 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optically pure heterobimetallic helicates from self-assembly and click strategies†
Suzanne E. Howson,
Guy J. Clarkson,
Alan D. Faulkner,
Rebecca A. Kaner,
Michael J. Whitmore and
Peter Scott*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: peter.scott@warwick.ac.uk; Fax: +44 (0) 24 7657 2710; Tel: +44 (0) 24 7652 3238
First published on 1st August 2013
Abstract
Single diastereomer, diamagnetic, octahedral Fe(II) tris chelate complexes are synthesised that contain three pendant pyridine proligands pre-organised for coordination to a second metal. They bind Cu(I) and Ag(I) with coordination geometry depending on the identity of the metal and the detail of the ligand structure, but for example homohelical (ΔFe,ΔCu) configured systems with unusual trigonal planar Cu cations are formed exclusively in solution as shown by VT-NMR and supported by DFT calculations. Similar heterobimetallic tris(triazole) complexes are synthesised via clean CuAAC reactions at a tris(alkynyl) complex, although here the configurations of the two metals differ (ΔFe,ΛCu), leading to the first optically pure heterohelicates. A second series of Fe complexes perform less well in either strategy as a result of lack of preorganisation.
Introduction
Metallo-helicate chemistry is almost exclusively confined to Dn-symmetric systems (n = 2–4) self-assembled from symmetric ditopic ligands as shown in Fig. 1(a). Heterobimetallic helicates, although relatively rare, provide an opportunity to synthesise Cn or lower symmetry systems in which two different bidentate coordinating groups are connected via a linker, and using two metals which have a preference for one end of the ligand over the other1–6 [Fig. 1(b)]. Alternatively, in a multi-step synthesis of heterobimetallics, one (inert) metal is coordinated before a second coordination site and metal are introduced.7 With fully inert systems, enantiomers can sometimes be resolved using e.g. chromatography to isolate (highly) optically enriched helicates2,7 but to our knowledge, the self-assembly of optically pure (or at least highly enantiomerically enriched) heterobimetallic complexes has not been reported. This could provide strategies for the synthesis of large complexes in which precise placement of functionality and charge can be achieved i.e. towards systems which mimic natural self-assembling systems such as protein α-helices.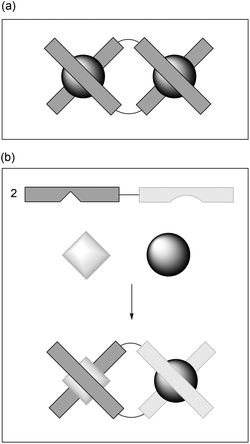 | ||
| Fig. 1 Showing (a) a conventional D2-symmetric helicate assembled from a bis(bidentate) ligand and two metal ions; and (b) a heterobimetallic C2-symmetric system. | ||
We have recently shown that a range of single diastereomer diimine complexes of Fe(II) can be made very readily using 2-pyridinecarboxaldehyde and simple phenylethanamines as the source of chirality (Fig. 2).8,9 The diastereomeric ratios were determined by 1H NMR spectroscopy to be >200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all cases and this unique system has since been exploited by ourselves10 and others11,12 to generate novel architectures. Since these monometallic Fe(II) units are relatively inert, the opportunity arises to carry out further reactions as one might with say a ruthenium tris(chelate).7
:1 in all cases and this unique system has since been exploited by ourselves10 and others11,12 to generate novel architectures. Since these monometallic Fe(II) units are relatively inert, the opportunity arises to carry out further reactions as one might with say a ruthenium tris(chelate).7
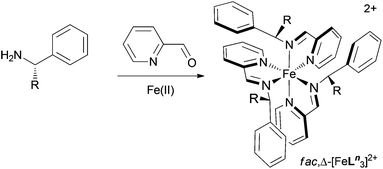 | ||
| Fig. 2 The self-assembling diastereomerically pure tris-chelate system used in this work.8,9 | ||
In this report we describe the coordination of a second metal to either a preformed second coordination site or one created in situ by copper(I)-catalysed Huisgen 1,3-dipolar cycloaddition (CuAAC).
Results and discussion
In principle, suitable trifunctional metallo-ligands based on the complexes of Fig. 2 might be formed by functionalising at the phenylglycinol-derived site R or at the pyridine. We address these two approaches with ligands L1–61–6 and L7–107–10 respectively (Fig. 3).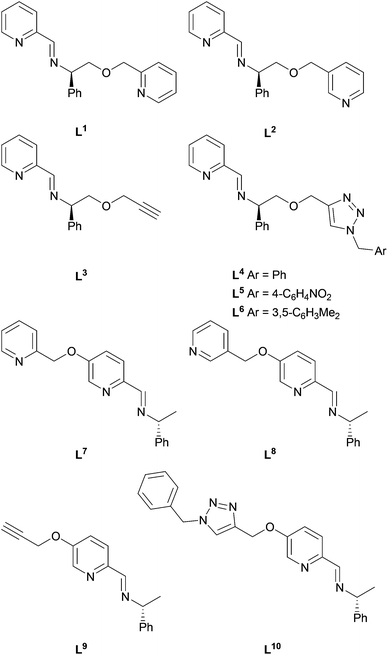 | ||
| Fig. 3 Ligands L11–L1010 assembled in situ via condensation or CuAAC reactions. | ||
Pyridinyl ethers L1–21–2
The use of three equivalents of 2-(pyridin-2-methoxy)-1-phenylethanamine in a one-pot synthesis with three equivalents of 2-pyridinecarboxaldehyde and one equivalent of Fe(ClO4)2·6H2O led to the formation of a complex of the type shown in Fig. 2 fac,ΔFe,RC-[FeL113](ClO4)2 (for L11 see Fig. 3). This complex formed as a single diastereomer (d.r. > 200:1) as shown by NMR spectroscopy.8,9 The complex fac,ΔFe,RC-[FeL223](ClO4)2 was synthesised in a similar manner.
An X-ray molecular structure of fac,ΔFe,RC-[FeL113](ClO4)2 was determined (Fig. 4). The Fe tris(diamine) unit is in the expected fac arrangement with three sets of π-stacks between phenyl and pyridine units.8,9 The pendant pyridine substituents are arranged in a 3-fold symmetric array but with opposite helicity to the Fe unit. This is as we expected since the stereogenic centres at C(7) act as “corners” effectively switching the sense of twist. A similar phenomenon was displayed in a bimetallic structure based on this tris(diimine) unit.10 It proved difficult with the X-ray data available to discriminate between the structure shown and an alternative with pyridine N atoms at the inward facing positions. Nevertheless we were encouraged that the system appeared to be preorganised to bind a second metal.
2·1.5(MeOH) (H atoms, counterions and solvent molecules omitted for clarity). Thermal ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.963(4), Fe(1)–N(2) 1.986(4); N(1)–Fe(1)–N(2) 80.78(16).](/image/article/2013/DT/c3dt51725j/c3dt51725j-f4.gif) | ||
| Fig. 4 Structure of the cation in the unit cell of fac,ΔFe,RC-[FeL113](ClO4)2·1.5(MeOH) (H atoms, counterions and solvent molecules omitted for clarity). Thermal ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.963(4), Fe(1)–N(2) 1.986(4); N(1)–Fe(1)–N(2) 80.78(16). | ||
While a range of reactions of fac,ΔFe,RC-[FeL113](ClO4)2 and fac,ΔFe,RC-[FeL223](ClO4)2 with Fe(II), Co(II/III), and Zn(II) sources failed to give stable bimetallic species, the addition of CuI or AgClO4 to acetonitrile solutions resulted in the formation of the four new crystalline heterobimetallic complexes ΔFe,ΔCu,RC-[FeL113Cu](ClO4)2I, ΔFe,ΔCu,RC-[FeL223CuI](ClO4)2, ΔFe,RC-[FeL113Ag(CH3CN)](ClO4)3 and ΔFe,RC-[FeL223Ag(CH3CN)](ClO4)3; the absolute configurations are discussed later.
Coordination of the second metal led to characteristic changes in the 1H NMR spectra. The starting material fac,ΔFe,RC-[FeL113](ClO4)2 [Fig. 5(a)] shows the two doublets separated by ca. 0.06 ppm for the CH2 group between the ether oxygen and the pyridine ring while in the bimetallic complex ΔFe,ΔCu,RC-[FeL113Cu](ClO4)2I this increases to ca. 0.20 ppm (b). These CH2 protons are held in a more rigid conformation compared with the monometallic structure and this leads to a less complete averaging of the magnetic environments. The 1H NMR spectrum of ΔFe,RC-[FeL113Ag(CH3CN)](ClO4)3 was similar. Low temperature (233 K) 1H NMR spectra of the two complexes revealed no further changes suggesting that in both cases essentially one diastereomer is present in solution at level detectable by NMR spectroscopy. Microanalyses were consistent with the presence of the second metal and appropriate counter-ions. In the case of ΔFe,RC-[FeL113Ag(CH3CN)](ClO4)3, microanalysis was consistent with a single molecule of acetonitrile per complex, thus making up the expected tetrahedral geometry at the Ag(I) ion.
2 and (b) ΔFe,ΔCu,RC-[FeL113Cu](ClO4)2I.](/image/article/2013/DT/c3dt51725j/c3dt51725j-f5.gif) | ||
| Fig. 5 1H NMR spectra in CD3CN of (a) fac,ΔFe,RC-[FeL113](ClO4)2 and (b) ΔFe,ΔCu,RC-[FeL113Cu](ClO4)2I. | ||
In contrast to the L11 system, the 1H NMR spectrum of ΔFe,ΔCu,RC-[FeL223CuI](ClO4)2 showed that the peaks relating to the protons close to the second binding site (i.e. OCH2Py) were broadened. At lower temperatures these peaks were sharper (Fig. 6, peaks marked *) indicating that some exchange process is being slowed. This may be due to the presence of two diastereomers (ΔFe,ΔCu and ΔFe,ΛCu) or fluxionality at the Cu(I) binding site either associated with conformers or iodide substitution (see calculations later). We also observe in the VT 1H NMR spectra that the peaks corresponding to the phenyl rings (Fig. 6, peaks marked #) broaden as the temperature decreases with the extent of broadening ortho > meta > para. This is consistent with restricted CH–Ph bond rotations which cause averaging of the magnetic environments at room temperature.13
2 in d3-acetonitrile (* OCH2Py, # Ph).](/image/article/2013/DT/c3dt51725j/c3dt51725j-f6.gif) | ||
| Fig. 6 VT 1H NMR spectra of [FeL223CuI](ClO4)2 in d3-acetonitrile (* OCH2Py, # Ph). | ||
The 1H NMR spectrum of ΔFe,RC-[FeL223Ag(CH3CN)](ClO4)3 is consistent with coordination of Ag(I); no broadening of signals associated with the second binding site was observed at accessible temperatures and only one diastereomer was observed.
