 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminescent biscyclometalated arylpyridine iridium(III) complexes with 4,4′-bi-1,2,3-triazolyl ancillary ligands†
Christine E. Welbya, Luke Gilmartina, Ryan R. Marriotta, Adam Zahida, Craig R. Ricea, Elizabeth A. Gibsonb and Paul I. P. Elliott*a
aDepartment of Chemical & Biological Sciences, University of Huddersfield, Huddersfield, HD1 3DH, UK. E-mail: p.i.elliott@hud.ac.uk; Tel: +44 (0)1484 472320
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG2 7RD, UK
First published on 18th July 2013
Abstract
The synthesis, characterization and photophysical investigation of complexes of the form [Ir(R-ppy)2(btz)]PF6 (1 to 3) are reported (btz = 1,1′-dibenzyl-4,4′-bi-1,2,3-triazolyl, R-ppy = 4-(pyrid-2-yl)benzaldehyde (1), 2-phenylpyridine (2) and 2-(2,4-difluorophenyl)pyridine (3)). Complexes 1, 2 and 3 are luminescent and exhibit structured emission bands with vibronic progressions at 532 & 568 nm (ϕ 0.28%), 476 & 508 nm (ϕ 0.82%) and 454 & 483 nm (ϕ 4.3%) respectively. The structuring of these emission bands is indicative of cyclometalated ligand centred emissive states and is further corroborated by the nearly identical emission spectra for 2 and 3 to previously reported analogous complexes with 4-(pyrid-2-yl)-1,2,3-triazole based ancillary ligands. Computational density functional theory calculations on these complexes show that the LUMOs of 2 and 3 are largely btz-centred but with some cyclometalated pyridine π* character. The LUMO of 1 on the other hand is localized primarily on the cyclometalated ligands. Spin population analysis of the lowest lying triplet excited states for these complexes indicate significant spin population over the iridium centres and the aryl and pyridyl moieties in these complexes with virtually no localization of unpaired electrons over the btz ancillary ligands. This is therefore in agreement with the assignment of the emissive state having largely cyclometalated 3LC character and being independent of the ancillary ligand.
Introduction
Cyclometalated iridium(III)1–3 and platinum(II)4,5 complexes have been the subject of a large amount of interest in the literature due to their attractive photophysical properties that results in their potential application in biological imaging, as the basis of luminescent molecular sensors6 and as the phosphors in organic light emitting diode (OLED) and light emitting electrochemical cell (LEEC) devices.7–10,11 Efforts to tune the wavelengths of emission in these complexes have focused on the modification of the cyclometalated and ancillary ligands in order to modulate the energies of the frontier orbitals.12–15 Synthetic routes that provide access to a wide range of ligands and hence allow facile tuning of electronic properties are therefore of great interest.The Huisgen–Sharpless copper catalysed alkyne/azide cycloaddition (CuAAC) to form 1,2,3-triazoles (commonly referred to as ‘click’ chemistry)16,17 has attracted enormous interest over the past decade in organic synthesis, as a linking moiety in novel polymer and dendrimer systems18–22 and in the modification of biological macromolecules.23–25 The past four years or so have since seen an explosion in the use of this versatile reaction in ligand design for metal complexes. Examples have appeared of monodentate N-donor triazole ligands, N-heterocyclic ‘click’ carbene complexes and triazole-containing chelate systems. This area has recently been the subject of a comprehensive review.26
Ligand architectures that have become ubiquitous in transition metal coordination chemistry due to the photophysical properties of their complexes include 2,2′-bipyridyl (bpy) and 2,2′:6′,2′′-terpyridyl (tpy). Several groups have reported analogous ligand systems constructed through CuAAC reactions where pyridyl moieties are replaced with N-donor 1,2,3-triazole rings.27–29 Several examples of ruthenium(II), rhenium(I) and iridium(III) complexes bearing 4-(pyrid-2-yl)-1,2,3-triazole (pytz) based ligands as bpy analogues have appeared and the resultant photophysical properties investigated. Replacement of bpy by pytz in hetero- and homoleptic complexes of the form [Ru(bpy)n(pytz)3–n]2+ was shown to lead to a blue-shifting in absorption and emission maxima but with significant quenching of emission intensity. Indeed, the homoleptic complex [Ru(pytz)3]2+ shows no observable luminescent emission.30,31 However, the complexes of the form [Re(pytz)(CO)3Cl]32 and [Ir(R-ppy)2(pytz)]+ (where R-ppy is a 2-arylpyridine cyclometalated ligand)33–35 are highly luminescent with appreciable emission quantum yields. Further, these heteroleptic iridium(III) complexes were shown to be amenable to the preparation of functional LEEC devices.36,37 Several groups have also investigated the use of aryl triazoles as precursors for the cyclometalated ligand itself.36,38–44
Symmetrical ligands analogous to pytz and based on a 4,4′-bi-1,2,3-triazolyl (btz) framework have received much less attention. Fletcher and co-workers30 along with Monkowius et al.45 prepared and characterized complexes of the form [Ru(btz)3]2+ with the latter group also preparing the rhenium complex [Re(btz)(CO)3Cl]. The homoleptic ruthenium complex displays no luminescent emission in solution at room temperature, nor does the rhenium complex despite its pytz analogue exhibiting significant emissive properties.
