 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of Cp2M (M = Ni, V) with dilithium diamido-aryl reagents; retention and oxidation of the transition metal ions†
Francesca A. Stokesa,
Mark A. Vincentb,
Ian H. Hillierb,
Tanya K. Ronsona,
Alexander Steinerc,
Andrew E. H. Wheatley*a,
Paul T. Wooda and
Dominic S. Wright*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: aehw2@cam.ac.uk; dsw1000@cam.ac.uk; Fax: +44 (0)1223 336362; Tel: +44 (0)1223 763966
bSchool of Chemistry, The University of Manchester, Manchester, M13 9PL, UK
cDepartment of Chemistry, University of Liverpool, Crown street, Liverpool, L69 7ZD, UK
First published on 29th July 2013
Abstract
The reactions of dilithium 1,2-diamidobenzene, [1,2-(HN)2C6H4]Li2 (L1111H2222)Li2, and dilithium 1,8-diamidonaphthalene, [1,8-(NH)2C10H6]Li2 (L222H222)Li2, with Cp2Ni and Cp2V have been used to obtain the new complexes (L222H222)2Ni{Li(THF)2}2 (3), (L222H222)3V{Li(THF)2}3 (4) and (L1111H2222)6Ni6·{[(L1111H2222)3(L11H)3Ni6Li(THF)]2−·2[Li(THF)4]+} (5), in which retention or oxidation of the initial metal(II) centre is observed. Whereas 3 and 4 contain one transition metal ion within ion-paired structures, 5 has a complicated co-crystalline composition which contains octahedral Ni6-cages constructed from six square-planar (16e) NiII centres.
Introduction
In contrast to ferrocene (Cp2Fe), in which the Cp–Fe interactions are predominantly covalent, the other first-row metallocenes are far more polar. As a result, these species have extensive synthetic utility as sources of the metal atoms in a range of organometallic and metallo-organic compounds. Extensive synthetic studies involving a broad range of N-, O- and C-based organic acids and nucleophiles have identified three main types of reactivity; (i) the deprotonation of organic acids (RH) by the anionic Cp ligands (pathway 1, Scheme 1),1 (ii) the addition of weaker nucleophiles (R−) to the transition metal centre (M) without displacement of the Cp ligands (pathway 2),2 and (iii) nucleophilic displacement of the Cp− ligands by stronger heteroatomic and organometallic nucleophiles (R−) (pathway 3).3 A common feature of all of these reactions is that the +2 oxidation state of the transition metal ion is maintained throughout, so that Cp2M has functioned as an organically-soluble source of M2+ ions. | ||
| Scheme 1 Common reactivity patterns of polar metallocenes with organic acids and nucleophiles. | ||
The ability to maintain the oxidation state of the transition metal in the course of each of the reactions in Scheme 1 is of particular interest in the formation of multiply-bonded transition metal compounds in which the retention of a low oxidation state is important for the participation of the metal d-orbitals in bonding. Some of our most recent studies have shown that the use of Cp2M (M = V, Cr, Mn, Ni) as metal precursors can indeed lead to formally multiply-bonded transition metal compounds. For example, the reaction of (hpp)Li (hppH = 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2,a]pyrimidine) with Cp2V gives the remarkable complex [{V2(hpp)4}Li(μ5-Cp)Li(μ5-Cp)Li{V2(hpp)4}]+·[(η5-Cp)Li(η5-Cp)Li(η5-Cp)]− in which a V![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) V triply-bonded fragment V2(hpp)43c functions as a metal-based Lewis base ligand in the trapping of a [{V2(hpp)4}Li(μ5-Cp)Li(μ5-Cp)Li{V2(hpp)4}]+ cation.2 Meanwhile, whereas reaction of the sterically-undemanding lithium amidinate [MeN
V triply-bonded fragment V2(hpp)43c functions as a metal-based Lewis base ligand in the trapping of a [{V2(hpp)4}Li(μ5-Cp)Li(μ5-Cp)Li{V2(hpp)4}]+ cation.2 Meanwhile, whereas reaction of the sterically-undemanding lithium amidinate [MeN![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) C(H)NMe]Li with Cp2Cr has given the classical, lantern-shaped compound Cr2[MeNC(H)NMe]4, which contains a Cr–Cr quadruple bond,3d the use of the dilithiate of 2,3-diphenyl guanidine [(PhNH)2C