Density functional theory (DFT) calculations using ADF2009 (version 2009.01)14 were used to investigate the structures of the Fe–Cu bimetallic complexes with L11 and L22. In addition to the helicity at Fe which is essentially fixed, the pitch sense of the propeller arrangement of the pendant pyridine ligands about the Cu(I) centre needs also to be taken into account, and it was found that both homohelical ΔFeΔCu and heterohelical ΔFeΛCu isomers for both the 2- and 3-pyridinyl complexes could be optimised (Table 1). In both cases the input geometries around Cu(I) were based on the related X-ray structure of [Cu(3-methylpyridine)3I] which has a distorted tetrahedral geometry.15
For the 2-pyridinyl (L11) complex, as the optimisations progressed the Cu–I bond was broken in both enantiomers, resulting in approximately trigonal planar geometries. As a result, the iodide anion was deleted from these calculations to allow convergence. The calculations reveal that the homohelical ΔFeΔCu isomer is lower in energy by ca. 4 kcal mol−1 compared to the heterohelical ΔFeΛCu isomer. Fig. 7 shows that the ΔFeΔCu isomer in (a) is more longitudinally extended than ΔFeΛCu (b) as further indicated by the Fe–Cu distances (ca. 6.15 Å vs. 6.44 Å) and this is accompanied by; (i) reduction of unfavourable steric interactions between the pyridinyl ethers and the phenyl groups on the neighbouring ligands; and (ii) an increase in the magnitude of the propeller pitch‡ of the (Py)3Cu units from ca. 36° to 49°. Whatever the origins of the energy difference – there are undoubtedly a number of competing factors – the calculations suggest the ΔFeΔCu isomer will be formed exclusively, at least at the level that can be determined by NMR spectroscopy, and indeed this is consistent with observations.
![Optimised structures and space-fill representations of (a) ΔFeΔCu,RC-[FeL113Cu]3+ where the propeller pitch of the pyridine units at the planar Cu(i) centre (blue) is most pronounced; and (b) ΔFeΛCu,RC-[FeL113Cu]3+.](/image/article/2013/DT/c3dt51725j/c3dt51725j-f7.gif) | ||
| Fig. 7 Optimised structures and space-fill representations of (a) ΔFeΔCu,RC-[FeL113Cu]3+ where the propeller pitch of the pyridine units at the planar Cu(I) centre (blue) is most pronounced; and (b) ΔFeΛCu,RC-[FeL113Cu]3+. | ||
We were interested to investigate further the unusual16 preference for trigonal planar Cu(I) geometry, specifically if this is a result of inherent chelate constraints in the ligand system [FeL113]2+ or from steric effects arising from 2-substitution of the pyridine. For these purposes we studied the model monometallic systems [Cu(2-methylpyridine)3I] and [Cu(3-methylpyridine)3I]. For both isomers, structures were optimised with a range of fixed Cu⋯I distances. The results in Fig. 8 show that for the 2-methylpyridine complex (filled squares) the energy decreases as the iodide ligand dissociates and the Cu(I) approaches the trigonal planar arrangement (i.e. as the out of plane angle θ → 0°). In contrast, the 3-methylpyridine system (open squares) increases in energy as the iodide dissociates, albeit more gradually, and indeed a tetrahedral structure is observed for [Cu(3-methylpyridine)3I] by X-ray crystallography.15 It would appear then that the preference for trigonal planar geometry about Cu(I) in the L11 system is expected from a steric effect of the 2-substitution alone, notwithstanding any influence of conformational strain.
![Optimised energies versus out of plane angle θ for [Cu(methylpyridine)3I] isomers on linear elongation of the Cu⋯I vector. Cu–I distances indicated at each point.](/image/article/2013/DT/c3dt51725j/c3dt51725j-f8.gif) | ||
| Fig. 8 Optimised energies versus out of plane angle θ for [Cu(methylpyridine)3I] isomers on linear elongation of the Cu⋯I vector. Cu–I distances indicated at each point. | ||
Consistent with the observed and calculated structure of [Cu(3-methylpyridine)3I], DFT calculations on [FeL223CuI]2+ isomers subsequently predicted tetrahedral geometries around Cu(I) in both ΔFeΔCu and ΔFeΛCu isomers, with the homohelical isomer being the more stable (ca. 3 kcal mol−1).
The electronic spectra of these L11 and L22 systems have some unusual features. UV-Vis absorbance spectra shown in Fig. 9 show familiar strong bands in the region 220–320 nm, corresponding to π–π* transitions within the ligands, three further peaks corresponding to MLCT bands are observed at 380, 520 and 540 nm respectively. In both cases, Cu(I) coordination results in an increase in intensity of π–π* region of the spectra, a decrease in the intensity of the MLCT bands between 460–650 nm and an increase at 360 nm. These Fe(II) based CT transitions are likely to be strongly affected by structural changes at the metal, but no such perturbation is evident from crystallography or calculations. We suggest that the presence of the nearby cationic charge of the Cu(I) is responsible for this change in relative intensity. For the π–π* region the change may be a result of ordering by coordination of the heterocycles to Cu(I). In the corresponding CD spectra measured on the same cuvette (Fig. 10) the picture is of a general reduction in intensity for reasons unknown, but evidently from the UV spectra this is not an error from concentration measurement.
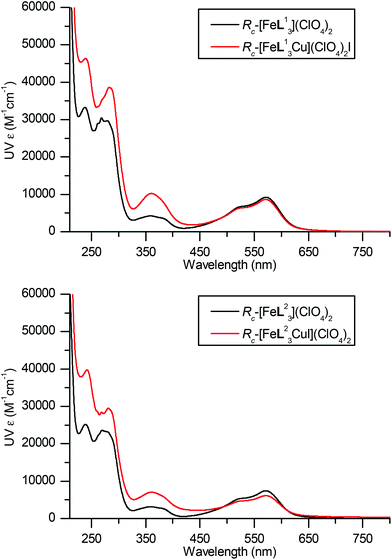 | ||
| Fig. 9 UV/vis spectra of Fe and Fe/Cu complexes of L113 and L22. Path length 1.0 cm and concentration 3.3 mM. | ||
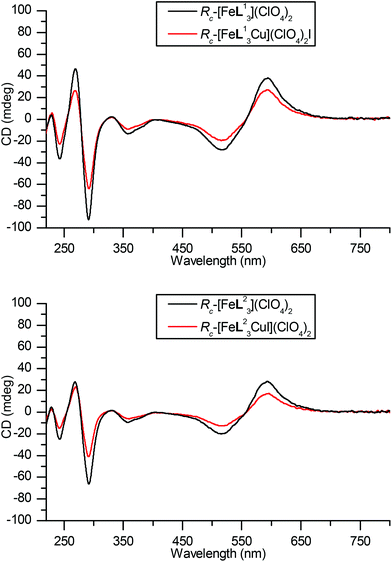 | ||
| Fig. 10 CD spectra of Fe and Fe/Cu complexes of L113 and L22. Path length 1.0 cm and concentration 3.3 mM. | ||
Click reactions of L33 alkynyl complex; complexes L4–64–6
Interestingly, despite the popularity of the CuAAC reaction in synthetic, biological and materials chemistry, relatively few examples have been reported on pre-formed metal complexes. The majority of these are found in the synthesis of rotaxanes and catenanes where a metal is used to template components (“gathering and threading”).17–25 Other examples exist for inert complex systems.26–32 In other cases [Zn(II), Mn(II), Fe(III), Co(II)] click reactions do not proceed cleanly; the metals are easily displaced by the Cu ‘click’ catalyst, either directly or via electron transfer.28The diastereomerically pure compound fac,ΔFe,RC-[FeL333](ClO4)2 (for L33 see Fig. 3) was synthesised using the standard one-pot synthesis with the propargylic ether derivative of (R)-2-phenylglycinol. This complex is of particular interest due to its potential to undergo copper(I)-catalysed Huisgen 1,3-dipolar cycloaddition (CuAAC) ‘click’ reactions between the three alkyne units and organic azides. Accordingly it was treated with benzyl azide in the presence of dry triethylamine and a catalytic amount of copper(I) iodide (0.3 eq. per complex) in dry acetonitrile to give a purple solid. The 1H NMR spectrum at ambient temperature contained only one set of ligand resonances, however at low temperature (233 K), the 1H NMR spectrum revealed that there were in fact two species present in the recrystallised sample in the ratio 1:1.7 [Fig. 11(a)]. The reaction was repeated using a stoichiometric amount of copper(I) iodide (1.0 equivalent per Fe). The resulting low temperature 1H NMR spectrum showed only one set of peaks [Fig. 11(b)] and microanalysis indicated that copper(I) iodide was present in a 1:1 ratio with the complex. Hence, the spectrum shown in Fig. 11(a) arises from an equilibrium between two complexes fac,ΔFe,RC-[FeL443]2+ and ΔFe,RC-[FeL443Cu]3+ as shown in Scheme 1.
2 and BnN3 with (a) 0.3 eq. CuI and (b) 1.0 eq. CuI.](/image/article/2013/DT/c3dt51725j/c3dt51725j-f11.gif) | ||
| Fig. 11 Imine/triazole region in the 1H NMR spectra in CD3CN at 233 K for the ‘click’ reactions between fac,ΔFe,RC-[FeL333](ClO4)2 and BnN3 with (a) 0.3 eq. CuI and (b) 1.0 eq. CuI. | ||
2 and BnN3 resulting in an equilibrium between fac,ΔFe,RC-[FeL443]2+ and ΔFe,RC-[FeL443Cu]3+.](/image/article/2013/DT/c3dt51725j/c3dt51725j-s1.gif) | ||
| Scheme 1 ‘Click’ reaction between fac,ΔFe,RC-[FeL333](ClO4)2 and BnN3 resulting in an equilibrium between fac,ΔFe,RC-[FeL443]2+ and ΔFe,RC-[FeL443Cu]3+. | ||
Slow vapour diffusion of ethyl acetate into an acetonitrile solution of ΔFe,RC-[FeL443Cu](ClO4)2I in air resulted in the formation of single crystals that were suitable for X-ray diffraction. The solid state structure is shown in Fig. 12 along with key bond lengths and angles. The asymmetric unit contains one complex and a mixture of perchlorate and triiodide (resulting from oxidation of iodide in air) as the counterions. The Fe(II) and Cu(I) cations lie on a threefold axis. The complex has an approximately octahedral arrangement around Fe(II) and a trigonal planar arrangement around Cu(I), with a distance between the metal centres of ca. 6.14 Å. Most interestingly, the sense of the helicity at the Cu(I) centre is opposite to that at the Fe(II) centre (i.e. ΔFe,ΛCu). Nevertheless this is not a mesocate since the metals are different and thus the system is chiral. Hence, we would describe this as the first example of an optically pure heterohelicate.33
2.66(I3)0.33 (H atoms and counterions omitted for clarity). Thermal ellipsoids are shown at 30% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.958(4), Fe(1)–N(2) 1.982(5), Cu(1)–N(3) 2.002(8); N(1)–Fe(1)–N(2) 81.46(19), N(3)#2–Cu(1)–N(3) 119.982(10).](/image/article/2013/DT/c3dt51725j/c3dt51725j-f12.gif) | ||
| Fig. 12 Structure of the cation in the asymmetric unit of ΔFe,ΛCu,RC-[FeCuL443](ClO4)2.66(I3)0.33 (H atoms and counterions omitted for clarity). Thermal ellipsoids are shown at 30% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.958(4), Fe(1)–N(2) 1.982(5), Cu(1)–N(3) 2.002(8); N(1)–Fe(1)–N(2) 81.46(19), N(3)#2–Cu(1)–N(3) 119.982(10). | ||
In addition to the usual8,9 three sets of inter-ligand parallel-offset π-stacking interactions in the Fe unit there are three sets of CH–π interactions34,35 present between the triazole benzyl substituent and the CHCH2O group on a neighbouring ligand. The Cu(I) is coordinated to the triazole rings via the 3-position i.e. the N(3) nitrogen atom, as expected on the basis of its higher basicity and perhaps also here a more favourable chelate conformation. There is nevertheless also some precedent for the coordination of metals to the 2-position [i.e. N(4)] in 1-substituted-1,2,3-triazole rings.36
The geometries of the homohelical ΔFeΔCu and heterohelical ΔFeΛCu isomers of ΔFe,RC-[FeL443Cu]3+ were optimised and their energies calculated (Table 2) from density functional calculations as above. While the benzyl substituents on the triazole rings point down towards Fe(II) in the X-ray structure, various rotamers were also considered and optimised. The observed heterohelical isomer was found to be the lower in energy than the homohelical diastereomer by at least 9.27 kcal mol−1 such that we expect this structure exist exclusively in solution at equilibrium. Low temperature NMR studies are in agreement and on the basis of these calculations and the X-ray structure we can assign the solution species as heterohelical ΔFe,ΛCu,RC-[FeL443Cu]3+.