We recently reported the preparation and characterization of the heteroleptic complexes [Ru(bpy)2(btz)]2+ and [Ru(bpy)(btz)2]2+.46 Our results revealed that replacement of bpy by btz yields an expected blue-shift in absorption bands and quenched emission. The quenching of luminescent emission in these systems arises through destabilization of the 3MLCT states of these complexes due to the much higher energy LUMO of the btz compared to that of bpy. The reduced separation between 3MLCT and 3MC then results in thermal population of the latter from the former and non-radiative deactivation to the ground state. This 3MLCT state destabilization is found to occur to such an extent for [Ru(btz)3]2+ that DFT calculations suggest that the lowest triplet excited state becomes 3MC in character. Romero et al. have prepared the complex [Ru(tap)2(btz)]2+ (tap = tetraazaphenazene) which is similarly non-emissive.47
Despite several examples of the use of pytz ligands, the btz framework has not been investigated as the ancillary ligand in cationic biscyclometalated iridium(III) complexes (we note, however, a report independently detailing iridium btz complexes by Zysman-Colman and co-workers48 has appeared whilst this manuscript was in the peer-review process). This is perhaps due to the presumption that this would similarly lead to deleterious effects on the photophysical properties of the resultant complexes. However, we were hopeful that complexes of the general structure [Ir(R-ppy)2(btz)]+ would make attractive luminescent complexes for LEEC device applications. The presence of a 5d metal and strongly donating anionic cyclometalate ligands will result in a larger ligand field splitting and higher energy 3MC states than in ruthenium complexes with neutral bpy ligands. Hence these may well become thermally inaccessible from 3MLCT/3LLCT and 3MLCT/3ILCT based excited states in these complexes thereby rendering them emissive. Indeed, we show here that in stark contrast to btz complexes of the ruthenium and rhenium complexes discussed previously, complexes of the form [Ir(R-ppy)2(btz)]PF6 do in fact show appreciable luminescent emission even in aerated solution at room temperature.
Results & discussion
The arylpyridines (R-ppy) 2-phenylpyridine (ppy), 4-(pyrid-2-yl)benzaldehyde (fppy) and 2-(2,4-difluorophenyl)pyridine (dfppy) were used to prepare biscyclometalated chloro-bridged dimers [(R-ppy)2Ir(μ-Cl)2Ir(R-ppy)2] by heating to reflux in aqueous ethoxyethanol solutions. The cationic btz complexes of were then prepared by refluxing these dimeric precursors with 2 equivalents of 1,1′-dibenzyl-4,4′-bi-1,2,3-triazolyl in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform–methanol. After concentration of the reaction mixtures under reduced pressure and treatment with aqueous NH4PF6, the target complexes 1 to 3 were isolated as their hexafluorophosphate salts (Scheme 1).
:1 chloroform–methanol. After concentration of the reaction mixtures under reduced pressure and treatment with aqueous NH4PF6, the target complexes 1 to 3 were isolated as their hexafluorophosphate salts (Scheme 1).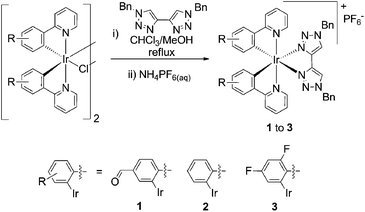 | ||
| Scheme 1 Synthesis of complexes in this study. | ||
1H NMR spectra of all complexes show a single set of resonances for the cyclometalated ligands as well as a single set of resonances for the btz triazole ring and methylene protons indicative of C2 symmetry of the cations. The aldehydic protons of 1 give rise to a singlet at δ 9.71 with a further singlet being observed for the triazole ring protons at δ 8.32, deshielded relative to that of the free ligand (δ 8.17). Three resonances are observed for the metalated aryl ring at δ 7.98, 7.51 and 6.75 along with a further four resonances for the pyridyl rings (δ 8.24, 8.04, 7.84 & 7.25). A pair of geminal doublets with a roofed AB pattern are observed at δ 5.59 and 5.54 dues to the methylene protons of the btz benzyl substituent which are diastereotopic due to the C2 symmetry. The 1H NMR spectrum of 2 displays the expected set of eight resonances for the ppy ligands, typical of C2 symmetric Ir(ppy)2 complexes of this type. The triazole protons give rise to a singlet at δ 8.32 whilst the methylene protons of the benzyl substituents again yield a geminal pair of doublets at δ 5.60 and 5.56. The 1H NMR spectrum of 3 again contains a singlet for the triazole protons (δ 8.33) whilst the benzylic methylene protons now result in a singlet resonance at δ 5.59. A total of six resonances are observed for the dfppy ligand with those of the aryl ring appearing at δ 6.67 and 5.77 whilst the fluorine substituents give rise to a pair of 19F19F-coupled doublets at δ −109.2 and −111.0 in the 19F NMR spectrum.