C(H)NMe]Li with Cp2Cr has given the classical, lantern-shaped compound Cr2[MeNC(H)NMe]4, which contains a Cr–Cr quadruple bond,3d the use of the dilithiate of 2,3-diphenyl guanidine [(PhNH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH = LH33] gives [Cr2(LH)4]{Li(THF)2}4(LiCp)2, which contains a quadruply-bonded CrII tetraanion.3
NH = LH33] gives [Cr2(LH)4]{Li(THF)2}4(LiCp)2, which contains a quadruply-bonded CrII tetraanion.3
Most recently, however, it was found that the tendency to maintain the metal +2 oxidation state in these reactions may be far more dependent on the particular organic acid or nucleophile employed than had been realised in earlier studies. The potential for complicated redox behaviour of the dianion of 1,2-diaminobenzene, [1,2-(HN)2C6H4]2− (L1111H2222), had already been observed in a number of main group4 and transition metal reactions.5 For example, the reaction of dilithium 1,2-diamidobenzene, [1,2-(HN)2C6H4]Li2 (L1111H2222)Li2 with the SnII base Sn(NMe2)2 is known to result in the formation of a mixed oxidation state SnII/SnIV complex.4 With this behaviour in mind we have begun to explore the reactions of (L1111H2222)Li2 with metallocene. We found that the reactions with Cp2V and Cp2Mn gave the VIII complex [(η5-Cp)(L1111H2222)2VV(L1111H2222)]−[Li(THF)4]+ (1) (Fig. 1a)6 and the oxo-capture MnII4MnIII2 cage Mn6(L1111H2222)6(μ6-O)(THF)4 (2) (Fig. 1b), respectively,6 in which (unlike in all other reactions of Cp2M observed so far) the metal centres are oxidised or partially oxidised during the course of reaction.
![(a) Structure of the VIII [(η5-Cp)(L1111H2222)2VV(L1111H2222)]− anionic component of 1 and (b) the MnII4MnIII2 cage 2.](/image/article/2013/DT/c3dt51632f/c3dt51632f-f1.gif) | ||
| Fig. 1 (a) Structure of the VIII [(η5-Cp)(L1111H2222)2VV(L1111H2222)]− anionic component of 1 and (b) the MnII4MnIII2 cage 2. | ||
We present here further synthetic and structural studies involving the reactions of Cp2Ni and Cp2V with dilithium 1,2-diamidobenzene, [1,2-(HN)2C6H4]Li2 (L1111H2222)Li2, and related dilithium 1,8-diamidonaphthalene, [1,8-(NH)2C10H6]Li2 (L222H222)Li2, in which we have explored the potential for (i) redox chemistry in these systems and (ii) molecular aggregation.
Results and discussion
Solid-state and solution studies
Initial reactions focused on use of the 1,8-diaminonaphthalene (L22H44). This was doubly deprotonated by its reaction with 2 equivalents of nBuLi in THF solution. Slow addition of the dilithiate, (L222H222)Li2, to a solution of Cp2Ni or Cp2V (1 equivalent) at −78 °C gave dark red and dark yellow solutions, respectively, from which the new compounds (L222H222)2Ni{Li(THF)2}2 (3) and (L222H222)3V{Li(THF)2}3 (4) were isolated in crystalline form (Scheme 2) (in 34 and 6% crystalline yields, respectively). Both compounds were characterised by 1H and 13C NMR spectroscopy and by elemental (C, H, N) analysis prior to obtaining their single-crystal X-ray structures. Although both complexes proved insoluble in most organic solvents (toluene, benzene and THF), the 1H NMR spectra of 3 and 4 could be obtained in DMSO-d6 at room temperature and showed the presence of a single environment for the L222H222 ligands and a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of THF:L222H222, which is consistent with the later structural characterisation of the complexes. Although no N–H resonance was observed in the 1H NMR spectrum of 4 at room temperature, the spectrum of 3 exhibited an N–H resonance at δ 1.36. The integration of this signal with respect to the aromatic resonances (in the region δ 5.26–6.12) showed that double deprotonation of the L22H44 precursor had occurred.