| ΔFe,RC-[FeL443Cu]3+ diastereomer | Rotamer typea | Energy (kcal mol−1) | Relative energy (kcal mol−1) | Sum of angles about Cu(1) (°) |
|---|---|---|---|---|
| a Type A is as shown in Fig. 10 whereas in type B a rotation of ca. 180° about the N–C bond in the benzyl groups was made before optimisation i.e. benzyl groups point “up”. | ||||
| Heterohelical (ΔFe,ΛCu) | A | −24144.94 |
0 | 360.01 |
| B | −24145.35 |
+0.41 | 359.96 | |
| Homohelical (ΔFe,ΔCu) | A | −24132.48 |
+12.46 | 359.99 |
| B | −24135.67 |
+9.27 | 359.97 | |
The origins of this stereoselection are not obvious, and indeed the sums of angles around Cu in the structures are all very close to 360° indicating a lack of strain here. While as for ΔFe,ΔCu,RC-[FeL113Cu]3+ there are likely to be a number of contributing factors, examination of space-filling models reveals an absence here of steric compression between the triazole and the ether units in the neighbouring ligands, thus allowing the system to adopt the more favourable conformations in the linker chain of the heterohelical system.
Further examples of heterohelical bimetallic complexes with L55 and L66 (Fig. 3) were synthesised from click reactions between fac,ΔFe,RC-[FeL333](ClO4)2 and the appropriate organic azides (3,5-dimethylbenzyl azide and 4-nitrobenzyl azide) in the presence of 1 equivalent of CuI per complex. In all cases, low temperature 1H NMR spectra show only a single set of peaks consistent with a single diastereomer in solution.
The ability to remove Cu(I) from these bimetallic complexes would be attractive so a number of methods were attempted to remove Cu(I) post-‘click’ reaction. The use of a copper scavenger, e.g. Smopex-111, Smopex 112, CupriSorb™, EDTA or sodium sulfide, either removed no copper or caused complete decomposition of the complex by removing both Cu(I) and Fe(II). As an alternative was also attempted the ruthenium(II) catalysed ‘click’ reactions37–39 but no reaction occurred. Conversely, the use of copper wire and PMDETA40,41 as the catalyst caused complete decomposition. These results led us to consider the scope for functionalisation from the pyridine side of the monometallic complex.
Pyridinyl ethers L7–87–8
In order to compare the coordination behaviour above with that of pendant units attached to the pyridine ligands in the tris(chelate)Fe(II), the ligands L7–87–8 (Fig. 3) were designed. Subsequently, the use of 5-(2-pyridinyloxy)picolinaldehyde in the standard one-pot synthesis with (R)-α-methylbenzylamine and Fe(ClO4)2·6H2O led to the formation of diastereomerically pure fac,ΛFe,RC-[FeL773](ClO4)2 (d.r. > 200:1). The X-ray molecular structure (Fig. 13) shows in contrast to that in Fig. 4 that the three pyridinyl ether groups are not preorganised for binding a second metal, but is otherwise conventional.
2·CH3CN (H atoms, counterions and solvent molecules omitted for clarity). Thermal ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.9745(14), Fe(1)–N(4) 1.9804(15), Fe(1)–N(7) 1.9748(15), Fe(1)–N(2) 1.9788(15), Fe(1)–N(5) 1.9789(15), Fe(1)–N(8) 1.9874(14); N(1)–Fe(1)–N(2) 81.58(6), N(4)–Fe(1)–N(5) 81.16(6), N(7)–Fe(1)–N(8) 81.22(6).](/image/article/2013/DT/c3dt51725j/c3dt51725j-f13.gif) | ||
| Fig. 13 Structure of the cation in the asymmetric unit of fac,ΛFe,RC-[FeL773](ClO4)2·CH3CN (H atoms, counterions and solvent molecules omitted for clarity). Thermal ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°): Fe(1)–N(1) 1.9745(14), Fe(1)–N(4) 1.9804(15), Fe(1)–N(7) 1.9748(15), Fe(1)–N(2) 1.9788(15), Fe(1)–N(5) 1.9789(15), Fe(1)–N(8) 1.9874(14); N(1)–Fe(1)–N(2) 81.58(6), N(4)–Fe(1)–N(5) 81.16(6), N(7)–Fe(1)–N(8) 81.22(6). | ||
Addition of 1 equivalent of CuI to an acetonitrile solution containing fac,ΛFe,RC-[FeL773](ClO4)2·CH3CN led to no significant changes in chemical shifts, suggesting that coordination of Cu(I) to the second binding site is weak in solution. The same observation was made on addition of AgClO4. In further support of this, crystallisation of fac,ΛFe,RC-[FeL773](ClO4)2 in the presence of AgClO4 resulted in a partially refined X-ray molecular structure showing an extended lattice structure with each tris(chelate)Fe(II) unit coordinated to three different silver(I) units; the formation of this structure is presumably driven by lattice enthalpy.
In contrast to the L77 case above, reaction of three equivalents of both 5-(3-pyridinyloxy)picolinaldehyde and (R)-α-methylbenzylamine with Fe(ClO4)2·6H2O gave a sample with broad NMR resonances. In solution the product slowly converted from purple to a paramagnetic red system on standing. This is likely to be due to the formation of a bis complex [FeL882]2+ as a result of the chelate effect; this 3-pyridinyl ligand is appropriately structured to coordinate in a tridentate fashion, as we have observed previously for a tert-leucinol derivative.8
Click reactions of L99 alkynyl complex; complexes of L1010
The diastereomerically pure compound fac,ΛFe,RC-[FeL993](ClO4)2 (for L99 see Fig. 3) was synthesised from the propargylic ether derivative of 5-hydroxy-2-picolinaldehyde. Copper(I)-catalysed Huisgen 1,3-dipolar cycloaddition (CuAAC) ‘click’ reactions between this complex and benzyl azide did not behave in the same way as for fac,ΔFe,RC-[FeL333](ClO4)2 and the reaction with sub-stoichiometric (catalytic) amounts of CuI led to mixtures of starting material [FeL993](ClO4)2 with [FeL992L1010](ClO4)2, [FeL99L10102](ClO4)2 and [FeL10103](ClO4)2 (Fig. 3) according to 1H NMR spectroscopy (see ESI†) and mass spectrometry. With one equivalent of CuI per Fe centre (i.e. the conditions that cleanly gave ΔFe,RC-[FeL443Cu](ClO4)2I) the majority product was [FeL10103](ClO4)2 but the without Cu+ coordination. Addition of three equivalents of CuI per Fe unit or more, essentially complete click conversion was apparent, and the chemical shift difference for the diastereotopic O–CH2–triazole group had widened to ca. 0.25 ppm indicating Cu+ coordination i.e. [FeL10103Cu]3+. Attempts to recrystallize this species for analysis led to slow decomposition and formation of red paramagnetic products. Presumably it suffers the same fate as the L88 system above, and for the same reasons.Conclusion
Optically pure (highly enantiomerically enriched) heterobimetallic Fe–Cu and Fe–Ag helicates can be synthesised via the pre-formed monometallic Fe(II) complexes of L11 and L22. In both cases only a single isomer was observed by 1H NMR spectroscopy and corresponding DFT calculations predict the ΔΔ homochiral isomer will form exclusively. Interestingly, formation of the trigonal planar geometry at Cu(I) is driven by the substitution pattern of the pyridine ligand, independently of any conformational effects in the bimetallic structure.CuAAC ‘click’ reactions are most efficient on the monometallic complex of L33 and give Fe–Cu helicates directly. Again, a single isomer is formed but in this case both X-ray crystallography and DFT calculations support the exclusive formation of the ΔFeΛCu heterochiral isomers. These compounds are the first examples of what might be called heterohelicates since they have opposite configurations at the metal but are not mesocates. Similar reactions on the Fe(II) complex of L99 are much less efficient presumably because the system, as demonstrated in the analogous pyridine series, is not preorganised for coordination of the Cu(I) ion; binding of the Cu(I) catalyst to the three triazoles appears to promote the reaction.
We are thus now in a position to confidently predict stereochemical properties in such bimetallic systems and will now move towards the synthesis of heterobimetallics designed for aqueous solubility.33
Experimental
General considerations
(R)-2-Phenylglycinol and 5-(hydroxy)picolinaldehyde were synthesised as previously described.9,42,43 Deuterated solvents were purchased from Sigma-Aldrich and Cambridge Isotope Laboratories. Sodium hydride dispersions in mineral oil were placed in a Schlenk vessel under an inert atmosphere and washed three times with diethyl ether to remove the oil. The sodium hydride powder was then dried and stored in the dry box. Suitable precautions should be taken in the synthesis of organic azides.44Where appropriate, reactions were carried out under argon using a dual manifold argon/vacuum line and standard Schlenk techniques or MBraun dry box. THF was pre-dried over sodium wire and then heated to reflux for 3 d under dinitrogen over potassium and degassed before use. Dried THF was stored in a glass ampoule under argon. All glassware and cannulae were stored in an oven at >375 K.
NMR spectra were recorded on Bruker Spectrospin 300/400/500 MHz spectrometers. Routine NMR assignments were confirmed by 1H–1H (COSY) and 13C–1H (HMQC) correlation experiments where necessary. The spectra were internally referenced using the residual protio solvent (CDCl3, CD3CN etc.) resonance relative to tetramethylsilane (δ = 0 ppm). ESI mass spectra were recorded on Bruker Esquire 2000 and Bruker MicroTOF spectrometers. Infra-Red spectra were measured using a Perkin-Elmer FTIR spectrometer. Elemental analyses were performed by Warwick Analytical Services, Coventry, UK and MEDAC Ltd, Surrey, UK.
