-coordinate Cu(I) complexes.
Mareque Rivas18 and others19 have carried out investigations into the hydrogen bond donor properties of complexes of tris(2-pyridylmethyl)amine (TPA) derivatives, with respect to their ability to interact with the auxiliary ligand occupying the final axial co-ordination site of the complex. Despite the heptadentate nature of this ligand, only five co-ordinate complexes have been reported to date. This may be due to the nature of the mixture of ligands available to the metal centre. With this in mind, we set out to determine whether, in the absence of other suitable ligands, seven co-ordinate species could be isolated from TPPA. In addition, as only Cu(II) complexes of TPPA have been crystallographically characterised, we investigated the co-ordination of this ligand with other transition metal ions.
Experimental
All reagents and solvents were purchased from commercial sources and used as received, unless noted otherwise. NMR spectra were measured using a Bruker AM 250–400 or Bruker Av-500 Plus FT-NMR spectrometer. Residual signals of the solvent were used for reference for 1H and 13C NMR. For infrared spectra, each compound was pressed into a disk with an excess of dried KBr and measured using a Jasco 660 FT-IR spectrophotometer. Electrospray (ES) and high-resolution (HR) mass spectra were measured using a Waters LCT Premier XE (oa-TOF) mass spectrometer. UV-vis absorption spectra were run in HPLC grade acetonitrile (Fisher) and measured using a Jasco V-570 spectrophotometer from 230 to 1100 nm (optical path length 1.0 cm). Elemental analyses were carried out by the Warwick Analytical Service, University of Warwick.
The cyclic voltammetry experiments were carried out using an AUTOLAB PGSTAT12 potentiostat in conjunction with the General Purpose Electrochemical System software (GPES version 4.7 for Windows) in a specially designed three-electrode glass cell with a Teflon-coated cell cap. A bioanalytical platinum working electrode (model no. MF2013) with a 1.6 mm disk was used for all experiments. The counter electrode was a platinum wire and the reference electrode an Ag/AgNO3 electrode. To regain electrochemical sensitivity and reproducibility the working electrode was polished, with 600 grid emery paper, to a mirror surface and then ultrasonicated. Prior to each experiment, the electrode was washed with high performance liquid chromatography (HPLC) grade CH3CN and dried in air for about 15 min. A 0.1 M [Bu4N][PF6] solution in CH3CN was used as the supporting electrolyte. In all cases, ferrocene was used as an internal reference. Solutions were degassed with nitrogen and a nitrogen atmosphere was maintained over the solution during the experiment.
Trisamide (TPPA) L1
Tris(6-pivaloylamino-2-pyridylmethyl)amine (TPPA) was prepared as reported by Harata et al.20
1H-NMR δH (250 MHz; CDCl3): 1.25 (27H, s); 3.69 (6H, s); 7.20 (3H, d, Ar, J = 7.4 Hz); 7.60 (3H, t, Ar, J = 7.8 Hz); 7.94 (3H, s, NH); 8.04 (3H, d, Ar, J = 8.1 Hz); 13C δC (62.5 MHz; CDCl3): 27.4, 39.7, 59.6, 112.1, 118.5, 138.8, 138.8, 150.9, 177.0; accurate ESMS (m/z): 610.3505 (100), [L + Na]+ [calculated 610.3481], 588.3676 (80) [L + H]+ [calculated 588.3676]; IR KBr/cm−1: ν = 3429m, 1691s; UV/Vis [λmax, nm (εM, M−1 cm−1)] in CH3CN: 320.5 (1811), 284.5 (17915), 243.5 (21865).
General procedure for the synthesis of metal complexes
The ligand (1 equivalent, typically 0.085 mmol) was dissolved in the minimum amount of acetonitrile (typically 3 mL). The solutions were warmed to ca. 60 °C to ensure that the ligand fully dissolved. To this stirring solution, the metal perchlorate, chloride or iodide salt (1 equivalent) dissolved in acetonitrile (typically ∼2 mL) was added dropwise. Recrystallisation of the compounds typically involved the diffusion of diethyl ether into acetonitrile solutions which were filtered through Celite. The yields from these reactions were moderate to high (25–97%). This crystalline material was then subsequently used for all spectroscopic measurements.
WARNING: Perchlorate salts of metal complexes are potentially explosive. Care should be taken while handling such complexes.
[MnC33H45N7O3]·[ClO4][Br]·CH3CN (1). Cream-coloured plate crystals (46% yield); accurate ESMS (m/z): 641.2864 (10) [Mn(L1) − H]+ [calculated 641.2886]; MnC35H48N8O3BrClO4 (found: C, 48.84; H, 5.72; N, 13.21% requires C, 48.76; H, 5.61; N, 13.00%); IR KBr/cm−1: ν = 3447br, 1617s, 1520s, 1424s, 1091s, 801s, 622s.
[FeC33H45N7O3]·[ClO4][Br]·CH3CN (2). Yellow needle crystals (66% yield); accurate ESMS (m/z): 642.2855 (20) [Fe(L1) − H]+ [calculated 642.2854]; FeC35H48N8O3BrClO4 (found: C, 48.92; H, 5.54; N, 13.12% requires C, 48.71; H, 5.61; N, 12.99%); IR KBr/cm−1: ν = 3445br, 1616s, 1533s, 1458s, 1443s, 1160s, 1086s, 622s; UV/Vis [λmax, nm (εM, M−1 cm−1)] in CH3CN: 321 (1180), 285 (20380), 237 (30170), 366 (315), 427 (220), 628 (3), 1087 (3).
[CoC33H45N7O3]·[Br]2·0.5(H2O)·2(CH3CN) (3). Pink plate crystals (29% yield); accurate ESMS (m/z): 745.2412 (15) [Co(L1) + ClO4] [calculated 745.2400], 645.2801 (10) [Co(L1) − H]+ [calculated 645.2837]; CoC37H52N9O3.50Br2 (found: C, 49.66; H, 5.93; N, 14.04% requires C, 49.50; H, 5.83; N, 14.04%); IR KBr/cm−1: ν = 3372s, 1618s, 1525s, 1420s, 1160s, 1097s, 622m; UV/Vis [λmax, nm (εM, M−1 cm−1)] in CH3CN: 236.2 (46100), 285.3 (27500), 501.5 (50), 618.4 (160), 685.5 (340).