:1 ratio of THF:L222H222, which is consistent with the later structural characterisation of the complexes. Although no N–H resonance was observed in the 1H NMR spectrum of 4 at room temperature, the spectrum of 3 exhibited an N–H resonance at δ 1.36. The integration of this signal with respect to the aromatic resonances (in the region δ 5.26–6.12) showed that double deprotonation of the L22H44 precursor had occurred.
 | ||
| Scheme 2 Reactions producing 3 and 4. The naphthalene ring systems in the products have been omitted for clarity. | ||
The low-temperature single-crystal structures of 3 and 4 show that molecules of both contain only a single transition metal atom. This contrasts with recent studies of the reactions of Cp2V and Cp2Cr with related ligands which give singly- or multiply-bonded (metal)2 compounds.1–3,6 In the case of 3, the structure consists of a central distorted square-planar NiII ion that is bonded to the four N-donor atoms of two dianionic L222H222 ligands (Ni(1)–N(1) 1.876(2), Ni(1)–N(2) 1.900(2) Å; N(1)–Ni(1)–N(2) 94.54(11), N(1)–Ni(1)–N(2A) 85.46(11)°). These N-atoms are, in turn, bonded to two symmetry-related, THF-solvated Li+ cations on either side of the centrosymmetric structure of 3. The asymmetry with the L222H222 dianions bond to the central Ni(II) atom is not discernable in the corresponding Li–N bond lengths (Li(1)–N(1) 2.081(5), Li(1)–N(2A) 2.100(5) Å), which are each as expected for essentially ionic Li–N interactions.7 Significant puckering is seen in the LiN2Ni ring units along the N(1)⋯N(2) vector (the puckering angle being ca. 115°). Overall, the structure of 3 is best regarded as an ion-paired complex of the [Ni(L222H222)2]2− dianion with two THF-solvated Li+ cations.
A number of compounds containing the dianionic L222H222 ligand have been structurally characterised previously, and the κ1,2N-κ1,2N′-bridging mode found in 3 has been seen in several dinuclear transition metal compounds (Fig. 2).8 Additionally, there exist examples of heterometallic transition metal/main group metal complexes in which 1,8-N-alkylated or -silylated naphthylamido ligands bridge the metal centres, examples being the MnII/Li complex (THF)Mn(Cl){1,8-(Me3SiN)2C10H6}Li(THF)9 and the TlI/Li complex Tl{1,8-(Me3SiN)2C10H6}Li(THF)2,10 both of which incorporate the κ1,2N-κ1,2N′-ligand mode to bridge the metals centres.
 | ||
| Fig. 2 Molecular structure of 3. H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°); Ni(1)–N(1) 1.876(2), Ni(1)–N(2) 1.900(2), Li(1)–N(1) 2.081(5), Li(1)–N(2) 2.100(5). Ni(1)⋯Li(1) 2.554(5), N(1)–Ni(1)–N(2) 94.54(11), N(1)–Ni(1)–N(2A) 85.46(11), Ni(1)–N(1)–Li(1) 80.20(17), Ni(1)–N(2)–Li(1) 79.19(17). | ||
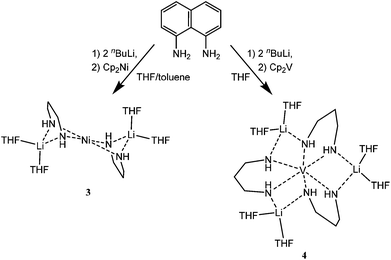
The solid-state structure of 4 (Fig. 3) shows that the complex consists of a [V(L222H222)3]3− anion that is coordinated by three [Li(THF)2]+ cations using a similar ligand bridging mode to that in 3 (now, however, with each of the ligand N-atoms bonding to separate Li+ ions). While the oxidation state of the NiII centre (unsurprisingly) remained unchanged in the formation of 3, the VII centre present in the Cp2V starting material has been oxidised to VIII in the case of 4. This is a similar outcome to that found in the formation of 1, discussed in the introduction (Fig. 1a).6
 | ||
| Fig. 3 Structure of the core of 4. H-atoms omitted for clarity. Selected bond lengths (Å) and angles (°); V(1)–N 2.047(2)–2.094(3), V(1)⋯Li 2.741(9)–2.792(5), N–Li 2.068(6)–2.117(6), Li–O 1.931(6)–1.959(6), N–V–N 82.4(1)–97.5(1), N–Li–N 93.5(2)–98.4(4). | ||
In contrast to the structure of 1, the L222H222 ligands in 4 are essentially symmetrically bound to the central metal ion with the V–N bond lengths being normal for such interactions (V(1)–N(1) 2.054(3), V(1)–N(2) 2.047(2), V(1)–N(3) 2.094(3) Å).7 The VIII ion adopts a distorted octahedral geometry (N(1)–V(1)–N(2) 97.46(10), N(3)–V(1)–N(3A) 96.78(15)°). Whereas puckered four-membered LiN2Ni metallocyclic units were noted in 3, the three LiN2V ring units in 4 are essentially planar (being puckered along the Li(1)⋯V(1) vector by no more than ca. 171°). Although complexes containing octahedral VIII ions are known for a range of ligands,11 complexes containing octahedral VIII which are tris-coordinated by chelating anionic N-ligands are relatively rare and 4 is the first example of a transition metal complex of the type [(L222H222)3M]n+ (or indeed of any anionic N-ligand related to L22).
