 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structural and magnetic characterisation of iron(II/III), cobalt(II) and copper(II) cluster complexes of the polytopic ligand: N-(2-pyridyl)-3-carboxypropanamide†
Mark E.
Russell
a,
Chris S.
Hawes
ab,
Alan
Ferguson
a,
Matthew I. J.
Polson
a,
Nicholas F.
Chilton
b,
Boujemaa
Moubaraki
b,
Keith S.
Murray
b and
Paul E.
Kruger
*a
aMacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, 8041, New Zealand. E-mail: paul.kruger@canterbury.ac.nz
bSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: keith.murray@monash.edu
First published on 19th July 2013
Abstract
Herein we describe the synthesis, structural and magnetic characterisation of three transition metal cluster complexes that feature the polytopic ligand N-(2-pyridyl)-3-carboxypropanamide (H2222L): [Fe3IIIFe2II(HL)6(O)(H2O)3][ClO4]5·3MeCN·4H2O, 1, [Co8(HL)8(O)(OH)4(MeOH)3(H2O)]-[ClO4]3·5MeOH·2H2O, 2, and [Cu6(Lox)4(MeOH)(H2O)3]·MeOH, 3. Complex 1 is a mixed valence penta-nuclear iron cluster containing the archetypal {Fe3IIIO} triangular basic carboxylate cluster at its core, with two Fe(II) ions above and below the core coordinated to three bidentate pyridyl-amide groups. The structure of the octanuclear Co(II) complex, 2, is based upon a central Co4 square with the remaining four Co(II) centres at the ‘wing-tips’ of the complex. The cluster core is replete with bridging oxide, hydroxide and carboxylate groups. Cluster 3 contains an oxidised derivative of the ligand, Loxox, generated in situ through hydroxylation of an α-carbon atom. This hexanuclear cluster has a ‘barrel-like’ core and contains Cu(II) ions in both square planar and square-based pyramidal geometries. Bridging between Cu(II) centres is furnished by alkoxide and carboxylate groups. Magnetic studies on 1–3 reveals dominant antiferro-magnetic interactions for 1 and 2, leading to small non-zero spin ground states, while 3 shows ferro-magnetic exchange between the Cu(II) centres to give an S = 3 spin ground state.
Introduction
The pyridyl, amide and carboxylate functional groups are commonly used ligand donor sets in coordination chemistry as they offer a broad range of potential coordination modes to metal ions from across the periodic table.1 While the monodentate pyridyl group is limited in the way it might coordinate to a metal ion, the amide and carboxylate functionalities offer a vast array of coordination capacity ranging from mono- and multi-dentate modes to bridging multiple metal ions in the formation of clusters and polymers. Carboxylate ligands have been used as key building blocks in supramolecular chemistry due to the many potential binding modes available to the carboxylate group.2 These various binding modes are observed often within metallo-clusters that may range in size from two to an impressive eighty-four metal ions.3 Many of these carboxylate containing cluster-complexes behave as single-molecule magnets (SMMs).4 In these rapidly evolving areas of research, the search for new polydentate ligands is an ongoing challenge. This has led increasingly to the design of ligands containing multiple functional groups that are capable of binding several metal ions.5 In this respect, the N-(2-pyridyl)-3-carboxy-propanamide ligand, H2222L, is an excellent candidate for the synthesis of new polynuclear transition metal complexes, Fig. 1.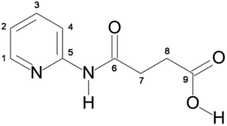 | ||
| Fig. 1 Structure of the ligand H2222L with the labelling scheme used for 1H and 13C NMR spectroscopy shown. | ||
Previous work with H2222L is limited and has been concerned primarily with its potential biological activity or medicinal application and in the formation of hydrogels.6 A few studies have reported the spectral characterisation of mononuclear Cu(II) and Ni(II) complexes of H2222L, although no solid-state structures of these species are known.7 Only one structurally characterised coordination compound has been reported: a discrete mono-nuclear complex, {[MnHL(phen)2]ClO4·1.25H2O}, where HL−− coordinates via a chelating carboxylate group to Mn(II) with 1,10-phenanthroline (phen) present as a co-ligand.8 Surprised by the dearth of structural reports featuring H2222L, and driven by the recognition of its potentially potent coordination capabilities, we begun an investigation into its coordination chemistry with a variety of 1st row transition metal ions. Here, we report the synthesis and structural characterisation of the first multi-nuclear cluster complexes of H2222L: a pentanuclear Fe(II/III), an octa-nuclear Co(II) and a hexanuclear Cu(II) species along with details of their magnetic properties.
Experimental
Materials and methods
All reagents were used as received from BDH or Sigma Aldrich. 1H and 13C NMR spectroscopy was carried out on a Varian INOVA 500 spectrometer operating at 500 MHz for 1H and 125 MHz for 13C. High resolution mass spectrometry was recorded on a Bruker maXis 3G UHR-TOF mass spectrometer. Infrared spectra were recorded on a Perkin Elmer Spectrum One FTIR spectrometer in the range 400–4000 cm−1. Samples were analyzed using diffuse reflectance in ground KBr. Elemental analyses (C, H, N) were conducted at Campbell Microanalytical Laboratories, Dunedin.The magnetic susceptibilities were measured using a Quantum Design Squid magnetometer, MPMS 5, at a dc field of 1 T, with the samples (ca. 20 mg) contained in gelatin capsules held at the centre of a drinking straw that was fixed to the end of the sample rod. Ligand diamagnetic corrections were obtained using Pascal's constants. Magnetization isotherms were measured using the same instrument under dc fields of 0 to 5 T at temperatures 2, 3, 4, 5.5, 10, 20 K with samples 1 and 2 dispersed in a Vaseline mull to prevent torquing effects.