The crystal data for ΔFe,ΛCu,RC-[FeL443Cu](ClO4)2I (CCDC 947140) and fac,ΛFe,RC-[FeL773](ClO4)2 (CCDC 947142) were collected using an Xcalibur Gemini diffractometer with a Ruby CCD area detector using CuKα (λ = 1.54184 Å) radiation source. The crystal data for fac,ΔFe,RC-[FeL113](ClO4)2 (CCDC 947141) was recorded by the National Crystallographic Service.45 The structures were solved with the XS structure solution program using Direct Methods and refined with the ShelXL46 refinement package using Least Squares minimisation.
Optical rotation measurements were performed on a Perkin Elmer Polarimeter 341 by Warwick Analytical Services, Coventry, UK. In all cases the following parameters were used: solvent methanol, temperature 20 °C, path length 100 mm, wavelength 589 nm.
Density functional optimisations were carried out using the Amsterdam Density Functional program (version 2009.01).14 Starting points for geometry optimisations were taken from crystallographic data where available, and where unavailable, starting structures were created from existing crystallographic fragments. Solution structures were optimised relative to acetonitrile (vide infra) using a triple-ζ plus polarisation basis set (TZP) on all atoms with the OPBE functional and Grimme's empirical correction for dispersion.47 Small frozen cores48 were used throughout. Calculations used integration level 5 (as defined by ADF) with convergence criteria of e = 0.0001 a.u., rad = 0.005 Å and grad = 0.001 a.u. Å−1 for the total binding energy, Cartesian displacement and energy gradient respectively. Acetonitrile solvent effects were included based on the conductor-like screening model (COSMO) implemented in ADF.49 Non-bonded radii used were H = 1.350 Å, C = 1.700 Å, N = 1.608 Å, Fe = 1.858 Å. A dielectric constant of 37.5 (acetonitrile) and an outer cavity radius of 2.76 Å were further used to parameterise the COSMO solvation cavity.
Synthesis
:0.15. This crude product was purified by Kügelrohr distillation at rotary pump vacuum to remove unreacted (R)-2-phenylglycinol (125 °C), and the product, a slightly yellow liquid (195 °C). Purified yield = 2.25 g, 9.8 mmol, 71%.1H NMR (400 MHz, 298 K, CDCl3) δH 8.56 (1H, d, 3JHH = 5.0 Hz, Py), 7.69 (1H, td, 3JHH = 8.0 Hz, 4JHH = 2.0 Hz, Py), 7.42–7.18 (7H, m, Ph/Py), 4.70 (2H, s, CH2Py), 4.31 (1H, dd, 3JHH = 4.5 Hz, 9.0 Hz, CH), 3.71 (1H, dd, 2JHH = 9.0 Hz , 3JHH = 4.5 Hz, CH2), 3.38 (1H, t, 2JHH/3JHH = 9.0 Hz, CH2), 1.83 (2H, s, NH2).
13C{1H} NMR (100 MHz, 298 K, CDCl3) δC 158.4 (Py), 149.2, 142.4, 136.6, 128.5, 127.5, 126.9, 122.4, 121.4 (Ar), 77.3 (CH2), 74.1 (CH2Py), 55.6 (CH).
MS (ESI) m/z 212.0 [M − NH2]+, 229.0 [M + H]+, 251.0 [M + Na]+.
IR v cm−1 3027 w, 2858 w, 1591 m, 1571 m, 1435 m, 1355 m, 1113 s, 755 s, 700 s.
Elemental Analysis found (Calculated for C14H16N2O) % C 73.76 (73.66), H 7.15 (7.06), N 12.21 (12.27).
Optical rotation −22.11° (6.640 g per 100 ml).
3-(Bromomethyl)pyridine hydrobromide50
3-(Hydroxymethyl)pyridine (2.00 g, 18.33 mmol) was dissolved in hydrobromic acid (20 ml, 48%). The mixture was stirred at reflux for 4 h. The solvent was then removed under reduced pressure to leave a light yellow oil which solidified on standing. The solid was washed with ethanol (3 × 50 ml) and the resulting white solid was dried in vacuo. Yield = 2.78 g, 10.99 mmol, 60%.1H NMR (400 MHz, 298 K, D2O) δH 8.92 (1H, s, Py), 8.75 (1H, d, 3JHH = 6.0 Hz, Py), 8.71 (1H, d, 3JHH = 8.0 Hz, Py), 8.08 (1H, dd, 3JHH = 6.0 Hz, 3JHH = 8.0 Hz, Py), 4.77 (2H, s, CH2).
13C{1H} NMR (100 MHz, 298 K, D2O) δC 147.3, 141.3, 140.6, 138.9, 127.5 (Py), 26.7 (CH2).
MS (ESI) m/z 172.0/174.0 [M + H]+.
Elemental Analysis found (Calculated for C6H7Br2N) % C 28.58 (28.49), H 2.82 (2.79), N 5.40 (5.54).
2-(Pyridin-3-methoxy)-1-phenylethanamine
(R)-2-Phenylglycinol (1.00 g, 7.29 mmol) was dissolved in dry THF (10 ml) and was added dropwise to a stirred suspension of sodium hydride (0.61 g, 25.5 mmol, 3.5 eq.) in dry THF (20 ml). This solution was stirred for 1 h at ambient temperature under partial vacuum. 3-(Bromomethyl)-pyridine hydrobromide (1.75 g, 6.93 mmol, 0.95 eq.) was added as a solid. The solution was stirred for 1 h at ambient temperature and then heated to reflux (65 °C) overnight, all under partial vacuum. The solution was then cooled to ambient temperature and brine (60 ml) was added slowly. The product was extracted into diethyl ether (3 × 150 ml), dried over sodium sulfate and the solvent was removed under reduced pressure to leave a yellow oil, which contained product and (R)-2-phenylglycinol in a ratio of 1:0.19. This crude product was purified by Kügelrohr distillation at rotary pump vacuum to remove unreacted (R)-2-phenylglycinol (125 °C), and the product, a slightly yellow liquid (195 °C). Purified yield = 1.16 g, 5.08 mmol, 73%.
1H NMR (300 MHz, 298 K, CDCl3) δH 8.49–8.46 (2H, m, Py), 7.57 (1H, dt, 3JHH = 8.0 Hz, 4JHH = 2.0 Hz, Py), 7.32–7.18 (6H, m, Ph/Py), 4.49 (2H, s, CH2Py), 4.17 (1H, dd, 3JHH = 4.0 Hz, 9.0 Hz, CH), 3.55 (1H, dd, 2JHH = 9.0 Hz, 3JHH = 4.0 Hz, CH2), 3.41 (1H, t, 2JHH/3JHH = 9.0 Hz, CH2), 1.68 (2H, s, NH2).
13C{1H} NMR (75 MHz, 298 K, CDCl3) δC 149.2, 142.3, 135.3, 133.5, 128.4, 127.5, 126.8, 123.5, 123.4 (Ar), 70.7 (CH2), 70.0 (CH2Py), 55.5 (CH).
MS (ESI) m/z 212.1 [M − NH2]+, 229.1 [M + H]+, 251.1 [M + Na]+.
Elemental Analysis found (Calculated for C14H16N2O) % C 73.81 (73.66), H 6.90 (7.06), N 11.95 (12.27).
Optical rotation −19.98° (6.244 g per 100 ml).
2-(Propargyloxy)-1-phenylethanamine
(R)-2-Phenylglycinol (0.50 g, 3.6 mmol) was dissolved in dry THF (15 ml) and was added dropwise to a stirred suspension of sodium hydride (0.17 g, 7.3 mmol, 2.0 eq.) in dry THF (10 ml). The solution was stirred for 1 h at ambient temperature under partial vacuum. Propargyl bromide (80 wt% in toluene, 0.43 ml, 3.8 mmol, 1.05 eq.) was added dropwise and the solution was stirred for 1 h at ambient temperature under argon. At this point, the solution was heated to reflux (65 °C) under partial vacuum overnight before cooling to ambient temperature, followed by the addition of brine (30 ml). The product was extracted into diethyl ether (4 × 50 ml), dried over sodium sulfate and the solvent was removed under reduced pressure to leave a yellow oil (crude yield = 0.52 g). This crude product was purified by Kügelrohr distillation at rotary pump vacuum to give the product as a clear liquid (95 °C). Purified yield = 0.45 g, 2.6 mmol, 71%.1H NMR (300 MHz, 298 K, CDCl3) δH 7.33–7.16 (5H, m, Ph), 4.17–4.10 (3H, m, CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and CH), 3.60 (1H, dd, 2JHH = 9.0 Hz, 3JHH = 4.0 Hz, CH2CHPh), 3.39 (1H, t, 2JHH/3JHH = 9.0 Hz, CH2CHPh), 2.36 (1H, t, 4JHH = 1.5 Hz, CCH), 1.70 (2H, s, NH2).
C and CH), 3.60 (1H, dd, 2JHH = 9.0 Hz, 3JHH = 4.0 Hz, CH2CHPh), 3.39 (1H, t, 2JHH/3JHH = 9.0 Hz, CH2CHPh), 2.36 (1H, t, 4JHH = 1.5 Hz, CCH), 1.70 (2H, s, NH2).
13C{1H} NMR (100 MHz, 298 K, CDCl3) δC 142.3, 128.5, 127.5, 126.9 (Ph), 79.6 (CCH), 76.3 (CH2CHPh), 74.6 (CCH), 58.5 (CH2–CC), 55.4 (CHPh).
MS (ESI) m/z 159.0 [M − NH2]+, 176.0 [M + H]+, 198.0 [M − Na]+, 214.0 [M + K]+.
IR v cm−1 3289 w, 2857 w, 1604 w, 1493 w, 1453 m, 1356 m, 1088 s, 1020 m, 860 m, 759 s, 699 s.
Elemental Analysis found (Calculated for C11H13NO) % C 75.46 (75.40), H 7.56 (7.48), N 7.95 (7.99).
Optical rotation −32.11° (6.082 g per 100 ml).
Benzyl azide51
Benzyl bromide (2.0 ml, 16.84 mmol, 1.0 eq.) was dissolved in DMSO (40 ml). Sodium azide (1.64 g, 25.26 mmol, 1.5 eq.) was added as a solid and the reaction was stirred overnight at ambient temperature. Water (75 ml) was added slowly (exothermic) before extracting the product into diethyl ether (3 × 150 ml). The combined diethyl ether layers were washed with brine (2 × 150 ml), dried over sodium sulfate and the solvent removed under reduced pressure to leave a clear colourless oil. Yield = 1.63 g, 12.24 mmol, 73%.1H NMR (400 MHz, 298 K, CDCl3) δH 7.42–7.32 (5H, m, Ph), 4.35 (2H, s, CH2).
13C{1H} NMR (100 MHz, 298 K, CDCl3) δC 135.4, 128.9, 128.3, 128.2 (Ar), 54.8 (CH2).
MS (EI/CI) m/z 105.1 [M − 2N]+.
IR v cm−1 2090 s, 1497 w, 1455 m, 1253 m, 876 w, 735 m, 696 s.
Elemental Analysis found (Calculated for C7H7N3) % C 63.53 (63.14), H 5.72 (5.30), N 31.34 (31.56).