Following on from our initial studies of the reactions using the 1,2-benzenediamine (L11H44) framework with Cp2Mn and Cp2V, in which oxidation or partial oxidation of the metal centres was observed,6 the dilithium salt (L1111H2222)Li2 (prepared in situ by the reaction of L11H44 with 2 equivalents of nBuLi) was reacted with Cp2Ni (2:1 equivalents, respectively) under N2 at −78 °C in THF. Subsequent storage of the reaction mixture at room temperature afforded highly air-sensitive purple crystals of a new complex, 5 (67% with respect to Cp2Ni), which exhibits a complicated co-crystalline structure in the solid state (Scheme 3).
 | ||
| Scheme 3 Formation of the two types of Ni6 cage arrangement, 5a and 5b2−, obtained by the reaction of (L1111H2222)Li2 with Cp2Ni. The three environments of the ligand backbones present in 5a are coloured black, red and blue. | ||
The single crystal X-ray diffraction study of 5 reveals an extensively disordered structure comprising a 1:1 co-crystalline mixture of two closely related octahedral NiII6 cages, (L1111H2222)6Ni6 cage (5a) (Fig. 4a) (solely containing the doubly-deprotonated [C6H4(NH)2]2− ligand) and [(L1111H2222)3(L11H)3Ni6Li(THF)]2−·2[Li(THF)4]+ 5b·2[Li(THF)4] (the dianionic part of which, 5b2−, is shown in Fig. 4b and which contains a mixture of doubly-deprotonated [C6H4(NH)2]2− and triply-deprotonated [C6H4(NH)(N)]3− ligands). In addition, there are several molecules of THF within the lattice, one of which forms a close contact to Ni(5) (ca. 2.55 Å) (solvent not shown in Fig. 4a). Triple deprotonation of L11H44 has been observed previously only in its reaction with the superbase mixture Sn(NMe2)2/nBuLi.4
![(a) The structure of 5a with the three types of chelating ligand highlighted (H-atoms and aromatic rings excluded) and (b) structure of the anion 5b2−2−2− incorporating its Li(THF) component (showing only the O-atom of THF, and with H-atoms and aromatic rings and the two [Li(THF)4]+ counter-ions omitted for clarity).](/image/article/2013/DT/c3dt51632f/c3dt51632f-f4.gif) | ||
| Fig. 4 (a) The structure of 5a with the three types of chelating ligand highlighted (H-atoms and aromatic rings excluded) and (b) structure of the anion 5b2−2−2− incorporating its Li(THF) component (showing only the O-atom of THF, and with H-atoms and aromatic rings and the two [Li(THF)4]+ counter-ions omitted for clarity). | ||
The crystallographic model for 5 is based on multiple data collections using conventional X-ray analysis as well as synchrotron radiation, all of which indicated the same ratio of 5a to 5b·2[Li(THF)4]. Despite the high R1 value of 14.3% (obtained using synchrotron radiation), which ostensibly results from subtle structural variations between the neutral and dianionic Ni6 cages 5a and 5b2−, the identity and essential connectivity of the separate cage components are not in doubt. Moreover, this formulation is also consistent with elemental C, H and N analysis on the bulk sample.