[NiC33H45N7O3]·[ClO4]1.67[Br]0.33·0.67(H2O) (4). Yellow plate crystals (63% yield); accurate ESMS (m/z): 644.2885 (60) [Ni(L1) − H]+ [calculated 644.2859]; NiC33H46.33Cl1.67Br0.33N7O10.33; IR KBr/cm−1: ν = 3354m, 1618s, 1531s, 1459s, 1422s, 1106s, 622s; UV/Vis [λmax, nm (εM, M−1 cm−1)] in CH3CN: 232 (26390), 285 (15235), 440 (30), 613 (8), 748 (7), 803 (8), 1092 (15).
[ZnC33H45N7O3]·[ClO4]2·2(CH3CN) (5). Colourless prism crystals (89% yield); 1H-NMR δH (400 MHz; CD3CN): 1.14 (27H, s); 4.09 (6H, s); 7.12 (3H, d, Ar, J = 7.1 Hz); 7.21 (3H, d, Ar, J = 7.8 Hz); 7.87 (3H, t, Ar, J = 7.8 Hz); 8.98 (3H, s, NH); 13C δC (62.5 MHz; CD3CN): 27.0, 41.2, 55.8, 114.7, 119.9, 141.9, 153.0, 153.3, 181.3; accurate ESMS (m/z): 650.2823 (10) [Zn(L1) − H]+ [calculated 650.2797]; ZnC33H45N7O3(CH3CN)2(ClO4)2 (found: C, 46.93; H, 5.51; N, 13.51 requires C, 47.67; H, 5.51; N, 13.53%); IR KBr/cm−1: ν = 3401br, 1617s, 1530s, 1464s, 1421s, 1120s, 1091s, 622s.
[ZnC33H45N7O3Cl2] (6). Colourless block crystals (80% yield); 1H-NMR δH (250 MHz; DMSO): 1.34 (27H, s); 4.12 (6H, s); 7.18 (3H, d, Ar, J = 7.1 Hz); 7.49–8.31 (6H, m, Ar); 9.59 (3H, s, NH); 13C-NMR δC (62.5 MHz; DMSO): 26.9, 54.9, 55.6, 114.8, 119.7, 140.6, 150.7, 151.0, 176.7; accurate ESMS (m/z): 650.2786 (40) [Zn(L1) − H]+ [calculated 650.2797]; ZnC33H45N7O3(Cl)2 (found: C, 54.54; H, 6.27; N, 13.32% requires C, 54.90; H, 6.28; N, 13.59%). IR KBr/cm−1: ν = 3426m, 1683s, 1608s, 1517s, 1455s, 1152s.
[ZnC33H45N7O3]·[ZnI4]0.5·[I] (7). White plate crystals (63% yield); 1H-NMR δH (250 MHz; DMSO): 1.16 (27H, s); 4.16 (6H, s); 7.24 (3H, d, Ar, J = 7.2 Hz); 7.49 (3H, d, Ar, J = 7.5 Hz); 8.07 (3H, t, Ar, J = 8.0 Hz); 10.44 (3H, s, NH); 13C-NMR δC (62.5 MHz; DMSO): 26.5, 54.9, 55.0, 113.8, 119.0, 141.1, 151.8, 152.3, 179.9; accurate ESMS (m/z): 778.1943 (10) [Zn(L1) + I]+ [calculated 778.1920]; ZnC66H90N14O6(ZnI4)I; IR KBr/cm−1: ν = 3260m, 1615s, 1528s, 1456s, 1434s, 1417s, 1155s, 801s.
[CdC33H45N7O3]·[ClO4]2·Na(ClO4)·H2O (8). Colourless plate crystals (96% yield); 1H-NMR δH (250 MHz; CDCl3): 0.96 (27H, s); 3.76 (6H, s); 6.89 (3H, d, Ar, J = 7.5 Hz); 6.98 (3H, d, Ar, J = 8.2 Hz); 7.61 (3H, d, Ar, J = 8.0 Hz); 8.62 (3H, s, NH); 13C-NMR δC: (62.5 MHz; CD3CN): 27.4, 41.7, 56.7, 116.6, 121.4, 142.3, 153.5, 154.0, 183.1; accurate ESMS (m/z): 700.2570 (70), [Cd(L1) − H]+ [calculated 700.2539]; CdC33H45N7O7NaClH2O(ClO4)2; IR KBr/cm−1: ν = 3370br, 1615s, 1533s, 1454s, 1419s, 1100m, 622s.
Results and discussion
Synthesis of the ligand and complexes
Tris(6-pivaloylamino-2-pyridylmethyl)amine (TPPA) (7.272 g, 66%) was synthesised according to a literature procedure.20 Initial attempts to obtain crystals of complexes of L1 were only successful for Zn(ClO4)2 and Cd(ClO4)2. However, on using the protonated ligand [HL1][Br] we were able to obtain single crystals for MnII, FeII, CoII and NiII complexes. However, using this ligand combination with Zn(ClO4)2 did not yield a crystalline species. With this in mind, further investigations were carried out with the ZnII species by using different starting materials, ZnCl2 and ZnI2. The complexation involved the use of either ligand L1 or [HL1][Br] (1 equivalent, typically 0.085 mmol) which was dissolved in the minimum amount of warm acetonitrile. The metal salt (1 equivalent) was dissolved in acetonitrile and added dropwise. The compound was isolated by crystallisation through diffusion of diethyl ether vapour into the acetonitrile reaction mixture. On some occasions (Mn(II), Fe(II)), slow evaporation was used to obtain crystals. The yields from these reactions ranged from moderate to good (25–97%), though, of course, this reflects the amount of product that precipitated from solution, not the yield of product from the initial reaction. The crystalline material was subsequently used for all spectroscopic measurements.