3,5-Dimethylbenzyl azide
Using a similar procedure to that for benzyl azide, 3,5-dimethylbenzyl bromide (4.29 g, 21.55 mmol, 1.0 eq.) was dissolved in DMSO (50 ml). Sodium azide (2.13 g, 32.76 mmol, 1.5 eq.) was added as a solid and the reaction was stirred overnight at ambient temperature. Water (80 ml) was added slowly (exothermic) before extracting the product into diethyl ether (3 × 150 ml). The combined diethyl ether layers were washed with brine (2 × 150 ml), dried over sodium sulfate and the solvent removed under reduced pressure to leave a clear colourless oil. Yield = 2.68 g, 16.63 mmol, 77%.1H NMR (300 MHz, 298 K, CDCl3) δH 6.85 (1H, s, Ph), 6.80 (2H, s, Ph), 4.11 (2H, s, CH2), 2.21 (6H, s, CH3).
13C{1H} NMR (75 MHz, 298 K, CDCl3) δC 138.4, 135.3, 129.9, 126.1 (Ph), 54.8 (CH2), 21.1 (CH3).
MS (EI/CI) m/z 119.1 [M − N3]+, 133.1 [M − N2]+, 162.0 [M + H]+.
IR v cm−1 3018 w, 2920 w, 2873 w, 2092 s, 1608 m, 1463 w, 1378 w, 1343 m, 1243 m, 1164 w, 1039 w, 940 w, 875 w, 840 s, 725 m, 689 m.
Elemental Analysis found (Calculated for C9H11N3) % C 67.11 (67.04), H 7.14 (6.89), N 26.19 (26.07).
4-Nitrobenzyl azide
Using a similar procedure to that for benzyl azide, 4-nitrobenzyl bromide (2.00 g, 9.26 mmol, 1.0 eq.) was dissolved in DMSO (25 ml). Sodium azide (0.90 g, 13.89 mmol, 1.5 eq.) was added as a solid and the reaction was stirred overnight at ambient temperature. Water (60 ml) was added slowly (exothermic) before extracting the product into diethyl ether (3 × 150 ml). The combined diethyl ether layers were washed with brine (2 × 150 ml), dried over sodium sulfate and the solvent removed under reduced pressure to leave a yellow-orange oil. Yield = 1.54 g, 8.64 mmol, 93%.1H NMR (400 MHz, 298 K, CDCl3) δH 8.24 (2H, d, 3JHH = 8.5 Hz, Ph), 7.50 (2H, d, 3JHH = 8.5 Hz, Ph), 4.50 (2H, s, CH2).
13C{1H} NMR (100 MHz, 298 K, CDCl3) δC 147.9, 142.8, 128.7, 124.2 (Ph), 53.9 (CH2).
MS (ESI) m/z 166.1 [M − N + H2]+.
Elemental Analysis found (Calculated for C7H6N4O2) % C 47.74 (47.19), H 3.35 (3.39), N 30.82 (31.45).
5-(2-Pyridinyloxy)picolinaldehyde
2-(Bromomethyl)pyridine hydrobromide (0.5 g, 1.97 mmol) was dissolved in DMF (10 ml). Potassium carbonate (0.68 g, 4.92 mmol) was added followed by 5-hydroxypicolinaldehyde (0.24 g, 1.97 mmol) and the mixture was stirred at 100 °C for 16 h. The solvent was removed under reduced pressure to give a dark brown solid which was extracted with chloroform (3 × 50 ml), washed with NaOH solution (1 M, 3 × 100 ml) and saturated brine solution (3 × 100 ml) and dried over MgSO4 before evaporation under reduced pressure to give a light brown solid (0.31 g, 1.44 mmol, 73%).1H-NMR (300 MHz, 298 K, CDCl3): δ 9.99 (s, 1H, CHO), 8.62 (d, 1H, 3JHH = 4.27 Hz, Py), 8.54 (d, 1H, 3JHH = 2.59 Hz, Py), 7.94 (d, 1H, 3JHH = 8.54 Hz, Py), 7.75 (td, 1H, 3JHH = 8.08 Hz, 4JHH = 1.52 Hz, Py), 7.51 (d, 1H, 3JHH = 8.08 Hz, Py), 7.39 (dd, 1H, 3JHH = 8.54 Hz, 4JHH = 2.74 Hz, Py), 7.29 (dd, 1H, 3JHH = 7.47 Hz, 4JHH = 4.73 Hz, Py), 5.33 (s, 2H, CH2).
13C-NMR (101 MHz, 298 K, CDCl3); δ 192.1 (CHO), 158.0, 155.5, 149.7, 146.8, 139.3, 137.2, 123.5, 123.4, 121.7, 121.2 (Py), 71.43 (CH2).
ESI-MS – 215 [M + H]+, 237 [M + Na]+.
Elemental Analysis found (Calculated for C12H10N2O2) C 66.16% (67.28%), H 4.48% (4.70%), N 12.86% (13.07%).
5-(3-Pyridinyloxy)picolinaldehyde
Using a similar procedure as for 5-(2-pyridinyloxy)picolinaldehyde, 3-(bromomethyl)pyridine hydrobromide (0.5 g, 1.97 mmol) gave a light brown solid (0.382 g, 1.78 mmol, 44%).1H NMR (400 MHz, 298 K, CDCl3): δH 10.00 (s, 1H, CHO), 8.72 (s, 1H, Py), 8.65 (1H, d, 3JHH = 5.0 Hz), 8.51 (1H, d, 3JHH = 2.5 Hz, Py), 7.97 (1H, d, 3JHH = 8.5 Hz, Py), 7.78 (1H, d, 3JHH = 8.0 Hz, Py), 7.37 (2H, m Py), 5.22 (2H, s, CH2).
13C NMR (101 MHz, 298 K, CDCl3): δC 196.0 (CHO), 192.0, 187.3, 162.6, 150.2, 149.1, 138.9, 135.4, 123.7, 123.3, 121.1 (Py), 68.24 (CH2).
ESI-MS: 215 [M + H]+, 237 [M + Na]+.
Elemental Analysis found (Calculated for C12H10N2O2) C 67.28% (67.28%), H 4.62% (4.70%), N 12.55% (13.07%).
5-(Propargyloxy)picolinaldehyde
Potassium carbonate (0.59 g, 4.26 mmol) was added to a solution of 5-(hydroxy)picolinaldehyde (0.50 g, 4.06 mmol) in acetonitrile (40 ml). Propargyl bromide (80 wt% in toluene, 0.475 ml) was added. The solution was stirred at reflux temperature (ca. 85 °C) overnight before allowing to cool to ambient temperature and passing a short column of silica, eluting with acetonitrile until the product was fully removed (TLC). This left a brown band at the top of the column. The solvent was evaporated and the crude product was dissolved in dichloromethane, filtered and evaporated under reduced pressure to leave a dark orange oil which was dried overnight in vacuo. Yield = 0.45 g, 2.79 mmol, 69%.1H NMR (400 MHz, 298 K, DMSO) δH 9.90 (1H, s, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 8.53 (1H, d, 4JHH = 3.0 Hz, Py), 7.97 (1H, d, 3JHH = 8.5 Hz, Py), 7.64 (1H, dd, 3JHH = 8.5 Hz, 4JHH = 3.0 Hz, Py), 5.05 (2H, d, 4JHH = 2.5 Hz, CH2–CC), 3.71 (1H, t, 4JHH = 2.5 Hz, CCH).
O), 8.53 (1H, d, 4JHH = 3.0 Hz, Py), 7.97 (1H, d, 3JHH = 8.5 Hz, Py), 7.64 (1H, dd, 3JHH = 8.5 Hz, 4JHH = 3.0 Hz, Py), 5.05 (2H, d, 4JHH = 2.5 Hz, CH2–CC), 3.71 (1H, t, 4JHH = 2.5 Hz, CCH).
13C{1H} NMR (100 MHz, 298 K, DMSO) δC 191.9 (CO), 156.7, 146.1, 138.8, 123.3, 121.7 (Py), 79.4 (CCH), 78.9 (CCH), 56.3 (CH2).
MS (ESI) m/z 162.2 [M + H]+, 184.1 [M + Na]+.
IR v cm−1 3213 w, 2127 w, 1692 s, 1569 s, 1490 w, 1474 w, 1379 w, 1308 m, 1282 w, 1259 s, 1203 s, 1132 m, 1006 s, 975 m, 916 w, 835 s, 800 s, 762 m, 732 m, 694 s, 659 s.
Elemental Analysis found (Calculated for C9H7NO2) % C 66.85 (67.07), H 4.02 (4.38), N 8.52 (8.69).
fac,ΔFe,RC-[FeL113](ClO4)2
2-Pyridinecarboxaldehyde (0.32 g, 3.0 mmol, 3.0 eq.) and (R)-2-(pyridin-2-methoxy)-1-phenylethanamine (0.68 g, 3.0 mmol, 3.0 eq.) were dissolved in acetonitrile (15 ml) to form a yellow solution. Iron(II) perchlorate hexahydrate (0.36 g, 1.0 mmol, 1.0 eq.) in acetonitrile (5 ml) was added to give a purple solution. This was stirred overnight at ambient temperature before the solvent was removed under reduced pressure. The product, [FeL113](ClO4)2, was recrystallised from acetonitrile and ethyl acetate and the resulting purple crystals were filtered and dried in vacuo. Yield = 0.59 g, 0.49 mmol, 49%. Single crystals were grown from acetonitrile–methanol.1H NMR (400 MHz, 298 K, CD3CN) δH 9.03 (3H, s, HCN), 8.60 (3H, d, 3JHH = 5.5 Hz, Py), 7.77 (3H, td, 3JHH = 8.0 Hz, 4JHH = 2.0 Hz, Py), 7.68 (3H, td, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, Py), 7.61 (3H, d, 3JHH = 8.0 Hz, Py), 7.40 (3H, d, 3JHH = 8.0 Hz, Py), 7.28 (3H, t, 3JHH = 6.0 Hz, Py), 7.16 (3H, td, 3JHH = 6.0 Hz, 4JHH = 1.5 Hz, Py), 7.03 (3H, t, 3JHH = 7.0 Hz, Ph), 6.86 (6H, t, 3JHH = 7.0 Hz, Ph), 6.75 (3H, d, 3JHH = 6.0 Hz, Py), 6.65 (6H, d, 3JHH = 7.0 Hz, Ph), 5.85 (3H, dd, 3JHH = 10.5 Hz, 3JHH = 3.5 Hz, CH), 4.95 (3H, d, 2JHH = 13.0 Hz, CH2Py), 4.89 (3H, d, 2JHH = 13.0 Hz, CH2Py), 4.35 (3H, t, 2JHH/3JHH = 10.5 Hz, CH2), 3.50 (3H, dd, 2JHH = 10.5 Hz, 3JHH = 3.5 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 171.6 (CN), 158.5, 157.2, 153.4, 149.4, 138.3, 136.9, 134.6, 129.0, 128.8, 127.8, 127.5, 125.8, 123.0, 122.4 (Ar), 74.2 (CH2Py), 72.5 (CHCH2), 71.1 (CH).