The structural analysis at 120 K was performed on an Agilent dual wavelength SuperNova with monochromated Cu-Kα (λ = 1.5418 Å) (1 and 3) or Mo-Kα (λ = 0.71073 Å) (2) radiation. CrysAlisPro9 was used for the data collection and data processing. The structure was solved using direct methods with SHELXS10 and refined on Olex211 using all data by full matrix least-squares procedures with SHELXL-97.12 Multi-scan absorption correction using SCALE3 ABSPACK.13 CCDC numbers: 939536 (1); 939537 (2); 939538 (3).
Synthesis of N-(2-pyridyl)-3-carboxypropanamide, H2222L
This synthesis was adapted from the method of Godina and co-workers,14 full details and spectroscopic data are reported here for the first time. Succinic anhydride (2.73 g, 2.73 × 10−2 mol) and 2-aminopyridine (2.57 g, 2.73 × 10−2 mol) were dissolved in ethyl acetate (150 ml) and heated at reflux temperature for 24 hours to produce a white solid, which was isolated by filtration, washed with hot ethyl acetate (50 ml) and air dried. Recrystallisation from methanol by slow evaporation afforded colourless needle crystals suitable for a single crystal X-ray diffraction study. Yield 2.08 g, 39%; mp. 180–182 °C (lit. 181–182 °C);14 HRMS-ESI m/z (MeOH): Found: 195.0766; C9H11N2O3 requires: 195.0764 [M + H+]; δH (500 MHz, CD3OD): 2.66 (2H, t, J = 6.1 Hz, 12.7 Hz, H8), 2.72 (2H, t, J = 6.1 Hz, 12.7 Hz, H7), 7.08 (1H, at, J = 6.1 Hz, 1.0 Hz, H2), 7.74 (1H, at, J = 8.1 Hz, 1.0 Hz, H3), 8.06 (1H, d, J = 8.1 Hz, H4), 8.26 (IH, d, J 6.1 Hz, 1.0 Hz, H1); δC (125 MHz, CD3OD): 28 (C7), 31 (C8), 114 (C4), 119 (C2), 138 (C3), 148 (C1), 152 (C5), 172 (C6), 175 (C9);![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) max/cm−1 (KBr): 3320 br, 3260 br, 3071 m, 2475 m, 1696 s br, 1590 m, 1438 m, 1357 m, 1313 m, 1248 s, 1168 s, 1006 s, 963 m, 916 w, 845 m, 784 s, 745 m, 634 s.
max/cm−1 (KBr): 3320 br, 3260 br, 3071 m, 2475 m, 1696 s br, 1590 m, 1438 m, 1357 m, 1313 m, 1248 s, 1168 s, 1006 s, 963 m, 916 w, 845 m, 784 s, 745 m, 634 s.
Synthesis and characterisation of the pentanuclear complex: [Fe3IIIFe2II(HL)6(O)(H2O)3][ClO4]5·3MeCN·4H2O, 1
To a MeCN solution (30 ml) of H2L (0.13 g, 0.67 mmol) was added Fe(ClO4)2·6H2O (0.25 g, 0.67 mmol) followed by the dropwise addition of a 10% Et3N solution (MeCN, 1 ml). The mixture was stirred for 12 h at 40 °C. Orange plate crystals suitable for a single crystal X-ray diffraction study were isolated after 1 week following the slow evaporation of the reaction mixture. A sample for analysis was dried in vacuo overnight to generate the partially desolvated phase of 1 as its mono-hydrate. Yield: 52 mg, 21%. Found: C, 32.20; H, 3.38; N, 8.08%; C54H60Cl5Fe5N12O43 requires: C, 32.07; H, 2.99; N 8.32%;max/cm−1 (KBr): 3322 br, 1677 s, 1601 s, 1540 s, 1478 s, 1319 s, 1219 s, 1095 s, 884 m, 788 s, 628 m.
Synthesis and characterisation of the octanuclear complex: [Co8(HL)8(O)(OH)4(MeOH)3(H2O)][ClO4]3·5MeOH·2H2O, 2
To a MeOH solution (30 ml) of H2L (0.13 g, 0.67 mmol) was added Co(ClO4)2·6H2O (0.245 g, 0.67 mmol) followed by the dropwise addition of a 10% Et3N solution (MeOH, 1 ml). Pink block crystals suitable for a single crystal X-ray diffraction study were isolated after 1 week following the slow evaporation of the reaction mixture. A sample for analysis was dried in vacuo overnight to generate the desolvated phase of 2. Yield: 23 mg, 10%. Found: C, 34.50; H, 3.38; N, 9.18%; C72H90Cl3Co8N16O45 requires: C, 34.89; H, 3.66; N 9.05%;max/cm−1 (KBr): 3318 w br, 1674 s, 1602 m, 1581 m, 1540 s, 1479 s, 1438 s, 1311 s, 1240 m, 1106 s, 783 m, 626 m, 512 m.
Synthesis and characterisation of the hexanuclear complex: [Cu6(Lox)4(MeOH)(H2O)3]·MeOH, 3
To a MeOH solution (30 ml) of H2L (0.13 g, 0.67 mmol) was added Cu(OAc)2·H2O (0.133 g, 0.67 mmol) followed by the dropwise addition of a 10% Et3N solution (MeOH, 1 ml). Large green block-like crystals suitable for a single crystal X-ray diffraction study were isolated after 2 weeks following the slow evaporation of the reaction mixture. A sample for analysis was dried in vacuo overnight to generate the desolvated phase of 3. Yield: 16 mg, 11%. Found: C, 33.80; H, 2.97; N, 8.63%; C36H34Cu6N8O19 requires: C, 34.19; H, 2.71; N 8.87%;max/cm−1 (KBr): 3340 w br, 2831 s, 1569 s, 1480 s, 1455 s, 1389 s, 1297 s, 1196 s, 1091 s, 1042 m, 969 s, 916 s, 886 s, 773 s, 746 m, 657 s, 581 m, 527 m.