Crystals of X-ray diffraction quality were obtained for complexes 2 and 3 from acetonitrile solution with slow diffusion vapour diffusion of diethyl ether. Complex 2 crystallises in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and exhibits two crystallographically unique cations in the unit cell. An ORTEP plot of the structure of one of the cations is depicted in Fig. 1 and selected bond distances and angles are provided in Table 1. The cations adopt distorted octahedral geometries with the pyridine rings of the ppy ligands occupying mutually trans coordination sites. Bond lengths and angles for the cyclometalated ligands are unremarkable. Ir–N(btz) bond lengths lie between 2.149 and 2.175 Å for the two cations with btz ligand bite angles of 75.77(7) and 75.27(7)° which are comparable to those of ruthenium complexes of the same ligand.45
and exhibits two crystallographically unique cations in the unit cell. An ORTEP plot of the structure of one of the cations is depicted in Fig. 1 and selected bond distances and angles are provided in Table 1. The cations adopt distorted octahedral geometries with the pyridine rings of the ppy ligands occupying mutually trans coordination sites. Bond lengths and angles for the cyclometalated ligands are unremarkable. Ir–N(btz) bond lengths lie between 2.149 and 2.175 Å for the two cations with btz ligand bite angles of 75.77(7) and 75.27(7)° which are comparable to those of ruthenium complexes of the same ligand.45
![ORTEP plot of the structure of one of two crystallographically unique cations (cation A in Table 1) in the unit cell for [Ir(ppy)2(btz)]PF6 (2) (cation B, hydrogen atoms and counter ions omitted for clarity, ellipsoids at 50% probability).](/image/article/2013/DT/c3dt51284c/c3dt51284c-f1.gif) | ||
| Fig. 1 ORTEP plot of the structure of one of two crystallographically unique cations (cation A in Table 1) in the unit cell for [Ir(ppy)2(btz)]PF6 (2) (cation B, hydrogen atoms and counter ions omitted for clarity, ellipsoids at 50% probability). | ||
| [Ir(ppy)2(btz)]PF6 | |||
|---|---|---|---|
| Cation A | |||
| Ir1–N1 | 2.1753(18) | Ir1–C36 | 2.003(2) |
| Ir1–N4 | 2.149(2) | N1–Ir1–N4 | 75.77(7) |
| Ir1–N7 | 2.051(2) | N7–Ir1–N8 | 175.03(7) |
| Ir1–N8 | 2.0437(19) | N7–Ir1–C25 | 80.63(9) |
| Ir1–C25 | 2.004(2) | N8–Ir1–C36 | 80.21(9) |
| Cation B | |||
| Ir2–N9 | 2.166(2) | Ir2–C76 | 2.007(2) |
| Ir2–N12 | 2.1630(19) | N9–Ir2–N12 | 75.27(7) |
| Ir2–N15 | 2.049(2) | N15–Ir2–N16 | 172.33(8) |
| Ir2–N16 | 2.041(2) | N15–Ir2–C65 | 80.79(9) |
| Ir2–C65 | 2.006(2) | N16–Ir2–C76 | 80.98(10) |
| [Ir(dfppy)2(btz)]PF6 | |||
|---|---|---|---|
| Ir1–C1 | 2.0057(14) | C1–Ir1–N4 | 80.68(6) |
| Ir1–N4 | 2.0443(14) | N4–Ir1–N4 | 170.87(7) |
| Ir1–N5 | 2.1540(12) | N4–Ir1–N5 | 95.96(5) |
| C1–Ir1–C1 | 87.33(8) | N5–Ir1–N5 | 75.77(6) |
An ORTEP plot of the structure of the cation for 3 is shown in Fig. 2. The cation sits on an axis of symmetry such that only half of it (one dfppy ligand and half of the btz ligand) is crystallographically unique. The complex again adopts a distorted octahedral geometry with mutually trans pyridine donors. The complex exhibits Ir–N(btz) bond lengths of 2.1540(12) Å with a btz N–Ir–N bite angle of 75.77(6)°, comparable to those observed for 2.