Spectroscopic properties of complexes
IR spectra. The free ligand reveals a very strong single band at 1691 cm−1, assigned to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration of the three equivalent amide carbonyl groups (Table 1). This signal is observed at lower energy in all complexes upon co-ordination to the metal. For complex 6, although the solid state structure shows two carbonyl environments, neither are directly co-ordinated to the metal centre, though two amides form strong hydrogen bonds to the co-ordinated chlorides. Interestingly, only one carbonyl stretch is observed at 1683 cm−1. Similarly, Mareque Rivas observed the carbonyl stretch of the amide in bppapa, hydrogen bonded to the Zn–Cl moiety at 1700 cm−1.21
O) vibration of the three equivalent amide carbonyl groups (Table 1). This signal is observed at lower energy in all complexes upon co-ordination to the metal. For complex 6, although the solid state structure shows two carbonyl environments, neither are directly co-ordinated to the metal centre, though two amides form strong hydrogen bonds to the co-ordinated chlorides. Interestingly, only one carbonyl stretch is observed at 1683 cm−1. Similarly, Mareque Rivas observed the carbonyl stretch of the amide in bppapa, hydrogen bonded to the Zn–Cl moiety at 1700 cm−1.21
Table 1 IR stretching frequencies of L11 (cm−1)
| Compound |
ν(CO) |
ν(Cl–O) |
| IR spectra measured as KBr discs. |
| L1 |
1691(s) |
— |
| 1 |
1617(s) |
1091(m), 622(s) |
| 2 |
1616(s) |
1086(m), 622(s) |
| 3 |
1618(s) |
1097(s), 622(m) |
| 4 |
1618(s) |
1106(m), 622(s) |
| 5 |
1617(s) |
1091(s), 622(s) |
| 6 |
1683(s) |
— |
| 7 |
1615(s) |
— |
| 8 |
1615(s) |
1100(m), 622(s) |
Compounds 1–5 and 8 reveal two characteristic unsplit infrared active bands at ∼1100 cm−1 and 622 cm−1 indicative of ionic perchlorate (Td symmetry).22,23 This is confirmed by X-ray diffraction studies.
Cyclic voltammetry
The cyclic voltammogram of the manganese compound, 1, reveals three irreversible oxidations within the anodic region at +0.420, +0.708 and +1.17 V (vs. Fc+/Fc). In comparison, the seven co-ordinate manganese(II) complex of N,N,N′-tris(2-pyridylmethyl)-N′-(2-salicylideneethyl)ethane-1,2-diamine and MnII(pyrdoxTPA)2 reveals a Mn(II/III) redox process at +0.10 V and +0.18 V (vs. Fc+/Fc) respectively (Table 2).24–26
Table 2 Electrochemical parameters for the redox processes exhibited by complexes 1, 2, 3 and 4 in acetonitrile solution (supporting electrolyte: [Bu4N][PF6] (0.1 mol dm−3); T = 20 °C). Measured at 0.1 V s−1
| Compound |
Ep/V (ΔE, mV)a,b |
| The potentials at which reversible processes occur are calculated as the average of the oxidative and reductive peak potentials (Eoxp + Eredp)/2. For irreversible processes, the anodic or cathodic peak potentials are given. Potentials are given in volts versus ferrocenium/ferrocene. |
| 1 |
0.420, 0.708, 1.17 |
| 2 |
0.253(140) |
| 3 |
0.354, 0.588(65) |
| 4 |
−2.14 |
The cyclic voltammogram of 2 exhibits a quasi-reversible wave at +0.253 V (vs. Fc+/Fc) which has been ascribed to the Fe(II/III) redox couple. The separation between the anodic and cathodic peaks, Ecp − Eap, is 140 mV (significantly larger than the 80 mV observed for the internal standard). The compound [Fe(k7N–L](ClO4)2 L = N,N,N′,N′-tetrakis(2-pyridylmethyl)-2,6-bis(aminomethyl)pyridine also exhibits a seven-coordinate environment around the Fe(II) ion and also reveals a quasi-reversible Fe(II)/Fe(III) couple at the higher potential of +0.51 V vs. Fc+/Fc27 which surprisingly suggests that L1 leads to a more electron rich metal centre in 2.
The cyclic voltammogram of the cobalt(II) complex, 3, reveals a quasi-reversible redox process in the anodic region at +0.588 V (65) which has been tentatively assigned to the Co(II)/Co(III) couple. There is also a further irreversible oxidation at +0.354 V.
No reversible processes are observed in the cyclic voltammogram of the nickel compound 4. Only one irreversible reduction at −2.14 V (vs. Fc+/Fc) is seen.
Electronic absorption spectra
The electronic spectra of L1 and its complexes have been measured and the data are presented in Table 3. All measurements were recorded from an acetonitrile solution of the respective complex. The free ligand shows typical π–π* transitions at high energy (243, 284 and 320 nm). Each of the complexes, 2–4, also reveals π–π* transitions at similar energy to those of the free ligand.
Table 3 Electronic spectral assignments for the ligand and complexesa
| Compound |
π–π* λ (nm) |
MLCT λ (nm) |
d–d λ (nm) |
| Performed in a CH3CN solution at room temperature; numbers in parentheses indicate molar absorption coefficients ε (M−1 cm−1). |
| L11 |
243(21870) |
— |
— |
| 284(17920) |
|
|
| 320(1810) |
|
|
| 2 |
237(30170) |
366(315) |
628(3) |
| 285(20380) |
427(220) |
1087(3) |
| 321(1180) |
|
|
| 3 |
236.2(46100) |
— |
501.5(50), 618.4(160), 685.5(340) |
| 285.3(27500) |
|
|
| 4 |
232(26390) |
440(30) |
613(8), 748(7) |
| 285(15235) |
|
803(8) |
| |
|
1092(15) |
The electronic spectrum of compound 2 reveals two MLCT transitions at 27325 and 23420 cm−1 and two d–d transitions within the visible region at 15925 and 9200 cm−1, assuming a high spin co-ordination environment in solution. The observation of two (rather than one observed in octahedral complexes) is consistent with the lower C3v symmetry of a capped octahedron, which results in the splitting of the T2g term into an A and an E term.
The CoII complex 3 has also been shown to contain a seven-coordinate environment around the metal centre and its absorption spectrum consists of three absorptions at 19940, 16170 and 14590 cm−1. While a seven co-ordinate species will in theory show extensive splitting of the free ion F terms (to 2A2, A1 and 2E) and P terms (to A2 and E), the observed spectrum suggests that the splitting is small and best approximated to an octahedral geometry. However, the lowest energy transition is significantly higher than that observed for most octahedral Co(II) complexes28 and it is likely that the first transition was not observed in the available spectral range (300–1100 nm).
The UV spectrum of NiII, 4, reveals two LMCT bands observed at 22730 and 16300 cm−1. In the solid state, the NiII cation lies at the centre of an N4O3 coordination environment of a mono capped octahedral geometry. The absorption pattern is at first glance indicative of an octahedral geometry in solution. Four transitions are observed at 16300, 13370, 12450 and 9160 cm−1 which suggest transitions from 3A2g to 3T1g(P), 3T1g(F), 1Eg and 3T2g, respectively. However, caution should be applied as, if Δ and B are calculated utilising the first two spin allowed transitions, the values obtained are not consistent with the energy of the third spin allowed transition. Therefore it seems likely that the spectrum reflects a lower symmetry than Oh.