The unusual cubeoctahedral arrangement of six, square-planar NiII ions and twelve N-atoms found in 5 is similar to the structure of the hexanuclear complex [Ni(NH2)2]6,12 and structurally equivalent to (PtCl2)6.13 Ni6 octahedra have also been observed in the cluster complex (CpNi)614 and hexanuclear, heteroatom-bridged cages such as Ph3PNi6Se5.15 Although within the range of potential Ni–Ni bonds,7 the Ni⋯Ni contacts within the core of 5 (range 2.74–2.82 Å; cf. 2.83 Å in [Ni(NH2)2]612) are unlikely to represent bonding interactions and it seems more appropriate to regard this cage within the context of conventional square-planar NiII (d8) coordination compounds. It can be noted in this regard that the NiII centres in 5 each have the expected 16e count.
In contrast to the straightforward spectroscopic analyses of 3 and 4 (see Experimental section) compound 5 exhibited complicated behaviour in the aromatic region of the 1H NMR spectrum in a variety of solvents (DMSO-d6, C5H5N-d5, THF-d8). Three different types of aromatic rings are clearly discernible in a 1:1:1 ratio by a combination of 1H NMR and COSY (in DMSO-d6, in which 5 is most soluble). This conclusion is re-enforced by HMBC NMR spectroscopy (in DMSO-d6) which indicates that three types of L1111H2222 ligand are present in solution in equal abundance and allows the assignment of each of the NH groups to their respective C6H4 ring units (Fig. 5a and 5b). The observation of six distinct NH groups of equal integral at very high field (δ 0.63 to −2.10) is consistent with only the neutral cage 5a being present in DMSO. The highly reactive cage 5b2−2−2− is presumably completely converted into 5a via the protonation reaction 5b2−2−2− + 3H+ → 5a; a reaction which is not unexpected given the known relatively high acidity of the Me-groups of DMSO compared to potent bases such as, for example, organolithium reagents.16
 | ||
| Fig. 5 The 1H NMR spectrum of 5; (a) the aromatic region and (b) the N–H region. The assignments are indicated by colour. | ||
Theoretical studies
To complement our experimental studies, density functional theory (DFT) calculations were carried out on 5a and 5b2−2−2− using Gaussian 09.17 The structures of both species were optimized for the S = 0 spin state using the M06-L and a 6-31+G** basis. This functional was chosen for its computational efficiency and has previously been shown to be suitable for describing metal clusters.18 The predicted structures of these species along with important inter-atomic distances are shown in Fig. 6. In the case of 5a, the predicted Ni⋯Ni and Ni–N lengths which span 2.74–2.91 and 1.93–1.99 Å, respectively, are in good agreement with our X-ray findings. Interestingly, 5a is predicted to have a significant electron affinity (153 kJ mol−1) so that in the solid state, electron transfer from 5b2−2−2− could well modify the geometric and electronic structure of the cluster. Moreover, when 5a is deprotonated we find that the energy of the HOMO of 5a− is negative, implying that this species does not readily undergo electron loss and that it thus represents the first step in the formation of 5b2−2−2−.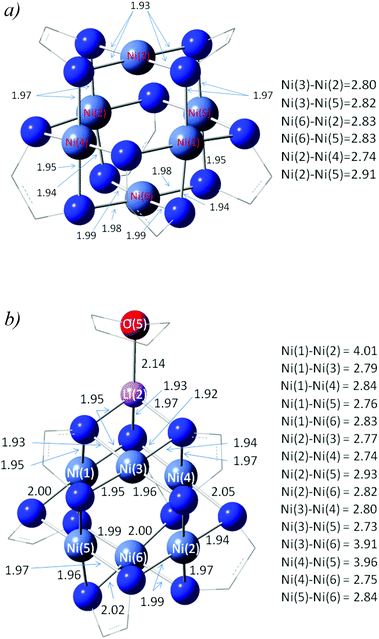 | ||
| Fig. 6 Structures of (a) 5a and (b) 5b2−2−2− at the M06-L/6-31+G** level. | ||