MS (ESI) m/z 503.70 [FeL113]2+.
IR v cm−1 1733 m, 1590 m, 1473 m, 1353 w, 1245 m, 1077 s, 760 s, 700 s.
Elemental Analysis found (Calculated for C60H57Cl2FeN9O11) % C 59.17 (59.71), H 4.88 (4.76), N 9.92 (10.44).
110 total reflections, 4777 unique (Rint = 0.0790), R1 = 0.0798 (obs. data), wR2 = 0.2101 (all data), GooF 1.027, Flack 0.07(4).ΔFe,ΔCu,RC-[FeL113Cu](ClO4)2I
fac,ΔFe,RC-[FeL113](ClO4)2 (0.20 g, 0.17 mmol) was dissolved in acetonitrile (15 ml). Copper(I) iodide (32 mg, 0.17 mmol) was dissolved in acetonitrile (10 ml) and was added. The solution was stirred overnight at ambient temperature. The solvent was then removed under reduced pressure and the crude purple solid was recrystallised from acetonitrile–ethyl acetate. Yield = 0.13 g, 0.093 mmol, 55%.1H NMR (400 MHz, 298 K, CD3CN) δH 9.05 (3H, s, HCN), 8.68 (3H, d, 3JHH = 5 Hz, Py), 7.81 (3H, t, 3JHH = 8 Hz, Py), 7.68 (3H, t, 3JHH = 8 Hz, Py), 7.62 (3H, d, 3JHH = 8 Hz, Py), 7.40 (3H, d, 3JHH = 8 Hz, Py), 7.33 (3H, t, 3JHH = 6 Hz, Py), 7.15 (3H, t, 3JHH = 6 Hz, Py), 7.04 (3H, t, 3JHH = 7 Hz, Ph), 6.88 (6H, t, 3JHH = 8 Hz, Ph), 6.73 (3H, d, 3JHH = 5 Hz, Py), 6.64 (6H, d, 3JHH = 7 Hz, Ph), 5.79 (3H, dd, 3JHH = 10 Hz, 3JHH = 2 Hz, CH), 5.02 (3H, d, 2JHH = 13 Hz, CH2Py), 4.82 (3H, d, 2JHH = 13 Hz, CH2Py), 4.34 (3H, t, 2JHH/3JHH = 11 Hz, CH2), 3.46 (3H, dd, 2JHH = 11 Hz, 3JHH = 2 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 172.5 (CN), 159.5, 158.1, 154.4, 150.5, 139.3, 138.0, 135.5, 130.0, 129.9, 128.9, 128.5, 126.8, 124.1, 123.6 (Py/Ph), 75.1 (CH2Py), 73.5 (CHCH2), 72.1 (CH).
MS (ESI) m/z 503.7 [FeL113]2+, 318.2 [L11 + H]+.
IR v cm−1 3055 w, 2868 w, 1641 w, 1592 m, 1571 w, 1495 w, 1475 m, 1437 m, 1388 w, 1358 w, 1302 w, 1239 w, 1048 s, 835 m, 759 s, 700 s.
Elemental Analysis found (Calculated for C60H57Cl2CuFeIN9O11) % C 51.79 (51.57), H 4.02 (4.11), N 9.31 (9.02).
ΔFe,RC-[FeL113Ag(CH3CN)](ClO4)3
fac,ΔFe,RC-[FeL113](ClO4)2 (0.05 g, 0.04 mmol) was dissolved in acetonitrile (15 ml). Silver(I) perchlorate (8.6 mg, 0.04 mmol) was added as a solid. The solution was stirred overnight at ambient temperature. The solvent was then removed under reduced pressure and the crude purple solid was recrystallised from acetonitrile–ethyl acetate. Yield = 0.04 g, 0.028 mmol, 71%.1H NMR (400 MHz, 298 K, CD3CN) δH 9.06 (3H, s, HCN), 8.75 (3H, dd, 3JHH = 5.0 Hz, 4JHH = 1.0 Hz, Py), 7.98 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, Py), 7.83 (3H, d, 3JHH = 7.5 Hz, Py), 7.64 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, Py), 7.50 (3H, m, Py), 7.42 (3H, d, 3JHH = 7.5 Hz, Py), 7.11 (3H, m, Py), 7.05 (3H, t, 3JHH = 7.5 Hz, Ph), 6.94 (6H, t, 3JHH = 7.5 Hz, Ph), 6.72 (3H, d, 3JHH = 5.5 Hz, Py), 6.49 (6H, d, 3JHH = 7.5 Hz, Ph), 5.84 (3H, dd, 3JHH = 11.5 Hz, 3JHH = 3.0 Hz, CH), 4.97 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.76 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.35 (3H, t, 2JHH/3JHH = 11.5 Hz, CH2), 3.46 (3H, dd, 2JHH = 11.5 Hz, 3JHH = 3.0 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 172.0 (CN), 159.4, 156.9, 154.4, 153.1, 140.5, 139.4, 134.9, 130.3, 130.2, 129.2, 128.6, 126.6, 126.5, 125.9 (Ar), 74.7 (CH2Py), 73.7 (CHCH2), 71.4 (CH).
MS (ESI) m/z 503.7 [FeL113]2+, 345.1 [FeL112]2+.
Elemental Analysis found (Calculated for C62H60AgCl3FeN10O15) % C 51.43 (51.17), H 4.01 (4.16), N 9.27 (9.62).
fac,ΔFe,RC-[FeL223](ClO4)2
2-Pyridinecarboxaldehyde (0.33 g, 3.0 mmol) and (R)-2-(pyridin-3-methoxy)-1-phenylethanamine (0.70 g, 3.0 mmol) were dissolved in acetonitrile (15 ml) to form a yellow solution. Iron(II) perchlorate hexahydrate (0.37 g, 1.0 mmol) in acetonitrile (5 ml) was added and immediately the solution turned purple. This was stirred overnight at ambient temperature before the solvent was removed under reduced pressure. The product, [FeL223](ClO4)2, was recrystallised from acetonitrile and ethyl acetate and the resulting purple crystals were filtered and dried in vacuo. Yield = 0.86 g, 0.71 mmol, 71%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.97 (3H, br s, Py), 8.94 (3H, s, HCN), 8.53 (3H, br s, Py), 7.96 (3H, d, 3JHH = 7.5 Hz, Py), 7.65 (3H, t, 3JHH = 7.5 Hz, Py), 7.39–7.35 (6H, m, Py), 7.11 (3H, t, 3JHH = 6.5 Hz, Py), 7.04 (3H, t, 3JHH = 7.5 Hz, Ph), 6.89 (6H, t, 3JHH = 7.5 Hz, Ph), 6.66 (3H, d, 3JHH = 5.5 Hz, Py), 6.53 (6H, d, 3JHH = 7.5 Hz, Ph), 5.72 (3H, dd, 3JHH = 11.0 Hz, 3JHH = 3.0 Hz, CH), 4.86 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.82 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.22 (3H, t, 2JHH/3JHH = 11.0 Hz, CH2), 3.18 (3H, dd, 2JHH = 11.0 Hz, 3JHH = 3.0 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 172.1 (CN), 159.5, 154.5, 147.0, 139.4, 137.4, 135.6, 130.1, 130.0, 129.1, 128.5, 127.8, 126.8, 126.7, 124.8 (Ar), 73.3 (CH2), 72.3 (CH), 72.1 (CH2Py).
MS (ESI) m/z 503.70 [FeLnnn3]2+, 318.1 [L + H]+.
Elemental Analysis found (Calculated for C60H57Cl2FeN9O11) % C 59.24 (59.71), H 4.85 (4.76), N 10.01 (10.44).
ΔFe,ΔCu,RC-[FeL223CuI](ClO4)2
fac,ΔFe,RC-[FeL223](ClO4)2 (50.0 mg, 0.0414 mmol) was dissolved in acetonitrile (10 ml). Copper(I) iodide (7.9 mg, 0.0414 mmol) was added as a solid to the reaction. The solution was stirred overnight at ambient temperature. The solvent was then removed under reduced pressure and the crude purple solid was recrystallised from acetonitrile–ethyl acetate. Yield = 48 mg, 0.0344 mmol, 83%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.94 (3H, s, HCN), 8.66 (3H, br s, Py), 8.00 (3H, br s, Py), 7.79 (3H, br s, Py), 7.68 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.0 Hz, Py), 7.41 (3H, d, 3JHH = 7.5 Hz, Py), 7.26 (3H, br s, Py), 7.16–7.13 (3H, m, Py), 7.06 (3H, t, 3JHH = 7.5 Hz, Ph), 6.92 (6H, t, 3JHH = 7.5 Hz, Ph), 6.69 (3H, d, 3JHH = 5.5 Hz, Py), 6.59 (6H, d, 3JHH = 7.5 Hz, Ph), 5.67 (3H, d, 3JHH = 9.5 Hz, CH), 5.02 (3H, br s, CH2Py), 4.78 (3H, br s, CH2Py), 4.14 (3H, t, 2JHH/3JHH = 10.0 Hz, CH2), 3.36 (3H, d, 2JHH = 10.0 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 172.9 (CN), 160.2, 155.1, 140.1, 136.1, 130.8, 130.7, 129.7, 129.2, 127.5 (Ar), 73.5 (CH2), 72.9 (CH), 72.3 (CH2Py). Note that the Cu-coordinated pyridine C atoms were not detected; the H signals are broad in 1H NMR spectrum.
MS (ESI) m/z 503.7 [FeL223]2+, 345.1 [FeL222]2+, 318.1 [L22+H]+.
Elemental Analysis found (Calculated for C60H57Cl2CuFeIN9O11) % C 51.41 (51.57), H 4.04 (4.11), N 9.19 (9.02).
ΔFe,RC-[FeL223Ag(CH3CN)](ClO4)3
fac,ΔFe,RC-[FeL223](ClO4)2 (50.0 mg, 0.0414 mmol) was dissolved in acetonitrile (10 ml). Silver(I) perchlorate (8.6 mg, 0.0414 mmol) was added as a solid to the reaction. The solution was stirred overnight at ambient temperature. The solvent was then removed under reduced pressure and the crude purple solid was recrystallised from acetonitrile–ethyl acetate. Yield = 52 mg, 0.0368 mmol, 89%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.93 (3H, s, HCN), 8.82 (3H, s, Py), 8.53 (3H, d, 3JHH = 3.5 Hz, Py), 7.94 (3H, d, 3JHH = 7.5 Hz, Py), 7.67 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, Py), 7.41–7.39 (6H, m, Py), 7.13 (3H, m, Py), 7.05 (3H, t, 3JHH = 7.5 Hz, Ph), 6.91 (6H, t, 3JHH = 7.5 Hz, Ph), 6.67 (3H, d, 3JHH = 5.5 Hz, Py), 6.57 (6H, d, 3JHH = 7.5 Hz, Ph), 5.68 (3H, dd, 3JHH = 10.5 Hz, 3JHH = 3.0 Hz, CH), 4.90 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.75 (3H, d, 2JHH = 11.5 Hz, CH2Py), 4.19 (3H, t, 2JHH/3JHH = 10.5 Hz, CH2), 3.28 (3H, dd, 2JHH = 10.5 Hz, 3JHH = 3.0 Hz, CH2).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 172.9 (CN), 160.2, 155.1, 153.4, 140.1, 138.8, 136.2, 130.8, 130.7, 130.6, 129.7, 129.2, 127.8, 127.4, 125.7 (Ar), 73.7 (CH2), 72.9 (CH), 72.4 (CH2Py).