Crystallographic studies
All single crystal X-ray data were collected on a Bruker/Nonius Kappa CCD diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å), equipped with an Oxford Cryostream cooling apparatus. Crystal parameters and details of the data collection, solution and refinement are presented in Table 4. The data were corrected for Lorentz and polarization effects and for absorption using SORTAV.29 Generally structure solution was achieved by direct methods (the Sir-92 program system)30 and refined by full-matrix least-squares on F2 (SHELXL-97)31 with all non-hydrogen atoms assigned anisotropic displacement parameters. Hydrogen atoms attached to carbon atoms were placed in idealised positions and allowed to ride on the relevant carbon atom. In the final cycles of refinement, a weighting scheme that gave a relatively flat analysis of variance was introduced and refinement continued until convergence was reached. Molecular structures in the figures were drawn with ORTEP-3.0 for Windows (version 2.02).32 The metal co-ordination geometry is given for all structures in Table 5. Continuous shape mapping (CShM) was used to calculate the deviation in the core geometry of crystal structures.33 Unfortunately, only poor quality crystals could be obtained for [NiL1][Br][ClO4], despite numerous attempts to grow better crystals.
| Compound |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
| Chemical formula |
MnC36H49.5N8.5O7BrCl |
FeC35H48N8O7BrCl |
CoC37H52N9O3.50Br2 |
NiC33H46.33Br0.33N7Cl1.67O10.33 |
ZnC37H51N9O11Cl2 |
ZnC33H45N7O3Cl2 |
Zn3C66H90N14O6I6 |
CdC33H47N7O16NaCl3 |
| Mr (g mol−1) |
883.64 |
864.02 |
897.63 |
850.87 |
934.14 |
724.03 |
2133.03 |
1039.52 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Hexagonal |
Monoclinic |
Monoclinic |
Trigonal |
Trigonal |
| Space group |
P21/m |
P21/m |
C2/c |
P63/m |
C2/c |
C2/c |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 c1 |
R32 |
| T (K) |
150(2) |
150(2) |
150(2) |
396(2) |
150(2) |
140(2) |
150(2) |
150(2) |
| a (Å) |
10.8160(2) |
10.730(5) |
11.1532(3) |
11.1018(7) |
37.4589(10) |
35.4990(4) |
11.2190(2) |
16.2935(5) |
| b (Å) |
40.8620(8) |
40.995(5) |
19.6165(5) |
11.1018(7) |
11.2206(3) |
9.1250(7) |
11.2190(2) |
16.2935(5) |
| c (Å) |
11.1620(2) |
11.101(5) |
37.6128(12) |
58.814(4) |
21.1473(8) |
24.5700(12) |
38.5920(4) |
28.8420(13) |
| α (°) |
90 |
90 |
90 |
90 |
90 |
90 |
90 |
90 |
| β (°) |
116.9550(10) |
117.050(5) |
90.6930(10) |
90 |
93.2280(10) |
119.271(2) |
90 |
90 |
| γ (°) |
90 |
90 |
90 |
120 |
90 |
90 |
120 |
120 |
| Z |
4 |
4 |
8 |
6 |
8 |
8 |
2 |
6 |
| Dc (Mg m−3) |
1.335 |
1.320 |
1.449 |
1.375 |
1.398 |
1.385 |
1.684 |
1.562 |
| μ(Mo Kα) (mm−1) |
1.322 |
1.378 |
2.407 |
0.875 |
0.740 |
0.906 |
3.102 |
0.760 |
| Collected reflections |
14579 |
10265 |
13749 |
2176 |
29824 |
16682 |
22993 |
6695 |
| Unique reflections |
8298 |
7092 |
7535 |
1987 |
10144 |
6270 |
3221 |
2772 |
| Rint |
0.0460 |
0.0477 |
0.0453 |
0.0735 |
0.1050 |
0.0696 |
0.0737 |
0.0821 |
| R1 [I > 2σ(I)] |
0.0619 |
0.0769 |
0.04256 |
0.2730 |
0.0779 |
0.0765 |
0.0456 |
0.1023 |
| wR2 (all data) |
0.1478 |
0.1749 |
0.0883 |
0.6453 |
0.1842 |
0.1891 |
0.1154 |
0.2514 |
| |
[MnL1]2+ 1 |
[FeL1]2+ 2 |
[CoL1]2+ 3 |
[NiL1]2+ 4 |
[ZnL1]2+ 5 |
[ZnL1]2+ 7 |
[CdL1]2+ 8 |
| For convenience, the following labels in the table should be replaced with those indicated for the corresponding metal complexes: For Ni(4), Zn(7) and Cd(8): N(4) = N(2)#1, N(6) = N(2)#2, O(2) = O(1)#1, O(3) = O(1)#2. For Zn(5): N(4) = N(3), N(6) = N(4). Symmetry operations represented by #1 and #2 are unique for each complex. |
| M(1)–N(1) |
2.264(4) |
2.220(6) |
2.184(3) |
2.53(2) |
2.221(4) |
2.180(6) |
2.354(12) |
| M(1)–N(2) |
2.309(4) |
2.256(5) |
2.217(3) |
2.264(7) |
2.244(5) |
2.233(4) |
2.362(5) |
| M(1)–N(4) |
2.316(4) |
2.257(5) |
2.236(3) |
2.264(7) |
2.182(4) |
2.233(4) |
2.362(5) |
| M(1)–N(6) |
2.309(4) |
2.256(6) |
2.205(3) |
2.264(7) |
2.205(4) |
2.233(4) |
2.362(5) |
| M(1)–O(1) |
2.173(3) |
2.143(5) |
2.143(2) |
2.033(11) |
2.148(4) |
2.127(3) |
2.249(8) |
| M(1)–O(2) |
2.161(3) |
2.158(5) |
2.119(2) |
2.033(11) |
2.149(3) |
2.127(3) |
2.249(8) |
| M(1)–O(3) |
2.185(3) |
2.132(5) |
2.114(2) |
2.033(11) |
2.137(3) |
2.127(3) |
2.249(8) |
| N(1)–M(1)–N(2) |
73.18(14) |
73.2(2) |
73.71(10) |
70.3(2) |
73.44(19) |
73.98(9) |
71.88(11) |
| N(1)–M(1)–N(4) |
71.78(14) |
73.0(2) |
73.03(9) |
70.3(2) |
74.42(15) |
73.98(9) |
71.88(11) |
| N(1)–M(1)–N(6) |
72.02(14) |
73.0(2) |
74.42(10) |
70.3(2) |
73.57(17) |
73.98(9) |
71.88(11) |
| N(2)–M(1)–N(4) |
111.38(14) |
113.0(2) |
112.58(10) |
109.3(2) |
110.18(15) |
112.69(7) |
110.79(11) |
| N(2)–M(1)–N(6) |
113.29(14) |
112.7(2) |
111.48(10) |
109.3(2) |
113.27(16) |
112.69(7) |
110.79(11) |
| N(6)–M(1)–N(4) |
108.96(14) |
109.86(19) |
113.33(10) |
109.3(2) |