Results and discussion
The ligand, H2222L, was synthesised through the condensation of succinic anhydride with 2-aminopyridine which, following recrystallisation from MeOH, produced colourless crystals. The IR spectrum of H2222L displays broad absorbance around 3300 cm−1 consistent with the presence of an amide (NH) and a stronger absorbance at around 2475 cm−1, indicating the formation of a hydrogen bond between pyridyl and carboxyl groups. The strong broad peak at 1696 cm−1 can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of free carboxylic groups and CO of both amide and hydrogen bonded carboxylic acid groups, respectively. These spectroscopic features are consistent with the interactions observed within the solid state structure as determined through X-ray crystallography (ESI†).
O of free carboxylic groups and CO of both amide and hydrogen bonded carboxylic acid groups, respectively. These spectroscopic features are consistent with the interactions observed within the solid state structure as determined through X-ray crystallography (ESI†).
Complexes 1–3 were obtained by adding M(II) salts to either MeCN or MeOH solutions of H2222L in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, followed by the addition of Et3N base, and allowing the resultant solutions to evaporate slowly at ambient temperature. Although isolated with modest yields, multiple crystalline batches were cropped from the mother liquor on further standing. The crystals of 1–3 were of sufficient quality to enable a single crystal X-ray diffraction study to be carried out from which their structures were determined unambiguously.
:1 stoichiometry, followed by the addition of Et3N base, and allowing the resultant solutions to evaporate slowly at ambient temperature. Although isolated with modest yields, multiple crystalline batches were cropped from the mother liquor on further standing. The crystals of 1–3 were of sufficient quality to enable a single crystal X-ray diffraction study to be carried out from which their structures were determined unambiguously.
X-ray crystal structure of 1
The atomic numbering scheme and atom connectivity for 1 are shown in Fig. 2. The structure was refined in the monoclinic P21/c space group and crystallographic data are detailed in Table 1. Complex 1 is a pentanuclear mixed valence iron cluster and displays unusual core geometry. Fe(II/III) oxidation states were assigned on the basis of bond valence sum calculations (ESI†), considering the bond lengths and charge balance for the cluster. At the centre of the complex resides the archetypal {Fe3IIIO} triangular basic carboxylate cluster. The coordination sphere around the Fe(III) ions is comprised of the six carboxylate groups of the ligand tail, which bind in a typical 1,3-bridging mode. The distorted octahedral geometry about each Fe(III) is completed by a central μ3-oxo-ligand and a coordinated water molecule. This {Fe3O} unit is common in iron-oxo chemistry and has served as a building block for many higher nuclearity clusters.15 At the periphery of the complex, above and below the triangular cluster, are two Fe(II) centres with pseudo-octahedral geometry. Coordination about these peripheral metal centres is furnished by three pyridyl nitrogen atoms and three amido oxygen atoms from three chelating HL− ligands to yield an N3O3 donor set.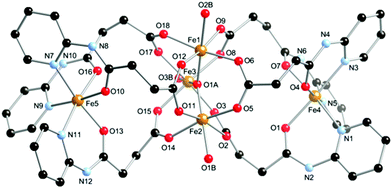 | ||
| Fig. 2 Molecular structure and atomic labelling scheme for 1. Hydrogen atoms, perchlorate anions and solvent molecules of crystallisation omitted for clarity. | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | |||
| Temperature/K | 120(1) | 120(1) | 120(1) |
| Chemical formula | C60H77Cl5Fe5N15O46 | C79.5H113Cl3Co8N16O52 | C38H42Cu6N8O21 |
| Formula mass | 2200.82 | 2702.65 | 1328.04 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| a/Å | 16.2556(2) | 25.9059(4) | 41.7229(8) |
| b/Å | 22.8571(4) | 14.9866(2) | 13.7165(3) |
| c/Å | 25.5874(4) | 27.3422(4) | 15.3592(3) |
| α/° | 90 | 90 | 90 |
| β/° | 102.0441(15) | 91.2539(15) | 91.5503(18) |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 9297.9(2) | 10612.8(3) |
8786.7(3) |
| Space group | P21/c | C2/c | C2/c |
| Z | 4 | 4 | 8 |
| λ/Å | 1.5418 | 0.7107 | 1.5418 |
| μ/mm−1 | 8.284 | 1.398 | 4.009 |
| N obs | 56674 |
20971 |
17949 |
| N unique | 16567 |
9391 | 8599 |
| R int | 0.0518 | 0.0220 | 0.0283 |
| R 1 | 0.0998 | 0.0481 | 0.0338 |
| wR2a | 0.3250 | 0.1029 | 0.0940 |
| Goodness of fit | 1.195 | 1.070 | 1.028 |