MS (ESI) m/z 503.7 [FeL223]2+, 345.1 [FeL222]2+, 318.1 [L22 + H]+.
Elemental Analysis found (Calculated for C62H60AgCl3FeN10O15) % C 51.24 (51.17), H 4.11 (4.16), N 9.41 (9.62).
fac,ΔFe,RC-[FeL333](ClO4)2
2-Pyridinecarboxaldehyde (0.33 g, 3.0 mmol) and (R)-2-(propargyloxy)-1-phenylethanamine (0.52 g, 3.0 mmol) were dissolved in acetonitrile (15 ml) to form a yellow solution. Iron(II) perchlorate hexahydrate (0.37 g, 1.0 mmol) in acetonitrile (5 ml) was added and immediately the solution turned purple. This was stirred overnight at ambient temperature before the solvent was removed under reduced pressure. The product, [FeL333](ClO4)2, was recrystallised from acetonitrile and ethyl acetate and the resulting purple crystals were filtered and dried in vacuo. Yield = 0.81 g, 0.77 mmol, 77%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.93 (3H, s, HCN), 7.70 (3H, td, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, Py), 7.40 (3H, d, 3JHH = 7.5 Hz, Py), 7.18 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, Py), 7.10 (3H, t, 3JHH = 7.0 Hz, Ph), 6.99 (6H, t, 3JHH = 7.0 Hz, Ph), 6.78 (9H, m, Py/Ph), 5.81 (3H, dd, 3JHH = 11.0 Hz, 3JHH = 3.5 Hz, CH), 4.60 (3H, dd, 2JHH = 16.0 Hz, 4JHH = 2.0 Hz, CH2–CC), 4.51 (3H, dd, 2JHH = 16.0 Hz, 4JHH = 2.0 Hz, CH2–CC), 4.38 (3H, t, 2JHH/3JHH = 11.0 Hz, CH2CHPh), 3.70 (3H, dd, 2JHH = 11.0 Hz, 3JHH = 3.5 Hz, CH2CHPh), 2.95 (3H, t, 4JHH = 2.0 Hz, CCH).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 170.9 (CN), 158.1, 153.3, 138.2, 134.1, 128.8, 128.8, 127.8, 127.4, 125.6 (Ph), 78.7 (CCH), 76.0 (CCH), 70.9 (CH2CHPh), 70.4 (CHPh), 58.0 (CH2–CC).
MS (ESI) m/z 424.16 [FeL333]2+.
IR v cm−1 3259 w, 1732 w, 1613 w, 1474 m, 1452 m, 1356 w, 1240 m, 1076 s, 758 s, 699 s.
Elemental Analysis found (Calculated for C51H48Cl2FeN6O11) % C 57.36 (58.47), H 4.65 (4.62), N 7.69 (8.02).
ΔFe,ΛCu,RC-[FeL443Cu](ClO4)2I
fac,ΔFe,RC-[FeL333](ClO4)2·H2O (0.25 g, 0.24 mmol) was dissolved in dry acetonitrile (30 ml) in a Schlenk vessel. Dry triethylamine (0.10 ml, 0.72 mmol) was added, followed by benzyl azide (95 mg, 0.72 mmol) in dry acetonitrile (10 ml). Finally, copper(I) iodide (45 mg, 0.24 mmol) was added as a solid. The solution remained purple in colour and was stirred for 24 h under argon at ambient temperature. The solvent was removed under reduced pressure and the resultant solid was recrystallised from acetonitrile and ethyl acetate. Yield = 0.21 g, 0.13 mmol, 53%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.85 (3H, s, HCN), 8.27 (3H, s, triazole CH), 7.58 (3H, t, 3JHH = 7.5 Hz, Py), 7.34 (3H, d, 3JHH = 7.5 Hz, Py), 7.15–6.91 (27H, m, Py/Ph), 6.64 (3H, d, 3JHH = 5.5 Hz, Py), 6.39 (6H, br m, Py/Ph), 5.53 (3H, d, 2JHH = 14.5 Hz, NCH2Ph), 5.48 (3H, d, 2JHH = 14.5 Hz, NCH2Ph), 5.39 (3H, dd, 3JHH = 11.0 Hz, 2.5 Hz, CH), 4.62 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 4.36 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 3.64 (3H, t, 2JHH/3JHH = 11.0 Hz, CH2CHPh), 1.39 (3H, br d, 2JHH = 10.0 Hz, CH2CHPh).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 171.5 (CN), 159.5, 154.2, 145.6, 139.1, 135.5, 135.1, 130.0, 129.8, 129.6, 129.4, 128.9, 128.2, 126.7, 126.6, 126.0 (Ar, triazole CH), 72.4 (CHCH2), 71.3 (CH), 63.2 (OCH2–triazole), 55.5 (NCH2Ph).
MS (ESI) m/z 623.75 [FeL443]2+.
IR v cm−1 3036 w, 1612 w, 1496 w, 1474 w, 1454 m, 1334 w, 1224 w, 1075 s, 821 w, 756 s, 720 s, 700 s.
Elemental Analysis found (Calculated for C72H69Cl2CuFeIN15O11) % C 52.75 (52.81), H 4.26 (4.25), N 12.55 (12.83).
683 total reflections, 5470 unique (Rint = 0.0552), R1 = 0.0876 (obs. data), wR2 = 0.2242 (all data), GooF 1.369, Flack 0.012(11).ΔFe,ΛCu,RC-[FeL553Cu](ClO4)2I
fac,ΔFe,RC-[FeL333](ClO4)2·H2O (0.25 g, 0.24 mmol) was dissolved in dry acetonitrile (25 ml) in a Schlenk vessel. Dry triethylamine (0.10 ml, 0.72 mmol) was added, followed by 3,5-dimethylbenzyl azide (0.12 g, 0.72 mmol) in dry acetonitrile (10 ml). Finally, copper(I) iodide (45 mg, 0.24 mmol) was added. The solution remained purple in colour and was stirred for 24 h under argon at ambient temperature. The solvent was removed under reduced pressure and the resultant solid recrystallised from acetonitrile and ethyl acetate. Yield = 0.23 g, 0.13 mmol, 56%.1H NMR (300 MHz, 298 K, CD3CN) δH 8.86 (3H, s, HCN), 8.27 (3H, s, triazole), 7.59 (3H, t, 3JHH = 7.5 Hz, Py), 7.33 (3H, d, 3JHH = 7.5 Hz, Py), 7.09–7.02 (6H, m, Py/Ph), 6.93–6.89 (6H, m, Py/Ph), 6.80–6.79 (9H, m, Py/Ph), 6.67 (3H, d, 3JHH = 7.5 Hz, Py), 6.31 (6H, br m, Py/Ph), 5.49–5.36 (9H, m, CH and NCH2-aryl), 4.63 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 4.30 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 3.80 (3H, t, 2JHH/3JHH = 11.0 Hz, CH2CHPh), 1.98 (18H, s, CH3), 1.73 (3H, br d, 2JHH = 11.0 Hz, CH2CHPh).
13C {1H} NMR (400 MHz, 298 K, CD3CN) δC 171.6 (CN), 159.4, 154.3, 145.5, 139.6, 139.2, 135.4, 134.9, 131.3, 130.1 130.0, 129.9, 128.9, 128.3, 127.2, 126.0 (Ar/triazole CH), 72.6 (CHCH2), 71.4 (CH2CHPh), 63.1 (OCH2–triazole), 55.7 (NCH2Ph), 21.2 (CH3).
MS (ESI) m/z 453.2 [FeL552]2+, 665.8 [FeL553]2+.
IR v cm−1 3016 w, 2864 w, 1728 m, 1610 m, 1473 m, 1452 m, 1390 m, 1334 m, 1239 m, 1160 w, 1071 s, 1007 m, 835 m, 796 m, 746 s, 701 s.
Elemental Analysis found (calculated for C78H81Cl2CuFeIN15O11) % C 54.72 (54.40), H 4.34 (4.74), N 11.86 (12.20).
ΔFe,ΛCu,RC-[FeL663Cu](ClO4)2I
fac,ΔFe,RC-[FeL333](ClO4)2·H2O (0.10 g, 0.095 mmol) was dissolved in dry acetonitrile (15 ml) in a Schlenk. Dry triethylamine (0.40 μl, 0.285 mmol) was added to the reaction, followed by 4-nitrobenzyl azide (0.051 g, 0.285 mmol) in dry acetonitrile (10 ml). Finally, copper(I) iodide (18 mg, 0.095 mmol) was added as a solid. The solution remained purple in colour and was stirred for 24 h under argon at ambient temperature. The solvent was removed under reduced pressure and the resultant solid was recrystallised from acetonitrile and ethyl acetate. Yield = 0.23 g, 0.13 mmol, 71%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.78 (3H, s, HCN), 8.38 (3H, s, triazole CH), 7.72 (6H, d, 3JHH = 8.5 Hz, Ph–NO2), 7.55 (3H, td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, Py), 7.40 (6H, d, 3JHH = 8.5 Hz, Ph–NO2), 7.29 (3H, d, 3JHH = 7.5 Hz, Py), 7.09 (3H, t, 3JHH = 7.5 Hz, Ph), 7.03–6.95 (9H, m, Py/Ph), 6.60 (3H, d, 3JHH = 5.5 Hz, Py), 6.33 (6H, br d, 3JHH = 7.0 Hz, Ph), 5.70 (3H, d, 2JHH = 14.5 Hz, NCH2Ph), 5.63 (3H, d, 2JHH = 14.5 Hz, NCH2Ph), 5.32 (3H, dd, 3JHH = 11.0 Hz, 3.0 Hz, CH), 4.70 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 4.46 (3H, d, 2JHH = 11.0 Hz, OCH2–triazole), 3.59 (3H, t, 2JHH/3JHH = 11.0 Hz, CH2CHPh), 1.43 (3H, br d, 2JHH = 10.0 Hz, CH2CHPh).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 171.6 (CN), 159.4, 154.1, 145.8, 142.4, 139.0, 135.0, 130.8, 130.1, 129.9, 129.0, 128.2, 127.8, 126.5, 126.3, 124.8 (Ar, triazole), 72.4 (CHCH2), 71.1 (CH), 63.4 (OCH2–triazole), 54.4 (NCH2Ph–NO2).
MS (ESI) m/z 691.23 [FeL663]2+.