114.16(16) |
112.69(7) |
110.79(11) |
| O(1)–M(1)–N(1) |
130.36(14) |
131.6(2) |
130.50(9) |
128.2(4) |
132.12(16) |
129.48(9) |
126.7(2) |
| O(1)–M(1)–N(2) |
76.88(13) |
78.81(19) |
78.89(9) |
81.9(4) |
78.98(17) |
78.34(12) |
73.5(4) |
| O(1)–M(1)–N(4) |
157.51(14) |
83.22(19) |
156.47(9) |
79.0(4) |
79.60(14) |
79.12(12) |
159.5(4) |
| O(1)–M(1)–N(6) |
85.07(14) |
155.3(2) |
78.56(9) |
161.8(5) |
154.28(15) |
156.54(13) |
84.5(5) |
| O(1)–M(1)–O(3) |
85.04(13) |
83.78(19) |
85.26(9) |
86.4(6) |
81.63(14) |
83.89(13) |
87.9(3) |
| O(2)–M(1)–O(1) |
84.92(13) |
82.54(18) |
83.15(9) |
86.4(6) |
83.62(14) |
83.89(13) |
87.9(3) |
| O(2)–M(1)–N(1) |
126.19(14) |
130.31(18) |
129.92(10) |
128.2(4) |
128.92(16) |
129.48(9) |
126.7(2) |
| O(2)–M(1)–N(2) |
79.75(13) |
156.5(2) |
80.18(9) |
161.8(5) |
157.64(16) |
156.54(13) |
85.5(5) |
| O(2)–M(1)–N(4) |
76.44(13) |
78.44(18) |
78.96(9) |
81.9(4) |
80.01 (14) |
78.34(13) |
73.5(4) |
| O(2)–M(1)–O(3) |
86.29(13) |
85.22(18) |
84.46(9) |
86.4(6) |
85.05(13) |
83.89(13) |
87.9(3) |
| O(2)–M(1)–N(6) |
161.19(14) |
79.91(19) |
155.66(10) |
79.0(4) |
77.97(14) |
79.12(12) |
159.5(4) |
| O(3)–M(1)–N(1) |
128.76(14) |
126.6(2) |
127.22(10) |
128.2(4) |
128.34(14) |
129.48(9) |
126.7(2) |
| O(3)–M(1)–N(2) |
157.98(14) |
78.7(2) |
159.07(10) |
79.0(4) |
78.65(15) |
79.12(12) |
159.5(4) |
| O(3)–M(1)–N(4) |
81.34(14) |
160.3(2) |
77.94(9) |
161.8(5) |
157.12(15) |
156.54(13) |
85.5(5) |
| O(3)–M(1)–N(6) |
77.00(13) |
77.6(2) |
78.18(9) |
81.9(4) |
79.06(14) |
78.34(13) |
72.5(4) |
[MnII(L1)][ClO4][Br]·CH3CN (1). The manganese compound crystallises in the monoclinic space group P21/m and contains one complex within the asymmetric unit (asu) (Fig. 2). The MnII ion lies at the centre of a slightly distorted capped octahedron co-ordination geometry which has been confirmed by CShM (Table 6). The MnII ion is surrounded by two types of donor atoms. There are four N-donors N1, N2, N4 and N6 and three oxygen donors O1, O2 and O3 (Fig. 3). The co-ordinative bond lengths range from 2.161(3) Å to 2.316(4) Å. The co-ordinative bond lengths of nitrogen donors N1, N2, N4 and N6 (Table 5) are slightly shorter than those of the seven coordinate compound [MnII(TPA)][NO3]2 (TPA = tris(2-pyridylmethyl)amine) which ranges from 2.2616(16) Å for the pyridyl donors to 2.3889(16) Å for the central nitrogen donor.34 This may reflect the steric constraints of L1, which leads to closer contact between the metal and the ligand.
 |
| | Fig. 2 The asymmetric unit of 1. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
 |
| | Fig. 3 A view of the core geometry of 1 of covalent bond radius less than 3 Å. The metal has been removed to help illustrate the geometry. | |
Table 6 Continuous symmetry mapping results of seven coordinate complexes show strong preferences of a mono capped octahedron
| Geometry |
Compound |
| 1 |
2 |
3 |
4 |
5 |
7 |
8 |
| Heptagon |
35.93 |
35.63 |
36.22 |
36.52 |
35.78 |
36.64 |
36.53 |
| Hexagonal pyramid |
20.02 |
20.40 |
19.78 |
19.60 |
20.18 |
19.79 |
19.29 |
| Pentagonal bipyramid |
7.84 |
7.68 |
7.75 |
8.75 |
7.79 |
8.28 |
8.53 |
| Capped octahedron |
0.33 |
0.34 |
0.23 |
0.53 |
0.29 |
0.24 |
0.48 |
| Capped trigonal prism |
1.53 |
1.50 |
1.48 |
2.02 |
1.25 |
1.64 |
1.89 |
| Johnson pentagonal bipyramid J13 |
11.67 |
11.42 |
11.49 |
12.08 |
11.37 |
12.05 |
12.40 |
| Johnson elongated triangular pyramid J7 |
18.85 |
19.44 |
20.31 |
18.05 |
19.69 |
20.19 |
17.31 |
[FeII(L1)][ClO4][Br]·CH3CN (2). The iron compound crystallises in the monoclinic space group P21/m and contains one complex within the asu. Similar to the Mn(II) complex, the FeII is co-ordinated via four N-donors and three oxygen donors. Again, the core geometry is best described as a capped octahedron, with S(OCF) and S(TPRS) values of 0.335 and 1.498, respectively (Table 6). The co-ordinative bond lengths are shorter than those observed for Mn(II) and range from 2.132(5) Å to 2.257(5) Å (Table 5). These values are similar to those observed for the 7-coordinate complex, [Fe(H2bppa)(HCOO)]2+ (bppa = bis(6-pivalamido-2-pyridylmethyl)(2-pyridylmethyl)amine), reported by Wada et al. where the Fe–N bond lengths range between 2.004(3) Å and 2.226(3) Å.35
[CoII(L1)][Br]2·0.5(H2O)·2(CH3CN) (3). The cobalt compound crystallises in the monoclinic space group C2/c and contains one complex within the asu (Fig. 4). There are four nitrogens and three oxygens within the co-ordinative sphere of the central Co(II) ion. In an attempt to grow crystals of 3, the perchlorate salt was used with no success. However, crystals were obtained by using HBrL1. The co-ordination geometry of the CoII ion is best described as a capped octahedron similar to all other structures (except compound 6). This is the first example of CoII being seven coordinate in a TPA based ligand.