The separation between the peripheral Fe(II) centres and the central cluster Fe(III) atoms ranges from 5.932(1) to 6.348(1) Å; intra-triangular cluster separations range from 3.269(1) to 3.304(1) Å and the distance between peripheral Fe(II) centres is 11.535 Å through the central cluster. These Fe(II) ions are offset from the centre of the triangular cluster toward the Fe2–Fe3 axis i.e. they are not positioned directly above/below the central μ3-oxo-ligand, O1A. Interestingly, the tris-chelated nature of the HL− ligands bestows handedness upon the terminal metal ions, which is Δ and Λ about Fe4 and Fe5, respectively. The cluster is therefore a cluster-mesocate; related to the cluster helicates which differ from 1 in that they contain peripheral metal centres that possess the same handedness.16 To the best of our knowledge this is only the second example of a cluster-mesocate to be structurally characterised.17
X-ray crystal structure of 2
The atomic numbering scheme and atom connectivity for 2 are shown in Fig. 3. The structure was refined in the monoclinic C2/c space group and crystallographic data are detailed in Table 1. Compound 2 consists of an octanuclear Co(II) cluster that is built around a {Co4OH} square assembly, with the four remaining Co(II) centres forming the ‘wing-tips’ of the complex. The Co(II) centres in the {Co4OH} unit (Co2, Co2′, Co3, Co3′) display a distorted octahedral geometry composed of two μ3-OH (O1H and O2H), one μ4-OH (O1A) and three carboxylate O atoms (O3, O5 and O11 for Co2; O2, O6 and O8 for Co3). The octahedral geometry of the remaining Co(II) ions (Co1, Co1′, Co4, Co4′) is also distorted. Co4 and Co4′ are bound by one μ3-OH (O1H), two carboxylate O atoms (O9 and O6), one amido O atom (O4), a pyridyl N atom (N3) and a coordinated MeOH molecule (O14). Co1 and Co1′ also possess a similar donor set: one μ3-OH (O2H), two carboxylate O atoms (O3 and O12), one amido O atom (O1), a pyridyl N atom (N1); however, the coordinated MeOH molecule (O19) is present with only 25% occupancy with the remaining occupancy filled by a coordinated water molecule.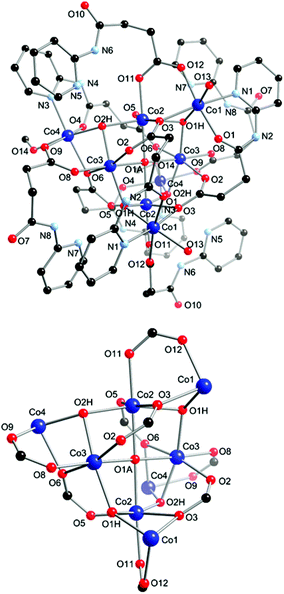 | ||
| Fig. 3 Molecular structure and partial atom labelling scheme for 2 (top) and the coordination core of 2 showing the potential pathways for magnetic exchange between the Co(II) centres (bottom). Hydrogen atoms and solvent molecules of crystallisation omitted for clarity. | ||
The HL− ligand is present in two distinct binding modes. Firstly, in the μ3:η2:η1:η1:η1:η0 mode the ligand chelates to one Co(II) centre at the wing-tip of the complex through the pyridyl N and amido O atoms, while the carboxylate group bridges three Co(II) ions: two from the central core and one from the wing-tip. Secondly, the μ2:η1:η1:η0:η0:η0 bonding mode is unbound at the pyridyl end and links a central Co(II) with a wing-tip Co(II) through the 1,3-bridging mode of the carboxylate group. The unbound pyridyl ring is stabilised by both intra- and inter-molecular interactions. Hydrogen bond interactions between the pyridyl nitrogen atom and a μ3-OH ligand (N5 to O2H and N7 to O1H) and from the adjacent amido NH to a carboxylate O atom (N6 to O11 and N8 to O8) act to stabilise the flexible unbound arm of the ligand. The pyridyl ring is stabilised further by offset intermolecular face-to-face π⋯π interaction (N5–C23 to N3–C14 = 3.475(13) Å, N5–C23 to N3′′–C14′′ = 3.664(14), N7–C32 to N1–C5 = 3.454(13) Å and N7–C32 to N7′′–C32′′ = 3.452(14) Å).
X-ray crystal structure of 3
The partial atomic numbering scheme and atom connectivity for 3 are shown in Fig. 4. The structure was solved and refined in the monoclinic C2/c space group and the crystallographic data are detailed in Table 1. The data identifies a hexanuclear Cu(II) complex and reveals that an in situ oxidation of the ligand has taken place via hydroxylation of the α-carbon atom adjacent to the amide carbonyl group, Fig. 4. Hydroxylation of this kind, although not common, is not without precedent and has been shown to proceed through metal directed oxidation involving molecular oxygen.18 When the reaction was performed under oxygen free conditions, and the solution left to stand under an inert atmosphere in an ‘open’ vial for several weeks, crystals of 3 failed to form. This is consistent with molecular oxygen being responsible for the hydroxylation, as no precaution was made to exclude water, and is in agreement with the previously proposed mechanism.18 The resultant ligand is in its triply deprotonated state and the [Cu:L] ratio of 6:4 generates a neutral hexanuclear cluster that is best described as having a distorted ‘barrel-like’ topology.