![ORTEP plot of the structure of the cation [Ir(dfppy)2(btz)]+ (hydrogen atoms and counter ion removed for clarity, ellipsoids at 50% probability).](/image/article/2013/DT/c3dt51284c/c3dt51284c-f2.gif) | ||
| Fig. 2 ORTEP plot of the structure of the cation [Ir(dfppy)2(btz)]+ (hydrogen atoms and counter ion removed for clarity, ellipsoids at 50% probability). | ||
UV-visible absorption spectra were recorded for dichloromethane solutions of complexes 1 to 3 and are shown in Fig. 3. A summary of the photophysical properties can be found in Table 2. Bands appear between 420 and 367 nm which are assigned to 1MLCT-based transitions with intense absorptions below 300 nm assigned to ligand centred π→π* transitions. The MLCT band of 1 appears at 420 nm and is red-shifted relative to that of 2 bearing unsubstituted ppy ligands (385 nm). This may due to the extended π-system associated with the formyl-substituted ligand which would result in stabilisation of the cyclometalated ligand centred unoccupied orbitals (vide infra) and a reduced HOMO–LUMO separation. The electron withdrawing fluorine substituents in 3 would be expected to lead to stabilization of the HOMO relative to that of 2 resulting in blue-shifted absorption and indeed the MLCT band appears at 367 nm.
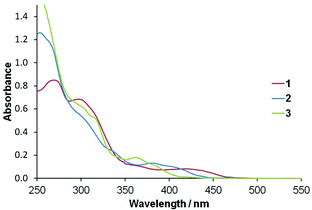 | ||
| Fig. 3 Absorption spectra for complexes 1 to 3 (in dichloromethane). | ||
In contrast to known rhenium and ruthenium btz complexes,30,45–47 complexes 1 to 3 exhibit luminescent emission upon excitation at 400 to 425 nm in aerated dichloromethane solutions at room temperature. Normalized emission spectra are shown in Fig. 4. All complexes display structured emission bands, often indicative of the presence of a large amount of ligand-centred character in the emissive excited states. Complex 3 exhibits bright blue luminescent emission with an intense band at 454 nm with almost equally intense lower energy vibronic progression at 483 nm and a shoulder at approximately 507 nm. The emission band of 3 is blue-shifted relative to that of 2 (476 and 508 nm with a shoulder at approximately 536 nm) mirroring the blue-shift in absorption bands. Introduction of the formyl substituents in 1 results in significantly red-shifted emission which again exhibits vibronic progressions with maxima at 532 and 568 nm.
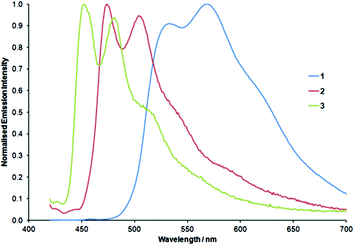 | ||
| Fig. 4 Normalised emission spectra for complexes 1 to 3 (in dichloromethane at room temperature). | ||
These emission wavelengths and the appearance of the spectra for 2 and 3 closely resemble those of related complexes of the form [Ir(dfppy)2(pytz)]+ and [Ir(ppy)2(pytz)]+ previously reported by De Cola and co-workers.33,37 In comparison, the analogous 2,2′-bipyridyl complex [Ir(ppy)2(bpy)]+ exhibits a broad unstructured band with an emission maximum at about 591 nm, significantly red-shifted relative to those of the corresponding pytz and btz complexes. These data confirm that the 3LC emissive excited states in heteroleptic biscyclometalated iridium complexes of the type described here have little or no contribution from, and are therefore largely independent of, the pytz or btz ancillary ligands. The excited state for the bpy complex is, on the other hand, predominantly localized on the ancillary ligand. We have previously shown that the LUMO of the btz ligand is significantly destabilized relative to that of bpy.46 The triazole moiety in the pytz ligand could therefore similarly lead to a large destabilization in the ligand-based LUMO bringing it to a comparable energy to those of vacant orbitals centred on the cyclometalate ligands leading to the observed switching in localization of the emissive state in these complexes.
As mentioned above, previously reported ruthenium and rhenium btz complexes were shown to be weakly or non-emissive in solution at room temperature, however, reasonably intense emission is observed at 77 K for [Ru(bpy)2(btz)]2+ and [Ru(bpy)(btz)2]2+.46 This is explained by the presence of the btz ligand resulting in a higher energy LUMO and hence elevated 3MLCT state from which non-radiative 3MC states can be thermally populated. The intense emission observed for the complexes described in the present study is a likely consequence of the presence of the strongly donating anionic cyclometalated ligands that result in a larger ligand field splitting. The 3MC states in complexes 1 to 3 are therefore elevated such that they become thermally inaccessible for population from the emissive excited states of these complexes.
Luminescent lifetimes were determined for each complex in aerated dichloromethane solutions at room temperature. Complex 2 exhibits a lifetime of 18 ns whereas that of 3 is slightly elongated at 24 ns. In contrast, the lifetime of 1 was measured as 792 ns. Quantum yields were determined from their integrated emission intensities and referenced to [Ru(bpy)3][PF6]2 in aerated acetonitrile (ϕ = 1.8%).49 Quantum yields of 2 and 3 are 0.82 and 0.28% respectively. Consistent with the much longer lifetime the quantum yield of 1 is much larger than those of the other btz complexes at 4.3%.