 |
| | Fig. 4 The asymmetric unit of 3. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
[NiII(L1)][ClO4]1.67[Br]0.33·0.67(H2O) (4). The nickel compound crystallises in the hexagonal space group P63/m and contains two unique complexes within the asu, of which one is disordered. Unfortunately, due to this disorder and the poor quality of the obtained crystals the solution to this structure has a high R factor, with the exact location of two counter-ions and a solvent molecule being unclear. However, the general co-ordination mode of the ligand to the nickel centre in one complex is well determined. In an analogous manner to the MnII (1) and FeII (2) complexes, the NiII ion is coordinated by four N-donors and three O donors (Fig. 5 and 6). The co-ordinative bond lengths are typical with the exception of the long NiII–N1 bond (Table 5). This long bond perhaps reflects the tendency of NiII towards a six co-ordinate octahedral geometry, and of the seven co-ordinate structures formed by L1, the nickel complex shows the greatest deviation from both mono capped octahedral and mono capped trigonal prismatic seven co-ordinate geometries, towards the six co-ordinate octahedral geometry.
 |
| | Fig. 5 The asymmetric unit of 4. Displacement ellipsoids are shown at 10% probability. H atoms are of arbitrary size. | |
 |
| | Fig. 6 A view of the core geometry of 4 of covalent bond radius less than 3 Å. The metal has been removed to help illustrate the geometry. | |
[ZnII(L1)][ClO4]2·2(CH3CN) (5). The zinc compound crystallises in the monoclinic space group C2/c and contains one complex within the asu (Fig. 7). The Zn(II) cation lies at the centre of a capped octahedral geometry (Table 6). There are four nitrogen donors (N1, N2, N3 and N4) and three oxygen donors (O1, O2 and O3) (Fig. 8). All four M–N bond lengths are very similar in length and vary from 2.182(4) Å to 2.244(5) Å. This is longer than those found in [ZnII(TPA)Cl]Cl36 which range from 2.062(2) to 2.083(2) Å for the M–N pyridyl bonds. All three oxygen donors are slightly shorter and range between 2.137(3) Å and 2.149(3) Å. The structure is similar to those observed with Mn(II), Fe(II), Co(II) and Zn(II) (7).
 |
| | Fig. 7 The asymmetric unit of 5. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
 |
| | Fig. 8 A view of the core geometry of 5 of covalent bond radius less than 3 Å. The metal has been removed to help illustrate the geometry. | |
[ZnII(L1)(Cl)2] (6). Compound (6) was synthesised by the reaction of L1 with ZnCl2. The zinc compound crystallises in the monoclinic space group C2/c and contains one complex within the asu (Fig. 9). The Zn(II) cation lies at the centre of a slightly distorted trigonal bipyramidal geometry (Tables 7 and 8). There are three nitrogen donors (N1, N2 and N4), two of which are pyridyl groups, the other being an aliphatic nitrogen donor (N1). The aliphatic nitrogen donor and two chloride donors occupy the equatorial positions. One arm of TPPA does not co-ordinate to the metal centre (Fig. 10). The co-ordinative bond lengths are typical of nitrogen donors and vary from 2.139(5) Å to 2.233(5) Å (Table 7) and again are similar to the compound [ZnII(TPA)Cl]Cl.36
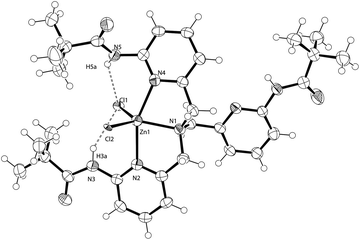 |
| | Fig. 9 The asymmetric unit of 6. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
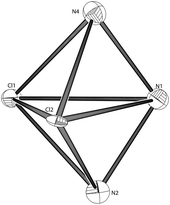 |
| | Fig. 10 A view of the core geometry of 6 of covalent bond radius less than 3 Å. The metal has been removed to help illustrate the geometry. | |
Table 7 Selected bond lengths (Å) and angles (°) for [ZnL1Cl2] 6
| Zn(1)–N(1) |
2.138(5) |
Zn(1)–Cl(1) |
2.3033(15) |
| Zn(1)–N(2) |
2.228(5) |
Zn(1)–Cl(2) |
2.3327(13) |
| Zn(1)–N(4) |
2.209(5) |
|
|
| |
| N(1)–Zn(1)–N(2) |
78.53(18) |
N(2)–Zn(1)–Cl(2) |
93.21(13) |
| N(1)–Zn(1)–N(4) |
78.31(18) |
N(4)–Zn(1)–N(2) |
156.82(18) |
| N(1)–Zn(1)–Cl(1) |
116.78(14) |
N(4)–Zn(1)–Cl(1) |
94.60(13) |
| N(1)–Zn(1)–Cl(2) |
123.81(14) |
N(4)–Zn(1)–Cl(2) |
99.04(14) |
| N(2)–Zn(1)–Cl(1) |
96.42(13) |
Cl(1)–Zn(1)–Cl(2) |
119.36(5) |
Table 8 Continuous shape mapping results for complexes 6
| Structure |
(PP) |
(VOC) |
(TBPY) |
(SPY) |
(JSPY) |
(JTBP) |
| PP: pentagon (D5h), VOC: vacant octahedron (C4v), TBPY: trigonal bipyramid (D3h), SPY: square pyramid (C4v), JSPY: Johnson square pyramid (C4v), JTBP: Johnson trigonal bipyramid (D3h). |
| L1_Zn(6) |
33.350 |
6.694 |
1.465 |
4.061 |
6.694 |
4.969 |
Hydrogen bond interactions are observed between H3a⋯Cl1 and H5a⋯Cl1 (Fig. 11) with bond lengths 2.637, 2.618 Å respectively. A weaker hydrogen bond is observed between H3a⋯Cl2 (3.208 Å). Previously, Mareque Rivas18 has shown how the formation of a CuL1Cl complex is stabilised by effective hydrogen bonding between the amide H-bond donor and the chloride. Surprisingly, in this case both chloride atoms co-ordinate to the metal leaving one arm of the ligand free. This might suggest that in the solid state for zinc, with no strong LFSE, there is no strong preference for a particular ligand set and strong hydrogen bonding interactions lead to both chlorides co-ordinating to the metal centre, each supported by an interaction with an amido group.