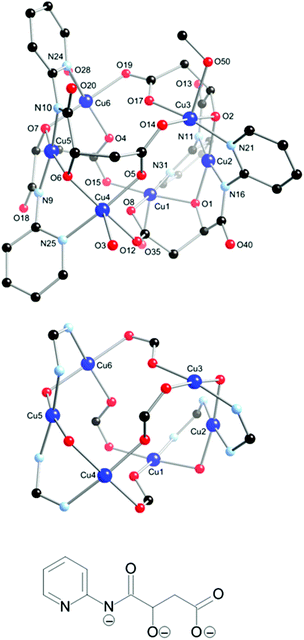 | ||
| Fig. 4 Molecular structure and partial atomic numbering scheme for 3 (top); the coordination core of 3 showing the potential pathways for magnetic exchange (middle) and the structure of the hydroxylated ligand generated in situ (bottom). Hydrogen atoms and solvent of crystallisation removed for clarity. | ||
Two square planar Cu(II) centres (Cu2 and Cu5) reside at the ends of the barrel, while four square-pyramidal Cu(II) ions (Cu1, Cu3, Cu4 and Cu6) occupy its sides. Square planar Cu2 and Cu5 are each bound by two amido nitrogen atoms (N11 and N16 for Cu2; N9 and N10 for Cu5) and two alkoxy oxygen atoms (O1 and O2 for Cu2; O6 and O7 for Cu5) from two different ligands to form two N,O-chelate rings around each Cu centre (four ligands in total). Each of the pyridyl rings adjacent to the coordinated amido groups are rotated by ca. 50° to the amido-alkoxy chelate ring plane and coordinate to square-pyramidal Cu(II) ions (Cu1, Cu3, Cu4 and Cu6). The basal plane of the square pyramidal Cu-centres is comprised of a pyridyl nitrogen atom, a bridging alkoxy oxygen atom and two bridging carboxylate oxygen atoms with either a coordinated water molecule (Cu1, Cu4 and Cu6) or methanol molecule (Cu3) located at the apex of the pyramid. The bridging and linking between each Cu-ion in 3 is such that the square or basal planes of the coordination spheres around adjacent Cu-centres subtend angles to each other between ca. 98–102° for the twelve angles formed by the six intersecting planes around the barrel.
Each hexanuclear complex is linked to four neighbouring clusters through eight hydrogen bond interactions that involve each amido oxygen atom and the neighbouring coordinated solvent molecules bound to the square-pyramidal Cu(II) ions. These connections create a 2D sheet in the crystallographic bc plane. Weak offset face-to-face π⋯π interactions between each pyridyl ring and its adjacent symmetry related partner (ring centroid–centroid distances range from 3.416(1)–3.763(1) Å) link the cluster to a further three neighbouring clusters along the crystallographic a-axis. The shortest intermolecular metal–metal separation is between Cu5 and Cu5′ at 3.683(1).
Magnetic properties
The bulk magnetic properties of 1–3 were probed via variable temperature, DC and AC susceptibility measurements on polycrystalline samples. For 1 the χMT value at 300 K is 10.1 cm3 mol−1 K, which is in good agreement with the expected value of ∼10.5 cm3 mol−1 K for a basic carboxylate {Fe3O} (S = 5/2 trimer)19 plus two non-coupled high-spin Fe(II) S = 2 centres, Fig. 5. As the temperature is lowered the χMT product decreases gradually to reach ∼7 cm3 mol−1 K at ca. 25 K, followed by a more rapid decrease to 1.9 cm3 mol−1 K at 2 K. This behaviour is consistent with dominant antiferromagnetic interactions within the cluster, while the sharp decrease at low temperature is due to zero-field splitting (ZFS).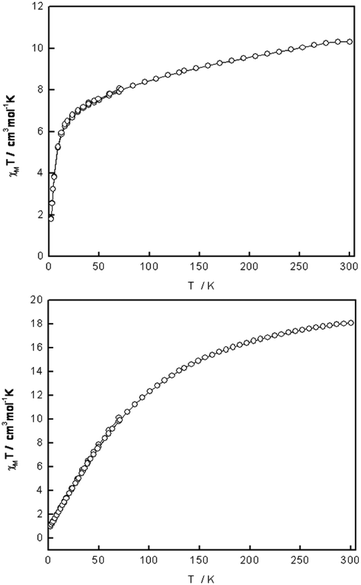 | ||
| Fig. 5 χ M T vs. T curves for 1 (top) and 2 (bottom). | ||
The magnetization (M) was measured as a function of the applied field at various temperatures (ESI†). It is clear the magnetisation curves are far from saturated. Even at 2 K and 5 T, the M vs. H curve is not saturated, but equal to 5.2Nβ, consistent with a small non-zero spin ground state for 1. The general shapes are indicative of antiferromagnetically coupled Fe(III) sites together with the two high-spin Fe(II) sites that have their contributing M values reduced because of spin–orbit coupling and orbital degeneracy effects. Antiferromagnetic coupling in Fe(III)-oxo complexes is common,20 however, larger complexes containing {Fe3O} building blocks have been shown to display large non-zero spin ground states.21
The χMT value of 2 at 300 K is 18 cm3 mol−1 K, which is consistent with eight uncoupled spin-only S = 3/2 Co(II) centres with g = 2.19. The χMT product decreases gradually as the temperature is lowered to reach 0.8 cm3 mol−1 K at 2 K, Fig. 5. This indicates antiferromagnetic exchange coupling between the metal centres. Changing the field in the range 2–70 K gave the same χMT value, thus magnetisable impurities and Zeeman depopulation effects appear to be absent. The magnetisation isotherms were measured using dc fields of 0 to 5 T and temperatures of 2 to 20 K (ESI†). The M values do not saturate and the values at 2 and 3 K at 5 T are similar, 2Nβ. These plots suggest the spin ground state is not zero and the low M value at 2 K and 5 T confirms antiferromagnetic coupling probably also influenced by spin–orbit coupling of the parent 4T1g single-ion states. Measurements of out-of-phase ac susceptibilities, χ′′M, for 2 in the range 2–10 K, gave zero values thus showing that no slow relaxation of magnetisation occurred and no SMM behaviour was observed, as expected for an antiferromagnetically coupled system.