Cyclic and square-wave voltammetry were performed on solutions of complexes 1 to 3. All three complexes show reversible oxidations at 1.05, 0.85 and 1.19 V respectively (referenced against ferrocene/ferrocenium, E = 0 V). The data for 3 is indicative of the commonly observed stabilization of the HOMO on inclusion of electron withdrawing substituents on the aryl rings. The observed stabilization in 1 compared to 2 is similarly likely due to the presence of the –M formyl groups. Complexes 2 and 3 exhibit irreversible reductions at −1.96 and −1.85 V, however, 1 displays a reversible reduction at much more positive potential centered at −1.38 V. We tentatively assign the reductions for 2 and 3 as arising from btz centred LUMOs and the reduction for 1 as arising from a cyclometalated ligand centered LUMO which is stabilized through the larger π-system provided by the formyl groups. This then accounts for the red shifting of absorption and emission in 1 relative to 2.
DFT studies were undertaken in order to further understand the photophysical and electronic properties of the ground and excited states of complexes 1 to 3. The ground state geometry of each complex was optimized without symmetry constraints in the gas phase at the B3LYP level of theory using the Stuttgart–Dresden relativistic small core potential for iridium and 6-311G* basis sets for all other atoms (optimized xyz coordinates for the complexes may be found in the ESI†). In order to minimize the computational expense required in these calculations the benzyl substituents of the btz ligands were simplified to methyl. In addition, the ground state geometries of the pytz and bpy complexes [Ir(ppy)2(pytz)]+ and [Ir(ppy)2(bpy)]+ were also calculated for comparison.
Energies of the frontier molecular orbitals were determined and plots of the HOMO and LUMO orbitals for each complex are depicted in Fig. 5. Associated energies are provided in Table 3 with a comparative energy level diagram shown in Fig. 6. As expected for complexes of this type, the HOMO of the parent complex 2 (which appears at −7.74 eV) has primarily phenyl π-character in an anti-bonding combination with an iridium d-orbital and is common for those of both 1 and 3 (−8.17 and −8.22 eV respectively). The stabilization of the HOMOs in 1 and 3 relative to that of 2 therefore mirrors the measured oxidation potentials for these complexes. The closely-spaced LUMO (−4.01 eV) and LUMO + 1 (−3.97 eV) orbitals of 2 are primarily centered on the ancillary btz ligand but have some additional pyridyl π* contribution. A similar involvement of the pyridyl moiety in the LUMO (−4.24 eV) and LUMO + 1 (−4.19 eV) of 3 is also observed. For both of these complexes, LUMO + 2 and LUMO + 3 appear solely btz-centered. The resultant HOMO–LUMO gap for 3 of 3.98 eV is larger than that for 2 (3.74 eV) in agreement with expectations and the observed blue-shift in experimental absorption and emission spectra.
![Plots of HOMO and LUMO orbitals for the ground states of complexes 1 to 3 and [Ir(ppy)2(L)]+ (L = pytz, bpy).](/image/article/2013/DT/c3dt51284c/c3dt51284c-f5.gif) | ||
| Fig. 5 Plots of HOMO and LUMO orbitals for the ground states of complexes 1 to 3 and [Ir(ppy)2(L)]+ (L = pytz, bpy). | ||
![Energy level diagram for the calculated molecular orbitals of the complexes 1 to 3 and [Ir(ppy)2(L)]+ (L = pytz, bpy).](/image/article/2013/DT/c3dt51284c/c3dt51284c-f6.gif) | ||
| Fig. 6 Energy level diagram for the calculated molecular orbitals of the complexes 1 to 3 and [Ir(ppy)2(L)]+ (L = pytz, bpy). | ||
| Complex | HOMO/eV | LUMO/eV | HOMO–LUMO gap/eV |
|---|---|---|---|
| 1 | −8.17 | −4.59 | 3.57 |
| 2 | −7.74 | −4.01 | 3.74 |
| 3 | −8.22 | −4.24 | 3.98 |
| [Ir(ppy)2(pytz)]+ | −7.88 | −4.77 | 3.01 |
| [Ir(ppy)2(bpy)]+ | −8.03 | −5.24 | 2.78 |
The LUMO of the analogous bpy complex [Ir(ppy)2(bpy)]+, calculated using the same exchange correlation functional and basis sets, is primarily bpy-based and resides at −5.24 eV, some 1.23 and 1.00 eV lower in energy than those of 2 and 3 respectively. This results in a much smaller HOMO–LUMO gap of 2.78 eV consistent with the largely red-shifted emission band for this complex relative to that of 2. Here, LUMO + 1 (−4.41 eV) is also localized on the ancillary bpy ligand without involvement of the cyclometalate ligands with the first unoccupied orbital with ppy pyridine π* character being LUMO + 2 (−4.22 eV).