 |
| | Fig. 11 A view of the hydrogen bonding interactions of 6. | |
[ZnII(L1)][ZnI4]0.5[I] (7). From the reaction of ZnI2 with L1, the resulting zinc compound crystallises in the trigonal space group Pc1 and contains one complex within the asu (Fig. 12). In the structure, there are two types of anionic counter-ions: a simple iodide anion and the dianion, [ZnI4]2−. There is one [ZnI4]2− counter-ion for every two cationic species, and the [ZnI4]2− species was found to be disordered within the lattice. Similar to 5 the metal ion lies within a slightly distorted capped octahedron (Table 6), although the complex now has threefold symmetry with all pyridyl donors and oxygen donors being symmetrically equivalent (Fig. 13). Comparison between complexes 5 and 7 shows very similar bond lengths and angles.
 |
| | Fig. 12 The asymmetric unit of 7. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
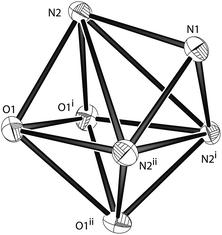 |
| | Fig. 13 A view of the core geometry of 7 of covalent bond radius less than 3 Å. The metal has been removed to help illustrate the geometry. | |
[CdII(L1)][ClO4]2·Na(ClO4)·H2O (8). The cadmium compound crystallises in the trigonal space group R32 and contains one complex within the asu (Fig. 14). The cadmium complex again has a seven co-ordinated, capped-octahedral geometry (Table 6). Unexpectedly, in the crystals obtained, a sodium perchlorate molecule co-crystallised. The sodium counter-ion was an impurity in the ligand starting material, included with the ligand when it was dried with sodium sulphate. Again, as seen in 4 and 7, there is threefold symmetry about the N(1)–M bond, making the three M–pyridine bond equivalent. The metal–donor bond lengths observed in this complex are similar to those observed in related species; for example in [CdII(TPA)(NO3)(H2O)]·NO33 the N–CdII bond lengths vary from 2.30(1) Å to 2.44(1) Å.
 |
| | Fig. 14 The asymmetric unit of 8. Displacement ellipsoids are shown at 50% probability. H atoms are of arbitrary size. | |
Structural analysis
Continuous shape mapping (CShM) quantifies the shape of a co-ordination geometry when compared to idealised polyhedra. It allows the identification of any distortion from the idealised geometry and the S value for a given geometry is a measure of how close it is to a given idealised geometry. Near-perfect shapes will have an S value close to zero and geometries which depart significantly from a perfect polyhedron will have higher S values. Table 6 lists the shape values for complexes 1–5, 7 and 8 for the various seven vertices structures.
Conclusions
A series of transition metal complexes with a trisamide ligand based on a TPA framework have been synthesised and structurally characterised. This ligand readily forms seven co-ordinate complexes when no additional potential ligands are present. However, in the case of 6, a five coordinate trigonal bipyramidal geometry is preferred due to the co-ordination of chloride anions to the metal centre. This formation of a five co-ordinate species in the presence of the chloride anion shows how the resulting co-ordination environment seems to be dependent on the identity of the counter-ion and suggests that the donor strength of the carbonyl in L1 is intermediate to chloride and bromide. In fact, bromide, iodide and perchlorate are all weaker donors than the carbonyl and result in seven co-ordinate structures. In addition, the ability of ternary ligands to bind to ML1 may also be modified by the ternary ligand's ability to hydrogen bond to L1 upon co-ordination to the metal.
With the exception of the five co-ordinate complex, 6, all the seven co-ordinate structures are best described as having a capped octahedral metal centre, though the shape analysis highlights how closely related the MCO and MCTP geometries are.
Table 9 shows the angles θ2 and θ5 (see Fig. 15) for all mono capped octahedron structures in relation to the ideal and Hoffman ideal angles.37 In this model, θ2 is the N–M–N angle between the capping N, the metal centre and any of the next three nearest N atom donors. θ5 is the N–M–N angle between the capping N donor and the furthest three N donors.
 |
| | Fig. 15 θ2 and θ5 in a mono capped octahedron. | |
Table 9 A comparison between θ2 and θ5 averaged angles for the structures and their relationship with Hoffman's angles
| |
θ2 |
θ5 |
| Ideal |
54.7° |
125.3° |
| Hoffman ideal |
75° |
138° |
| MnII (1) |
72.4° |
128.5° |
| FeII (2) |
73.7° |
129.2° |
| CoII (3) |
73.1° |
129.6° |
| NiII (4) |
70° |
128° |
| ZnII (5) |
73.9° |
129.8° |
| ZnII (7) |
73.9° |
129.5° |
| CdII (8) |
71.9° |
126.6° |
It would appear that the ML1 structures are sterically constrained and all angles lie between the ideal CO and Hoffman ideal angles.37 In addition, due to the long bond for Ni–N1, in comparison to all other M–N1 bonds, we can see that θ2 (N1–Ni–N2, N1–Ni–N4, N1–Ni–N6) is relatively acute compared to θ2 in the other complexes; however, in general terms, it is interesting to note that such a different structure only produces a ∼3–4° change in angle when compared to the other late transition metal ions, while the larger cations, Mn(II) and Cd(II), are different again and the angles lie between those observed for nickel and the other metals.