Modelling Co(II) systems in any situation is extremely difficult due to the strong orbital moment and important crystal field effects. This case with eight such octahedral Co(II) ions is at present much too complicated to make any attempt at quantifying the magnetic data. A previously reported Co8 complex has a similar core to 2, however, it does not contain a bridging μ4-OH moiety and the Co(II) ions display trigonal bipyramidal geometry compared to the pseudo octahedral geometry observed in complex 2.22 Despite these differences both complexes display similar magnetic behaviour, with antiferromagnetic interactions dominating. Whilst Co(II)-based clusters often display antiferro-magnetic exchange interactions,23 there are several examples where ferromagnetic interactions lead to the observation of SMM behaviour.24
The χMT value of 3 at 300 K is 2.5 cm3 mol−1 K, which is in good agreement with the expected value for six uncoupled S = 1/2 Cu(II) centres with g = 2.10, Fig. 6. As the temperature is decreased towards 50 K, χMT increases gradually to 3.3 cm3 mol−1 K. Then below 50 K there is a more rapid increase in χMT to a maximum of 6.1 cm3 mol−1 K at 4 K, before finally decreasing to 4.9 cm3 mol−1 K at 2 K. Such behaviour is indicative of ferromagnetic exchange coupling with an S = 3 ground spin state. The magnetisation isotherms confirm this behaviour with the 2 K data saturating at M = 6Nβ as expected for an isolated S = 3 ground state (ESI†). Simultaneous fitting of the susceptibility and magnetisation data was performed with the program PHI,25 using the exchange spin Hamiltonian −2J1(Ŝ1·Ŝ2 + Ŝ2·Ŝ3 + Ŝ4·Ŝ5 + Ŝ5·Ŝ6) − 2J2(Ŝ1·Ŝ4 + Ŝ1·Ŝ6 + Ŝ3·Ŝ4 + Ŝ3·Ŝ6). The J1 pathways involve {(OR)(1,3-NCN)} bridging while the J2 pathways involve {anti-1,3-OCO} bridging. This two J model reproduced the data extremely well and led to a fit of the magnetic properties giving J1 = 15.1 cm−1, J2 = 5.18 cm−1 and g = 2.10, Fig. 6. It is more common to observe antiferromagnetic exchange interactions in polynuclear Cu(II) complexes.26 Complex 3 is one of the few examples of a Cu(II) cluster displaying ferromagnetic interactions, leading to a spin ground state of S > 1.27 Possible reasons for ferromagnetic coupling occurring include the Cu–OR–Cu bridging angles being low, in the range 102–104°, with Cu–Cu separations of ∼3.04 Å, and, or combined with, the equatorial planes of individual Cu centres (having Cu(dx2−y2) magnetic orbitals) being orthogonal to those observed for neighbouring centres as one traverses the barrel-shaped cluster; vide supra (Fig. 4). The Cu–N–C–N–Cu bridges, from the 2-pyridyl-amido ligand moieties, are not well known for their magnetic exchange features and these might also contribute to ferromagnetic coupling.
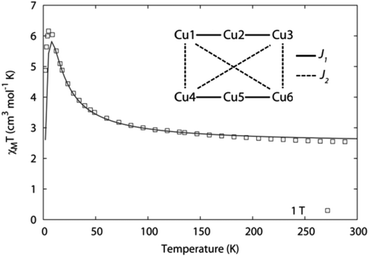 | ||
| Fig. 6 Fitting of the magnetic data for 3 with PHI,25 using J1 = 15.1 cm−1, J2 = 5.18 cm−1 and g = 2.10 with proposed coupling scheme (inset). | ||
Conclusions
This study has identified for the first time some of the potential coordination capability possessed by the polytopic ligand N-(2-pyridyl)-3-carboxy-propanamide, H2222L. Reaction of H2222L with either Fe(II), 1, Co(II), 2, or Cu(II), 3, led to the formation of three new transition metal cluster complexes possessing different and novel core geometries which were structurally and magnetically characterised. Pentanuclear complex 1 and octa-nuclear 2 show dominant antiferromagnetic interaction that leads to small non-zero spin ground states, whereas hexanuclear 3 displays ferromagnetic interaction between the Cu(II) centres yielding an S = 3 spin ground state. Interestingly, an in situ oxidation of H2222L occurred during the synthesis of 3via the hydroxylation of the α-carbon adjacent to the amide carbonyl group to form Loxox3−3−. H2222L and its oxidised derivative H33Loxox are extremely flexible in the nature of their binding to the metal centres, which is highlighted by the presence of four distinct binding modes across the three complexes, Fig. 7. In complexes 1 and 2, the ligand is mono-deprotonated and binds via the pyridyl group and the amido-O atom with the carboxylate group in various bridging modes. However, in complex 3 coordination involves the pyridyl group, and now the amido-N atom (which is deprotonated) and an O-atom from the hydroxyl group in combination with a bridging carboxylate group.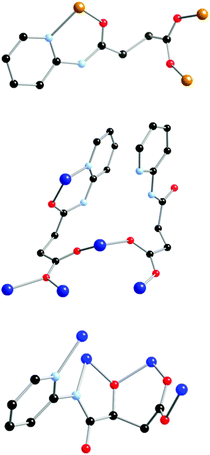 | ||
| Fig. 7 The different binding modes adopted by the ligands in 1 (top), 2 (middle) and 3 (bottom). | ||
The results presented here suggest that H2222L has great potential in the synthesis of new polynuclear complexes containing paramagnetic metal ions and we are currently extending this family of complexes to include different metals from across the transition and lanthanide metal ion series and will report upon these examples in the future.