In the case of the pytz complex [Ir(ppy)2(pytz)]+ the HOMO is similarly localized on the metal and the cyclometalated phenyl rings with a greater contribution from the ppy ligand trans to the pyridyl donor of the ancillary ligand. The LUMO and LUMO + 1 orbitals are both pytz π* in character and are localized to a greater extent on the pyridyl ring. Here, the LUMO lies between those of 2 and [Ir(ppy)2(bpy)]+, some 0.47 eV above that of the bpy analogue. LUMO + 2 (−4.11 eV) and LUMO + 3 (−4.03 eV) are the lowest ppy-based pyridyl π* orbitals.
This change in the localization of the unoccupied frontier orbitals in complexes 2 and 3, in which there is some localization on the cyclometalated ligands, when compared to those of [Ir(ppy)2(bpy)]+ is due to the much higher energy of the LUMO of the btz ligand compared to that of bpy. Indeed, in separate calculations on the free btz and bpy ligands, the LUMO of btz is some 1.02 eV higher in energy relative to that of bpy.46 Hence, the btz LUMO lies close in energy to orbitals of the cyclometalated ligands allowing mixed R-ppy/btz character in the unoccupied frontier orbitals of these complexes.
In contrast to 2 and 3, the LUMO and LUMO + 1 orbitals of 1 are primarily centered on the pyridyl rings and formyl groups of the cyclometalated ligand and have no significant contribution from the btz ligand. Consistent with experimentally observed red-shifted spectra, the HOMO–LUMO gap for 1 is smaller than that of 2 at 3.57 eV. Here, LUMO + 2, some 0.32 eV above the LUMO, is the first btz-centered unoccupied orbital.
Time-dependent DFT (TDDFT) calculations were performed at the ground state geometries each complex to derive vertical excitation energies and hence simulated optical absorption spectra. TDDFT derived spectra for 1 to 3 (with experimental spectra overlaid) are presented in Fig. 7. From a simple visual analysis of the positions of the major transitions depicted in Fig. 3 it can be seen that the energies of the calculated transitions are in good agreement with the experimentally recorded spectra. Consistent with experimental data these major transitions are also observed to blue-shift from complex 1 to 3. Indeed, the S1 states appear at 426 nm (f = 0.054), 409 nm (f = 0.030) and 381 nm (f = 0.014) for 1, 2 and 3 respectively, are relatively intense and are primarily HOMO → LUMO in character. Hence, the nature of these transitions is R-ppy-based MLCT/LC for 1 and MLCT/LLCT to btz for 2 and 3.
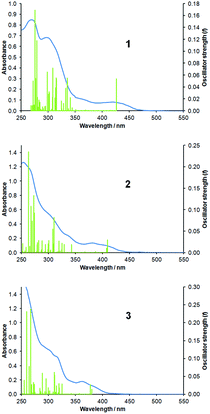 | ||
| Fig. 7 Overlaid experimental absorbance and calculated TDDFT spectra for complexes 1 to 3. | ||
In order to probe the nature of the emissive states of these complexes the lowest triplet excited states of 1 to 3 were optimized, along with those of [Ir(ppy)2(pytz)]+ and [Ir(ppy)2(bpy)]+, starting from their S0 geometries using the constraint of the spin multiplicity of 3. Table 4 collates the calculated energies of the T1 states for complexes 1 to 3 and those of [Ir(ppy)2(pytz)]+ and [Ir(ppy)2(bpy)]+ quote relative to their respective S0 ground state energies. Consistent with the experimentally observed spectroscopic data which shows a blue-shift in emission maxima, the T1 state is destabilized on going from complex 1 to 3. The T1 state of [Ir(ppy)2(pytz)]+ (2.59 eV) is noted to have almost the same energy as that of 2 (2.60 eV) consistent with emission occurring from the same ppy-based 3LC state. The T1 state of [Ir(ppy)2(bpy)]+, however, is observed to be at significantly lower energy (2.25 eV).
| Complex | T1/eV | S0*/eV | T1 − S0*/eV | λemcalc/nm |
|---|---|---|---|---|
| 1 | 2.35 | 0.28 | 2.07 | 600 |
| 2 | 2.60 | 0.28 | 2.32 | 534 |
| 3 | 2.75 | 0.30 | 2.45 | 507 |
| [Ir(ppy)2(pytz)]+ | 2.59 | 0.26 | 2.33 | 531 |
| [Ir(ppy)2(bpy)]+ | 2.25 | 0.30 | 1.95 | 634 |
Estimated Frank–Condon emission energies were calculated for each complex through the Δ-SCF method; having optimized the geometries of the T1 states for the complexes, the energies of the singlet ground states at these T1 geometries were calculated in single point calculations. The difference in energy between the optimized T1 states and these non-equilibrium S0* states at the same geometries are therefore the calculated emission energies which are then used to derive the calculated emission maximum. As can be seen from Table 4 the calculated emission wavelengths are over estimated by approximately 25–30 nm with respect to the lower energy vibronic progressions observed in the emission spectra of complexes 1 to 3 and that of [Ir(ppy)2(pytz)]+. These calculations are however conducted in the gas phase and hence in the absence of solvent interaction which will have an influence on the absolute energies of the states under investigation. Nevertheless, the calculated data clearly reproduce the observed experimental trends with a progressive blue shift in emission on going from 1 (600 nm) to 3 (507 nm) and near identical emission wavelengths for 2 and [Ir(ppy)2(pytz)]+ (534 and 531 nm respectively) (Table 4).