We subsequently used the computed structure of 5a to predict the corresponding 1H NMR chemical shifts using the Gauge Independent Atomic Orbital (GIAO) method in conjunction with both the B3LYP and M06-L functionals and a large basis set (6-311++G(2df,2pd)). We have also investigated the effect of solvent (DMSO), which has been included by a continuum (bulk) model (SMD), on the M06-L 1H NMR spectroscopic shifts. We find that the aromatic signals are predicted to fall in the range δ 6.84–6.21 and δ 6.89–6.13 for the B3LYP and M06-L functionals, respectively. These data are in reasonable agreement with the experimentally observed range of δ 6.45–5.45. The predicted chemical shifts of the twelve NH groups (six pairs in C2 symmetry) are shown in Table 1, and are to be compared to the experimental spectrum, which has five peaks in the range δ 0.63 to −0.05 with a sixth peak well separated at δ −2.10. According to the calculations, and in line with expectation, the hydrogen atoms are all protic rather than hydridic in nature. B3LYP and initial M06-L calculations reported in Table 1 (lines 1 and 2) predict the same general pattern of two groups (Ha, Hb, He and Hc, Hd, Hf) of three fairly closely spaced lines, and thus do not reproduce the experimentally observed pattern of shifts well. However, we can see from the geometric structure that due to their close proximity to the face of the aromatic rings, Hc/d/f are shielded from solvent to a greater extent than are Ha/b/e. This should be reflected in the effect of solvent on the calculated chemical shifts and, in line with this, the overall spread of the NH resonances is increased when the SMD method is used. Hence, the inclusion of solvent effects in the calculations raises the spread of the computed NH resonances from a value of δ 1.55 (Table 1, line 2) to one of δ 2.37 (Table 1, line 3), bringing it significantly closer to the experimentally seen spread of δ 2.73 (see above). Moreover, in addition to giving an improved range of computed shifts, the inclusion of solvent effects starts to distinctly separate one high-field NH (Hc) from the other peaks – again in line with experimental observation.

Conclusions
The combined studies of the reactions of Cp2M (M = V, Mn, Ni) with the related amido-aryl salts (L1111H2222)Li2 and (L222H222)Li2 reported by us in our previous communication6 and in the current paper reveal that retention or increase of the transition metal oxidation state can occur depending on the transition metal present in the precursor. Whereas V and Mn show partial or complete oxidation of the metal centres with this type of ligand, Ni proves to be considerably more redox stable. The mechanism and associated redox chemistry involved in these systems is not currently understood. However, the use of L1111H2222 and L222H222 as supporting ligands clearly has the capacity to yield high nuclearity transition metal complexes. In the current study this feature is seen through formation of the Ni6 compound 5.Experimental section
Synthesis
All reactions were performed under dry, O2-free N2. Solvents (THF and toluene) were dried using Na/benzophenone. Products were handled and stored in a glove box (Saffron, type β). Cp2Ni and Cp2V were obtained using literature procedures and were purified by sublimation.19,20 nBuLi in hexanes was acquired from Aldrich and was used without standardising. NMR spectra were recorded on a Bruker DRX 500 FT-NMR spectrometer using deuterated NMR solvents (THF-d8 and C5H5N-d5 were dried using a Na mirror, DMSO-d6 was used as supplied). 1H and 13C NMR spectra were referenced to the solvent peaks. 7Li NMR spectra were referenced to 1 M LiOH–D2O. Elemental analysis (C, H and N) was obtained in-house using an Exeter Analytical CE-440 Elemental Analyzer.X-ray crystallography