Acknowledgements
The authors thank the University of Canterbury (College of Science Scholarship to CSH), the Royal Society of New Zealand Marsden Fund (PEK), the MacDiarmid Institute for Advanced Materials and Nanotechnology (PEK) and the Australian Research Council (KSM) for financial support. We also thank Dr Anthea Lees (UC) for helpful discussions.Notes and references
- W. B. Blanton, S. W. Gordon-Wylie, G. R. Clark, K. D. Jordan, J. T. Wood, U. Geiser and T. J. Collins, J. Am. Chem. Soc., 1999, 121, 3551–3552 CrossRef CAS; V. Maurizot, G. Linti and I. Huc, Chem. Commun., 2004, 924–925 RSC; E. J. L. McInnes, S. Piligkos, G. A. Timco and R. E. P. Winpenny, Coord. Chem. Rev., 2005, 249, 2577–2590 CrossRef; B. H. Ye, M. L. Tong and X. M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef; C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef ; Terpyridine, Oligopyridine, and Polypyridine Ligands; R. P. Thummel, in Comprehensive Coordination Chemistry II, ed. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 1, pp. 41–53 CrossRef; H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91–119 CrossRef; H. Hou, Y. Wei, Y. Song, L. Mi, M. Tang, L. Li and Y. Fan, Angew. Chem., Int. Ed., 2005, 44, 6067–6074 CrossRef; M. Y. Huang, C. Y. Yeh, G. H. Lee and S. M. Peng, Dalton Trans., 2006, 5683–5690 RSC; H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef; D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers. Design, Analysis and Application, The Royal Society of Chemistry, 2009 CrossRef CAS; M.-L. Hu, A. Morsali and L. Aboutorabi, Coord. Chem. Rev., 2011, 255, 2821–2859 CrossRef CAS; C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef; E. K. Brechin, C. Boskovic, W. Wernsdorfer, J. Yoo, A. Yamaguchi, E. C. Sañudo, T. R. Concolino, A. L. Rheingold, H. Ishimoto, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 2002, 124, 9710–9711 CrossRef; M. Moragues-Canovás, C. E. Talbot-Eeckelaers, L. Catala, F. Lloret, W. Wernsdorfer, E. K. Brechin and T. Mallah, Inorg. Chem., 2006, 45, 7038–7040 CrossRef; C. Cañada-Vilalta, T. A. O'Brien, M. Pink, E. R. Davidson and G. Christou, Inorg. Chem., 2003, 42, 7819–7829 CrossRef; G. Chaboussant, R. Basler, H.-U. Güdel, S. Ochsenbein, A. Parkin, S. Parsons, G. Rajaraman, A. Sieber, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Dalton Trans., 2004, 2758–2766 RSC.
- B. N. Figgis and R. L. Martin, J. Chem. Soc., 1956, 3837 RSC; A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS; L. F. Jones, E. K. Brechin, D. Collison, M. Helliwell, T. Mallah, S. Piligkos, G. Rajaraman and W. Wernsdorfer, Inorg. Chem., 2003, 42, 6601–6603 CrossRef; E. K. Brechin, Chem. Commun., 2005, 5141–5153 RSC; R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC; G. E. Kostakis, A. M. Ako and A. K. Powell, Chem. Soc. Rev., 2010, 39, 2238–2271 RSC.
- Y. S. Moroz, S. Demeshko, M. Haukka, A. Mokhir, U. Mitra, M. Stocker, P. Müller, F. Meyer and I. O. Fritsky, Inorg. Chem., 2012, 51, 7445–7447 CrossRef CAS; H. Tian, L. Zhao, Y. N. Guo, Y. Guo, J. Tang and Z. Liub, Chem. Commun., 2012, 48, 708–710 RSC; M. U. Anwar, L. K. Thompson, L. N. Dawe, F. Habibb and M. Murugesu, Chem. Commun., 2012, 48, 4576–4578 RSC; P. E. Kruger, B. Moubaraki, G. D. Fallon and K. S. Murray, J. Chem. Soc., Dalton Trans., 2000, 713–718 RSC; P. E. Kruger, B. Moubaraki, K. S. Murray and E. R. T. Tiekink, J. Chem. Soc., Dalton Trans., 1994, 2129–2134 RSC; D. Pelleteret, R. Clérac, C. Mathonière, E. Harté, W. Schmitt and P. E. Kruger, Chem. Commun., 2009, 221–223 RSC; R. J. Archer, C. S. Hawes, G. N. L. Jameson, V. McKee, B. Moubaraki, N. F. Chilton, K. S. Murray, W. Schmitt and P. E. Kruger, Dalton Trans., 2011, 40, 12368–12373 RSC; J. Carranza, H. Grove, J. Sletten, F. Lloret, M. Julve, P. E. Kruger, C. Eller and D. P. Rillema, Eur. J. Inorg. Chem., 2004, 4836–4848 CrossRef; N. R. Kelly, S. Goetz, S. R. Batten and P. E. Kruger, CrystEngComm, 2008, 10, 68–78 RSC.
- A. Nisonoff and D. Pressman, J. Am. Chem. Soc., 1957, 79, 5565–5572 CrossRef CAS; M. Habash and M. O. Taha, Bioorg. Med. Chem., 2011, 19, 4746–4771 CrossRef; J. Wu, L. Tang, K. Chen, L. Yan, F. Li and Y. Wang, J. Colloid Interface Sci., 2007, 307, 280–287 CrossRef; Y. J. Wang, L. Yan, L. M. Tang and J. Yu, Chin. Chem. Lett., 2007, 18, 1009–1012 CrossRef.