Elemental Analysis found (calculated for C72H66Cl2CuFeIN18O17) % C 49.23 (48.79), H 3.84 (3.75), N 14.01 (14.22).
fac,ΛFe,RC-[FeL773](ClO4)2·3H2O
(R)-(+)-α-Methylbenzylamine (0.30 ml, 2.33 mmol) was dissolved in acetonitrile (10 ml). 5-(2-Pyridinyloxy)picolinaldehyde (0.5 g, 2.33 mmol) was added, followed by iron(II) perchlorate hexahydrate (0.282 g, 0.78 mmol) to give an immediate colour change to dark pink/purple. After stirring at ambient temperature for 16 h, addition of ethyl acetate (10 ml) caused precipitation of a dark purple powder (0.667 g, 0.55 mmol, 70%) that contained water of crystallisation according to IR spectroscopy. Single crystals were grown by layering a solution in acetonitrile onto ethyl acetate.1H-NMR (300 MHz, 298 K, CD3CN): δ 8.51 (bs, 6H, CHN, Py), 7.78 (t, 3H, 3JHH = 7.68 Hz, Py/Ph), 7.25–7.41 (m, 12H, Py/Ph), 7.05 (t, 3H 3JHH = 7.13 Hz, Py/Ph), 6.92 (t, 3H, 3JHH = 7.68 Hz, Py/Ph), 6.51 (d, 3H, 3JHH = 7.68 Hz, Py/Ph), 6.35 (d, 3H, 3JHH = 1.83 Hz, Py/Ph), 5.05–5.20 (m, 9H, PhCHCH3, CH2), 1.87 (d, 9H, 3JHH = 6.46, CH3).
13C NMR (75 MHz, CD3CN) δ 170.3 (CHN), 157.9, 155.7, 152.5, 150.5, 143.0, 141.1, 138.2, 130.8, 130.0, 128.3, 125.4, 124.6, 124.5, 123.2, 72.2 (CH2), 69.3 (CHPh).
ESI-MS: 345 [ML2]2+, 503 [ML3]2+, 789 [ML2·ClO4]+.
Elemental Analysis found (calculated for C60H63Cl2FeN9O14) % C 57.16 (57.15), H 4.58 (5.04), N 9.79 (10.00).
fac,ΔFe,SC-[FeL993](ClO4)2
5-(Propargyloxy)picolinaldehyde (0.50 g, 3.1 mmol) and (S)-α-methylbenzylamine (0.38 g, 3.1 mmol) were dissolved in acetonitrile (15 ml) to form a yellow solution. Iron(II) perchlorate hexahydrate (0.38 g, 1.0 mmol, 1.0 eq.) in acetonitrile (5 ml) was added to give a purple solution. This was stirred overnight at ambient temperature before the solvent was removed under reduced pressure. The product, [FeL993](ClO4)2, was recrystallised from acetonitrile and ethyl acetate and the resulting purple crystals were filtered and dried in vacuo. Yield = 0.65 g, 0.59 mmol, 59%.1H NMR (400 MHz, 298 K, CD3CN) δH 8.62 (3H, s, HCN), 7.42–7.35 (6H, m, Py), 7.09 (3H, t, 3JHH = 7.5 Hz, Ph para), 7.00 (6H, t, 3JHH = 7.5 Hz, Ph meta), 6.62 (6H, d, 3JHH = 7.5 Hz, Ph ortho), 6.35 (3H, d, 4JHH = 2.5 Hz, Py), 5.22 (3H, q, 3JHH = 6.5 Hz, CH), 4.72 (3H, dd, 2JHH = 16.5 Hz, 4JHH = 2.5 Hz, CH2–CC), 4.64 (3H, dd, 2JHH = 16.5 Hz, 4JHH = 2.5 Hz, CH2–CC), 2.94 (3H, t, 4JHH = 2.5 Hz, CCH), 1.92 (9H, d, 3JHH = 6.5 Hz, CH3).
13C{1H} NMR (100 MHz, 298 K, CD3CN) δC 170.5 (CN), 156.7, 152.9, 142.9, 140.9, 130.9, 130.0, 128.4, 125.5, 124.5 (Ar), 79.0 (CCH), 77.7 (CCH), 69.4 (CH), 57.3 (CH2), 26.3 (CH3).
MS (ESI) m/z 424.2 [FeL993]2+.
IR v cm−1 3256 w, 1592 w, 1560 m, 1496 m, 1452 w, 1383 w, 1297 w, 1278 m, 1229 m, 1070 s, 1007 s, 928 m, 838 m, 760 m, 740 w, 700 s.
Elemental Analysis found (calculated for C51H48Cl2FeN6O11) % C 58.61 (58.47), H 4.52 (4.62), N 7.94 (8.02).
Notes and references
- M. Albrecht and R. Fröhlich, J. Am. Chem. Soc., 1997, 119, 1656–1661 CrossRef CAS
.
- M. Cantuel, G. Bernardinelli, G. Muller, J. P. Riehl and C. Piguet, Inorg. Chem., 2004, 43, 1840–1849 CrossRef CAS
- F. E. Hahn, M. Offermann, C. Schulze Isfort, T. Pape and R. Fröhlich, Angew. Chem., Int. Ed., 2008, 47, 6794–6797 CrossRef CAS
- V. C. M. Smith and J.-M. Lehn, Chem. Commun., 1996, 2733–2734 RSC
- M. Albrecht, I. Janser, A. Lützen, M. Hapke, R. Fröhlich and P. Weis, Chem.–Eur. J., 2005, 11, 5742–5748 CrossRef CAS
- G. Canard and C. Piguet, Inorg. Chem., 2007, 46, 3511–3522 CrossRef CAS
- N. C. Fletcher, R. T. Brown and A. P. Doherty, Inorg. Chem., 2006, 45, 6132–6134 CrossRef CAS
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. van Gorkum and P. Scott, Chem. Commun., 2009, 1727–1729 RSC
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416–10433 RSC
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS
- N. Ousaka, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 1464–1468 CrossRef CAS
- N. Ousaka, S. Grunder, A. M. Castilla, A. C. Whalley, J. F. Stoddart and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 15528–15537 CrossRef CAS
- C. P. Sebli, S. E. Howson, G. J. Clarkson and P. Scott, Dalton Trans., 2010, 39, 4447–4454 RSC
- E. J. B. A. Baerends, C. Bo, P. M. Boerrigter, L. Cavallo, L. Deng, R. M. Dickson, D. E. Ellis, L. Fan, T. H. Fischer, C. Fonseca Guerra, S. J. A. van Gisbergen, J. A. Groeneveld, O. V. Gritsenko, F. E. Harris, P. van den Hoek, H. Jacobsen, G. van Kessel, F. Kootstra, E. van Lenthe, V. P. Osinga, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, P. Ros, P. R. T. Schipper, G. Schreckenbach, J. G. Snijders, M. Sola, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, O. Visser, E. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and T. Ziegler, adf2008.01, Scientific Computing and Modelling NV: Free University, Amsterdam, 2008 Search PubMed
- J. C. Dyason, P. C. Healy, C. Pakawatchai, V. A. Patrick and A. H. White, Inorg. Chem., 1985, 24, 1957–1960 CrossRef CAS
- G. Wilkinson, R. D. Gillard and J. A. McCleverty, in Comprehensive Coordination Chemistry Volume 5 – Late Transition Elements, Pergamon Press, Oxford, 1987, pp. 538–551 Search PubMed
- P. Mobian, J. P. Collin and J. P. Sauvage, Tetrahedron Lett., 2006, 47, 4907–4909 CrossRef CAS
- I. Aprahamian, O. S. Miljanic, W. R. Dichtel, K. Isoda, T. Yasuda, T. Kato and J. F. Stoddart, Bull. Chem. Soc. Jpn., 2007, 80, 1856–1869 CrossRef CAS
- A. I. Prikhod'ko, F. Durola and J.-P. Sauvage, J. Am. Chem. Soc., 2007, 130, 448–449 CrossRef
- S. Durot, P. Mobian, J. P. Collin and J. P. Sauvage, Tetrahedron, 2008, 64, 8496–8503 CrossRef CAS
- J. D. Megiatto and D. I. Schuster, J. Am. Chem. Soc., 2008, 130, 12872–12873 CrossRef CAS
- J.-P. Collin, J. Frey, V. r. Heitz, J.-P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609–5620 CrossRef CAS
- J.-P. Collin, J.-P. Sauvage, Y. Trolez and K. Rissanen, New J. Chem., 2009, 33, 2148–2154 RSC
- J. D. Megiatto and D. I. Schuster, Chem.–Eur. J., 2009, 15, 5444–5448 CrossRef CAS
- A. I. Prikhod'ko and J.-P. Sauvage, J. Am. Chem. Soc., 2009, 131, 6794–6807 CrossRef CAS
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS
- S.-i. Fukuzawa, H. Oki, M. Hosaka, J. Sugasawa and S. Kikuchi, Org. Lett., 2007, 9, 5557–5560 CrossRef CAS
- D. Gonzalez Cabrera, B. D. Koivisto and D. A. Leigh, Chem. Commun., 2007, 4218–4220 RSC
- C. Haensch, M. Chiper, C. Ulbricht, A. Winter, S. Hoeppener and U. S. Schubert, Langmuir, 2008, 24, 12981–12985 CrossRef CAS
- M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283–6285 RSC
- A. R. McDonald, H. P. Dijkstra, B. M. J. M. Suijkerbuijk, G. P. M. van Klink and G. van Koten, Organometallics, 2009, 28, 4689–4699 CrossRef CAS
- E. C. Constable, C. E. Housecroft, M. Neuburger and P. Rosel, Chem. Commun., 2010, 46, 1628–1630 RSC
- S. E. Howson and P. Scott, Dalton Trans., 2011, 40, 10268–10277 RSC
- S. Paliwal, S. Geib and C. S. Wilcox, J. Am. Chem. Soc., 1994, 116, 4497–4498 CrossRef CAS
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC
- J. L. M. Abboud, C. Foces-Foces, R. Notario, R. E. Trifonov, A. P. Volovodenko, V. A. Ostrovskii, I. Alkorta and J. Elguero, Eur. J. Org. Chem., 2001, 3013–3024 CrossRef CAS
- L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS
- B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS
- J. R. Johansson, P. Lincoln, B. Nordén and N. Kann, J. Org. Chem., 2011, 76, 2355–2359 CrossRef CAS
- C. N. Urbani, C. A. Bell, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 1057–1060 CrossRef CAS
- C. Ornelas, J. Broichhagen and M. Weck, J. Am. Chem. Soc., 2010, 132, 3923–3931 CrossRef CAS
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. van Gorkum and P. Scott, Chem. Commun., 2009, 1727–1729 RSC
- M. Seredyuk, A. B. Gaspar, V. Ksenofontov, Y. Galyametdinov, J. Kusz and P. Gütlich, J. Am. Chem. Soc., 2008, 130, 1431–1439 CrossRef CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS
- E. J. Baerends, D. E. Ellis and P. Ros, Theor. Chim. Acta, 1972, 27, 339–354 CrossRef CAS
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 CrossRef CAS
- R. L. Bixler and C. Niemann, J. Org. Chem., 1958, 23, 575–584 CrossRef CAS
- E. J. O'Neil, K. M. DiVittorio and B. D. Smith, Org. Lett., 2007, 9, 199–202 CrossRef CAS
Footnotes |
| † CCDC 947140–947142. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51725j |
| ‡ The propeller pitch is defined as the angle between mean plane of the three pyridine N atoms and that of a pyridine ligand. |
| This journal is © The Royal Society of Chemistry 2013 |