Finally, a CCDC database search shows how rare this coordination number is for transition metal complexes. For example, among all manganese structures only 6.1% are seven coordinate, iron ∼4.0%, cobalt ∼2.3%, nickel ∼0.7%, zinc ∼0.4%, and cadmium ∼7.3%. This tripodal ligand favours the formation of seven co-ordinate capped octahedral complexes when no additional ligands are present. Such geometries are extremely rare and more typically seen in complexes of the larger cations, such as Mn(II) and Cd(II). It is particularly unusual to observe seven co-ordinate structures of Ni(II) and Zn(II). The crystallographic data highlight how the pre-organisation of the ligand leads to what may typically be regarded as an unfavourable geometry of the transition metal ions and the formation of seven co-ordinate species with labile amide donors may be useful for the formation of catalyst precursors.
Acknowledgements
We wish to thank Dr R. J. Jenkins and R. Hicks for performing the mass spectrometry experiments. We also thank Umm Al-Qura University of the Kingdom of Saudi Arabia for financial support.
References
- G. Anderegg and F. Wenk, Helv. Chim. Acta, 1967, 50, 2330 CrossRef CAS.
- A. Hazell, K. B. Jensen, C. J. McKenzie and H. Toftlund, Inorg. Chem., 1994, 33, 3127 CrossRef CAS.
- C. S. Allen, C.-L. Chuang, M. Cornebise and J. W. Canary, Inorg. Chim. Acta, 1995, 239, 29–37 CrossRef CAS.
- B. G. Gafford and R. A. Holwerda, Inorg. Chem., 1989, 28, 60 CrossRef CAS.
- C. X. Zhang, S. Kaderli, M. Costas, E. Kim, Y.-M. Neuhold, K. D. Karlin and A. D. Zuberbühler, Inorg. Chem., 2003, 42, 1807 CrossRef CAS PubMed.
- A. Diebold and K. S. Hagen, Inorg. Chem., 1998, 37, 215 CrossRef CAS.
- D. G. Lonnon, D. C. Craig and S. B. Colbran, Dalton Trans., 2006, 3785 RSC.
- Z. H. Zhang, X. H. Bu, Z. A. Zhu and Y. T. Chen, Polyhedron, 1996, 15, 2787 CrossRef CAS.
- J. Bjernemose, A. Hazell, C. J. McKenzie, M. F. Mahon, L. P. Nielsen, P. R. Raithby, O. Simonsen, H. Toftlund and J. A. Wolny, Polyhedron, 2003, 22, 875 CrossRef CAS.
- A. Diebold and K. S. Hagen, Inorg. Chem., 1998, 37, 215 CrossRef CAS.
- Y. Gultneh, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, Inorg. Chim. Acta, 1993, 211, 171 CrossRef CAS.
- Z. He, D. C. Craig and S. B. Colbran, Dalton Trans., 2002, 13 Search PubMed.
- Z. He, P. J. Chaimungkalanont, D. C. Craig and S. B. Colbran, Dalton Trans., 2000, 1419 RSC.
- Y. Zang, J. Kim, Y. H. Dong, E. C. Wilkinson, E. H. Appelman and L. Que, J. Am. Chem. Soc., 1997, 119, 4197 CrossRef CAS.
- H. Hayashi, S. Fujinami, S. Nagatomo, S. Ogo, M. Suzuki, A. Uehara, Y. Watanabe and T. Kitagawa, J. Am. Chem. Soc., 2000, 122, 2124 CrossRef CAS.
- H. Hayashi, K. Uozumi, S. Fujinami, S. Nagatomo, K. Shiren, H. Furutachi, M. Suzuki, A. Uehara and T. Kitagawa, Chem. Lett., 2002, 416 CrossRef CAS.
- C.-l. Chuang, K. Lim, Q. Chen, J. Zubieta and J. W. Canary, Inorg. Chem., 1995, 34, 2562 CrossRef CAS.
- J. C. Mareque Rivas, S. L. Hinchley, L. Metteau and S. Parsons, Dalton Trans., 2006, 2316 RSC; D. Natale and J. C. Mareque Rivas, Chem. Commun., 2008, 425 RSC.
- S. Yamaguchi, A. Wada, Y. Funahashi, S. Nagatomo, T. Kitagaw, K. Jitsukawa and H. Masuda, Eur. J. Inorg. Chem., 2003, 4378 CrossRef CAS.
- M. Harata, K. Jitsukawa, H. Masuda and H. Einaga, Chem. Lett., 1995, 61 CrossRef CAS.
- J. C. Mareque Rivas, R. T. Martin de Rosales and S. Parsons, Dalton Trans., 2003, 2156 RSC.
- M. G. B. Drew, J. Nelson, F. Esho, V. McKee and S. M. Nelson, J. Chem. Soc., Dalton Trans., 1982, 1837 RSC.
- P. Dapporto, G. De Munno, A. Sega and C. Mealli, Inorg. Chim. Acta, 1984, 83, 171 CrossRef CAS.
- O. Horner, J.-J. Girerd, C. Philouze and L. Tchertanov, Inorg. Chim. Acta, 1999, 290, 139 CrossRef CAS.
- P. Gili, M. G. Martín Reyes, P. Martín Zarza, M. F. C. Guedes da Silva, Y. Y. Tong and A. J. L. Pombeiro, Inorg. Chim. Acta, 1997, 255, 279 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- D. G. Lonnon, G. E. Ball, I. Taylor, D. C. Craig and S. B. Colbran, Inorg. Chem., 2009, 48, 4863 CrossRef CAS PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, 1984 Search PubMed.
- R. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33 CrossRef.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693 CrossRef CAS PubMed.
- S. M. Baldeau, C. H. Slinn, B. Krebs and A. Rompel, Inorg. Chim. Acta, 2004, 357, 3295 CrossRef CAS PubMed.
- A. Wada, S. Ogo, S. Nagatomo, T. Kitagawa, Y. Watanabe, K. Jitsukawa and H. Masuda, Inorg. Chem., 2002, 41, 616 CrossRef CAS PubMed.
- C. Duboc, T. Phoeung, D. Jouvenot, A. G. Blackman, L. F. McClintock, J. Pécaut, M.-N. Collomb and A. Deronzier, Polyhedron, 2007, 26, 5243 CrossRef CAS PubMed.
- R. Hoffmann, B. F. Beier, E. L. Muetterties and A. R. Rossi, Inorg. Chem., 1977, 16, 511 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a