Mulliken population analyses were carried on to determine the localization of spin density for these T1 states and summed atomic spin populations for the iridium atom and the aryl, pyridyl and btz moieties of 1 to 3 are listed in Table 5. The data clearly show significant spin populations on the iridium atom and the cyclometalated ligands but which do not extend to the btz ligands. Hence, the emissive triplet states in these complexes are predicted to be largely 3LC in character with little or no involvement of the ancillary btz ligand. Hence, tuning of the emissive properties of these complexes is entirely dependent on the nature of the substituents of the cyclometalated ligands. Similarly, and in agreement with the near identical emission characteristics to those of complex 2, analysis of the spin population in [Ir(ppy)2(pytz)]+ shows that there is no significant population of the unpaired electrons on the pytz ligand and that again the T1 state of this complex is also 3LC in character. On the other hand, the spin populations for [Ir(ppy)2(bpy)]+ reveal that the excited electron is localized on the bpy ligand confirming the 3MLCT/3LLCT character of the emissive state for this complex.
| Complex | Ir | Aryl | Pyridyl | Ancillary |
|---|---|---|---|---|
| 1 | 0.22 | 1.24 | 0.54 | 0.007 |
| 2 | 0.24 | 0.97 | 0.79 | 0.008 |
| 3 | 0.14 | 0.91 | 0.94 | 0.005 |
| [Ir(ppy)2(pytz)]+ | 0.27 | 0.93 | 0.93 | 0.054 |
| [Ir(ppy)2(bpy)]+ | 0.53 | 0.41 | 0.02 | 1.02 |
The difference in the frontier orbital localization for 1 compared to those of 2 and 3 along with the 3LC character in the emissive states of these btz complexes may account for the greater quantum yield of emission observed for 1 and the greatly elongated luminescent lifetime. Since both the HOMO and LUMO in this complex are largely centered in the cyclometalate ligands, redistribution of the unpaired electron density occurs in 1 to a much lesser extent during the process of the excitation of an electron to the LUMO, subsequent inter-system crossing and relaxation of the resultant T1 state than would be the case for 2 and 3. This may therefore lead to the greater rate of non-radiative decay rate through interactions with ligand vibrational oscillators for the latter complexes relative to their radiative decay, kr, when compared to 1.
The computational data presented here is therefore in agreement with the 3LC centered assignment of the emissive states for the pytz complexes [Ir(ppy)2(pytz)]+ and [Ir(dfppy)2(pytz)]+33,37 and complexes 2 and 3 based on the near identical emission spectra and the observed vibronic structure therein. Hence, the tuning of the photophysical properties of these btz complexes and their pytz analogues are entirely dependent on the nature of cyclometalated ligands. These complexes provide the basis of further development of LEEC phosphors.
Conclusions
We have reported the synthesis, characterization and the photophysical and theoretical study of iridium(III) cyclometalated complexes with 4,4′-bi-1,2,3-triazolyl ancillary ligands. We have shown that these are luminescent with emission wavelengths are tunable via through variation of the aryl substituents of the cyclometalated ligands but are independent of the ancillary btz ligand. Complexes of this type are promising candidates for further development as phosphors in light emitting devices and in other applications such as biological imaging.Experimental section
General methods
The btz ligand,46 the iridium dimers [Ir(fppy)2Cl]2,50 [Ir(ppy)2Cl]251 and [Ir(dfppy)2Cl]252 and the complex [Ir(ppy)2(bpy)][PF6]53 were all prepared by previously reported procedures. NMR spectra were recorded on Bruker 500 Avance and 400 AVIII spectrometers and mass spectrometry data were obtained on a Bruker Micro-Q-TOF instrument. UV-visible absorption data were recorded on a Varian Cary 4000 UV-visible spectrophotometer and emission spectra were recorded on a Jobin-Yvon Fluoromax instrument. Excited state lifetimes were obtained using an Edinburgh Instrument Mini-tau spectrometer. Quantum yields, ϕ, were determined using the equationwhere A is the absorbance at the wavelength of excitation, I is the integrated emission intensity, η is the refractive index of the solvent and the subscripts u and r refer to the unknown and reference ([Ru(bpy)3][PF6]2 in aerated acetonitrile, ϕ = 0.01849) samples respectively. The radiative and non-radiative decay constants were derived by the equations kr = ϕ/τ and knr = (1 − ϕ)/τ respectively.