Data on 3·2/3PhMe and 4·2THF were collected on a Nonius KappaCCD diffractometer equipped with an Oxford cryostream low-temperature device. Crystals were mounted directly from solution using a perfluorohydrocarbon oil that freezes at low temperature.21 Data were solved by direct methods and refined by full-matrix least squares on F2.22 Data for 5 were collected at Beamline I19 of the Diamond Light Source23 employing silicon double crystal monochromated synchrotron radiation (0.6889 Å) with ω scans at 100(2) K.24 Data were first treated with ECLIPSE25 and integration and reduction were undertaken with SAINT and XPREP.26 A multi-scan empirical absorption correction was applied to the data using SADABS.26 Crystals of 3·2/3PhMe contain 2/3 of a molecule of toluene per formula unit, disordered across the −3 positions and refined with constraints. All non-hydrogen atoms were refined anisotropically with the exception of toluene molecules, which were refined isotropically with common thermal parameters per molecule. Crystals of 4·2THF contain non-coordinated THF molecules which are disordered and these were refined by splitting atoms over two positions. All non-hydrogen atoms were refined anisotropically with the exception of atoms of the disordered THF molecules, which were refined isotropically. The data for 5·5THF were rather diffuse and of limited resolution due to the superimposition of complexes 5a and 5b2−. Therefore the data were truncated at 0.92 Å. Ligands and THF molecules in 5 were treated with similar distance and U restraints during the refinement. However, the diffuse character of the electron density did not allow the refinement of disordered atoms over split positions. All atoms were refined isotropically, except Ni atoms which were treated anisotropically. Hydrogen atoms were not considered in the refinement. CCDC 943106–8 contain the supplementary crystallographic data for 3–5. Details of the data collections and refinements for the compounds are given in Table 2.| Compound | 3·2/3PhMe | 4·2THF | 5·5THF |
|---|---|---|---|
| Chemical formula | C40.6H23.33Li2N4NiO4 | C62H82Li3N6O8V | C128H181N24O14Ni12Li3 |
| FW | 734.80 | 1111.10 | 3005.31 |
| Crystal system | Trigonal | Monoclinic | Monoclinic |
| Space group | P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
C2/c | P21 |
| a (Å) | 20.0120(6) | 25.7117(6) | 13.2275(3) |
| b (Å) | 20.0120(6) | 14.1550(4) | 17.0537(7) |
| c (Å) | 8.1348(2) | 18.9518(6) | 16.5692(6) |
| α (°) | −90 | −90 | −90 |
| β (°) | −90 | 119.140(1) | 95.948(3) |
| γ (°) | −120 | −90 | −90 |
| V (Å3) | 2821.36(14) | 6024.5(3) | 3717.5(2) |
| Z | 3 | 4 | 1 |
| ρcalc (Mg m−3) | 1.297 | 1.225 | 1.342 |
| μ (Mo-Kα) (mm−1) | 0.562 | 0.222 | 1.543 |
| Reflections collected | 15927 |
21086 |
27906 |
| Independent reflections (Rint) | 3334 (0.053) | 6087 (0.064) | 10066(0.114) |
| R1 [I > 2σ(I)] | 0.043 | 0.069 | 0.138 |
| wR2 (all data) | 0.114 | 0.206 | 0.366 |
Acknowledgements
We thank the UK EPSRC (F. A. S., T. K. R.). We also thank Dr J. E. Davies for collecting X-ray data on 3 and 4 and the Diamond Light Source (UK) for synchrotron beam time on Beamline I19 (MT7569). We thank Dr Julian J. Holstein and Colm Browne for assistance with X-ray data collection for 5.Notes and references
- A. D. Bond, E. A. Harron, R. A. Layfield, C. Soria-Álvarez and D. S. Wright, Organometallics, 2001, 20, 4135 CrossRef CAS; C. Soria-Álvarez, D. Cave, A. D. Bond, E. A. Harron, R. A. Layfield, M. E. G. Mosquera, C. M. Pask, M. McPartlin, J. M. Rawson, P. Wood and D. S. Wright, Dalton Trans., 2003, 3002 RSC; F. A. Stokes, R. J. Less, J. Haywood, R. L. Thompson, A. E. H. Wheatley and D. S. Wright, Organometallics, 2012, 31, 23 CrossRef CAS.
- R. A. Layfield, J. A. McAllister, M. McPartlin, J. M. Rawson and D. S. Wright, Chem. Commun., 2001, 1956 RSC; C. Soria-Álvarez, A. Bashall, E. McInnes, R. A. Layfield, M. McPartlin, J. M. Rawson and D. S. Wright, Chem.–Eur. J., 2006, 3053 CrossRef.