- B. S. Garg, V. Kumar and M. J. Reddy, Transition Met. Chem., 1993, 18, 364–368 CrossRef CAS; B. S. Garg, M. J. Reddy and V. Kumar, J. Coord. Chem., 1993, 29, 33–43 CrossRef; B. S. Garg, V. Kumar and M. J. Reddy, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1993, 32, 726–729 Search PubMed; B. S. Garg, V. Kumar and M. J. Reddy, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1996, 35, 598–600 Search PubMed.
- X. Shen, B. Kang, Y. Tong, X. Shi, Y. Li and X. Huang, J. Coord. Chem., 1998, 46, 105–114 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Version 1.171.35.19 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef.
- CrysAlisPro, Agilent Technologies, Version 1.171.35.19; Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm Search PubMed.
- N. V. Kolotova, A. V. Dolzhenko, V. O. Koz'minykh, V. P. Kotegov and A. T. Godina, Pharm. Chem. J., 1999, 33, 635–637 CrossRef CAS.
- L. F. Jones, A. Batsanov, E. K. Brechin, D. Collison, M. Helliwell, T. Mallah, E. J. L. McInnes and S. Piligkos, Angew. Chem., Int. Ed., 2002, 41, 4318–4321 CrossRef CAS; A. Ferguson, J. McGregor, A. Parkin and M. Murrie, Dalton Trans., 2008, 731–733 RSC.
- R. W. Saalfrank, N. Löw, S. Trummer, G. M. Sheldrick, M. Teichert and D. Stalke, Eur. J. Inorg. Chem., 1998, 559–563 CrossRef CAS; R. Ishikawa, M. Nakano, A. Fuyuhiro, T. Takeuchi, S. Kimura, T. Kashiwagi, M. Hagiwara, K. Kindo, S. Kaizaki and S. Kawata, Chem.–Eur. J., 2010, 16, 11139–11144 CrossRef; S. Romain, J. Rich, C. Sens, T. Stoll, J. Benet-Buchholz, A. Llobet, M. Rodriguez, I. Romero, R. Clérac, C. Mathonière, C. Duboc, A. Deronzier and M. N. Collomb, Inorg. Chem., 2011, 50, 8427–8436 CrossRef.
- X. Bao, W. Liu, J. L. Liu, S. Gómez-Coca, E. Ruiz and M. L. Tong, Inorg. Chem., 2013, 52, 1099–1107 CrossRef CAS.
- E. Amadei, E. H. Alilou, F. Eydoux, M. Pierrot, M. Reglier and B. Waegell, J. Chem. Soc., Chem. Commun., 1992, 1782–1784 RSC; S. Itoh, H. Nakao, L. M. Berreau, T. Kondo, M. Komatsu and S. Fukuzumi, J. Am. Chem. Soc., 1998, 120, 2890–2899 CrossRef CAS.
- F. E. Mabbs and D. J. Machin, Magnetism and Transition Metal Complexes, Chapman and Hall, 1973, 196–199 Search PubMed.
- S. Koizumi, M. Nihei, M. Nakano and H. Oshio, Inorg. Chem., 2005, 44, 1208–1210 CrossRef CAS; K. Graham, F. J. Douglas, J. S. Mathieson, S. A. Moggach, J. Schnack and M. Murrie, Dalton Trans., 2011, 40, 12271–12276 RSC.
- C. Benelli, J. Cano, Y. Journaux, R. Sessoli, G. A. Solan and R. E. P. Winpenny, Inorg. Chem., 2001, 40, 188–189 CrossRef CAS; C. Cañada-Vilalta, T. A. O'Brien, M. Pink, E. R. Davidson and G. Christou, Inorg. Chem., 2004, 43, 5505–5521 CrossRef.
- S. Akine, W. Dong and T. Nabeshima, Inorg. Chem., 2006, 45, 4677–4684 CrossRef CAS.
- G. E. Kostakis, S. P. Perlepes, V. A. Blatov, D. M. Proserpio and A. K. Powell, Coord. Chem. Rev., 2012, 256, 1246–1278 CrossRef CAS.
- M. Murrie, Chem. Soc. Rev., 2010, 39, 1986–1995 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS.
- L. Y. Wang, S. Igarashi, Y. Yukawa, Y. Hoshino, O. Roubeau, G. Aromí and R. E. P. Winpenny, Dalton Trans., 2003, 2318–2324 RSC.
- J. A. Real, G. De Munno, R. Chiappetta, M. Julve, F. Lloret, Y. Journaux, J. C. Colin and G. Blondin, Angew. Chem., Int. Ed. Engl., 1994, 33, 1184–1186 CrossRef CAS; L. Zhao, Z. Xu, L. K. Thompson, S. L. Heath, D. O. Miller and M. Ohba, Angew. Chem., Int. Ed., 2000, 39, 3114–3117 CrossRef; P. E. Kruger, G. D. Fallon:, B. Moubaraki, K. J. Berry and K. S. Murray, Inorg. Chem., 1995, 34, 4808–4814 CrossRef; R. P. Doyle, M. Julve, F. Lloret, M. Nieuwenhuyzen and P. E. Kruger, Dalton Trans., 2006, 2081–2088 RSC; X. Zhang, B. Li, J. Tang, J. Tian, G. Huang and J. Zhang, Dalton Trans., 2013, 42, 3308–3317 RSC; L. Merz and W. Haase, J. Chem. Soc., Dalton Trans., 1980, 875–879 RSC; C. J. Calzado, Chem.–Eur. J., 2013, 19, 1254–1261 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum and single crystal X-ray diffraction data for H2222L; hydrogen bonding diagrams and bonding parameters for H2222L; magnetisation isotherms for 1–3. CCDC 939536–939538. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51301g |
| This journal is © The Royal Society of Chemistry 2013 |