- (a) C. Soria-Álvarez, S. R. Boss, J. Burley, S. M. Humphrey, R. A. Layfield, R. A. Kowenicki, M. McPartlin, J. M. Rawson, A. E. H. Wheatley, P. T. Wood and D. S. Wright, Dalton Trans., 2004, 3481 RSC; (b) C. Brinkmann, F. García, J. V. Moray, M. McPartlin, S. Singh, A. E. H. Wheatley and D. S. Wright, Dalton Trans., 2007, 1570 RSC; (c) C. Fernández, F. García, J. V. Morey, M. McPartlin, S. Singh, A. E. H. Wheatley and D. S. Wright, Angew. Chem., Int. Ed., 2007, 46, 5425 CrossRef CAS; (d) J. Haywood, F. A. Stokes, R. J. Less, M. McPartlin, A. E. H. Wheatley and D. S. Wright, Chem. Commun., 2011, 47, 4120 RSC.
- R. J. Less, V. Naseri, M. McPartlin and D. S. Wright, Chem. Commun., 2011, 47, 6129 RSC.
- S.-M. Peng and D.-S. Liaw, Inorg. Chim. Acta, 1986, 113, L11 CrossRef; S. K. Brownstein and G. D. Enright, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1579 CrossRef; S. Stanković and D. Lazar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51–1581 Search PubMed; B. Milliken, L. Borer, M. Russell, M. Bilich and M. M. Olmstead, Inorg. Chim. Acta, 2003, 348, 212 CrossRef CAS.
- F. A. Stokes, Y. Lv, A. E. H. Wheatley and D. S. Wright, Chem. Commun., 2012, 48, 11298 RSC.
- Search of the Cambridge crystallography data base, May 2013; Li–N mean 2.08 Å (3312 hits), V–N mean 2.12 Å (2424 hits), Ni–N mean 2.04 Å (14905 hits), Ni–Ni mean 2.61 (range 2.19–3.44) Å (628 hits).
- For example, L. A. Oro, M. J. Fernandez, J. Modrego, C. Foces-Foces and F. H. Cano, Angew. Chem., Int. Ed. Engl., 1984, 23, 913 Search PubMed; M. J. Jimenez, E. Sola, A. P. Martinez, F. J. Lahoz and L. A. Oro, Organometallics, 1999, 18, 1125 CrossRef.
- A. J. Blake, M. L. Gillbrand, G. J. Moxey and D. L. Kays, Inorg. Chem., 2009, 48, 10837 CrossRef CAS.
- K. W. Helmann, C. Galka, L. H. Gade, A. Steiner, D. S. Wright, T. Kottke and D. Stalke, Chem. Commun., 1998, 549 RSC.
- F. A. Cotton, L. M. Daniels and C. A. Murillo, Inorg. Chem., 1993, 32, 2881 CrossRef CAS; B. S. Lim, A. Rahtm, J.-S. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951 CrossRef CAS; S. Hao, P. Berno, R. K. Minhas and S. Gambarotta, Inorg. Chim. Acta, 1996, 37, 244 Search PubMed; K. R. Gust, J. E. Knox, M. J. Heeg, H. B. Schlegel and C. H. Winter, Eur. J. Inorg. Chem., 2002, 2327 CrossRef CAS.
- A. Tenten and H. Jacobs, J. Less-Common Met., 1991, 170, 145 CrossRef.
- H. G. von Schnering, J.-H. Chang, K. Peters, E.-M. Peters, F. R. Wagner, Y. Grin and G. Thiele, Z. Anorg. Allg. Chem., 2003, 629, 516 CrossRef.
- M. S. Paquette and L. F. Dahl, J. Am. Chem. Soc., 1980, 102, 6621 CrossRef CAS.
- D. Fenske and J. Ohmer, Angew. Chem., Int. Ed. Engl., 1987, 26, 148 CrossRef.
- The pKa of DMSO is ca. 35 (vs. ca. 50 for nBuLi), W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision b.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 4, 157 Search PubMed.
- C. Floriani and V. Mange, Inorg. Synth., 1990, 28, 263 Search PubMed.
- E. O. Fischer, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1952, 7, 377.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435 Search PubMed.
- Crystalclear, Rigaku Americas and Rigaku Corporation, TX, USA, 1997–2009 Search PubMed.
- S. Parson, ECLIPSE, The University of Edinburgh, Edinburgh, UK, 2004 Search PubMed.
- G. M. Sheldrick, University of Göttingen, Germany, 1996–2008.
Footnote |
| † Electronic supplementary information (ESI) available: DFT coordinates for 5a and 5b2−2−2−. CCDC 943106–943108. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51632f |
| This journal is © The Royal Society of Chemistry 2013 |