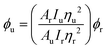
For electrochemical measurements complexes were 1 mM in 0.1 M solution of [Bu4N][PF6] in acetonitrile. The reference electrode was a silver wire in contact with 0.3 M KCl in ethanol. The working electrode was glassy carbon whilst the counter electrode was a platinum foil. The solutions were purged with nitrogen and potentials are calibrated using FeCp2+/0 in 0.1 M solution of [Bu4N][PF6] in acetonitrile as an external standard. The potentiostat used was an Autolab PGSTAT.
:1 chloroform–methanol (8 cm3) and degassed with N2. The mixture was then heated to reflux for 5 hours after which the solution was concentrated under reduced pressure. Diethyl ether was added (10 cm3) and the volume of the mixture reduced by rotary evaporation and the resulting yellow precipitate was filtered and recrystallized from acetonitrile–diethyl ether. Yield 32.2 mg (44.3%).1H NMR (500 MHz, CD3CN) δ 9.71 (s, 2H, CHO); 8.32 (s, 2H, tz); 8.24 (d, JHH 8.2 Hz, 2H, py-H3); 8.04 (t, JHH = 7.7 Hz, py-H4); 7.98 (d, JHH = 8.0 Hz, Ar-H2); 7.84 (d, JHH = 5.6 Hz, py-H6); 7.51 (dd, JHH = 8.0 & 1.4 Hz, Ar-H3); 7.42–7.36 (m, 6H, Ph); 7.25 (t, JHH = 7.1 Hz, py-H5); 7.22–7.18 (m, 4H, Ph); 6.75 (d, JHH 1.1 Hz, Ar-H5); 5.59 (d, JHH = 15.5 Hz, 2H, CH2); 5.54 (d, JHH = 15.5 Hz, 2H, CH2). 13C NMR (CD3CN) δ 192.7; 165.9; 150.6; 150.1; 146.1; 140.2; 139.2; 136.4; 133.6; 131.6; 129.1; 129.0; 128.2; 124.9; 124.7; 124.7, 123.7; 121.3; 55.4. MS (ESI) m/z 873.2 ([IrC42O2N8H32]+). HRMS (ESI) calcd 873.22800, found 873.22842.
:1 chloroform–methanol (20 cm3) and degassed with N2. The mixture was then heated to reflux for 2.5 hours after which the solvent was removed under reduced pressure. The residue was redissolved in dichloromethane (5 cm3) and excess ammonium hexafluorophosphate was added in methanol (10 cm3). The volume of the mixture reduced by rotary evaporation and the resulting yellow crystalline precipitate was filtered. Yield 50 mg (35%).1H NMR (500 MHz, CD3CN) δ 8.32 (s, 2H, tz); 8.07 (d, JHH = 8.2 Hz, 2H, py-H3); 7.91 (td, JHH = 7.6 & 1.6 Hz, 2H, py-H4); 7.77–7.72 (m, 4H, py-H6 & Ar-H2); 7.42–7.37 (m, 6H, Ph); 7.21–7.19 (m, 4H, Ph); 7.10 (ddd, JHH = 7.3, 5.8 & 1.4 Hz, 2H, py-H5); 6.99 (td, JHH = 7.5 & 1.1 Hz, 2H, Ar-H3); 6.85 (td, JHH = 7.5 & 1.3 Hz, 2H, Ar-H4); 6.27 (dd, JHH = 7.7 & 1.1 Hz, 2H, Ar-H5); 5.60 (d, JHH = 15.2 Hz, 2H, CH2); 5.56 (d, JHH = 15.2 Hz, 2H, CH2). 13C NMR (CD3CN) δ 167.6; 149.5; 146.3; 144.5; 140.2; 138.5; 133.8; 131.7; 129.6; 129.1; 129.0; 128.1; 124.3; 123.5; 123.2; 122.3; 119.6; 55.3. MS (ESI) m/z 817.2 ([IrC40H32N8]+). HRMS (ESI) calcd 817.237368, found 817.236011.
:1 chloroform–methanol (20 cm3) and the mixture degassed. The solution was then heated to reflux at 75 °C for 3.5 hours. NH4PF6 (124 mg) was then added and the reaction heated to reflux for a further 2.5 hours. On cooling, the bright yellow precipitate was filtered and washed with small portions of diethyl ether (10 cm3). Yield 303 mg (87.1%.)1H NMR (500 MHz, CD3CN) δ 8.33 (d, JHH = 8.3 Hz, 2H, py-H3); 8.33 (s, 2H, tz); 7.96 (t, JHH = 8.0 Hz, 2H, py-H4); 7.75 (d, JHH = 5.9 Hz, py-H6); 7.44–7.36 (m, 6H, Ph); 7.24–7.18 (m, 4H, Ph); 7.15 (ddd, JHH = 1.3, 5.9 & 7.4 Hz, 2H, py-H5); 6.67 (ddd, JHF = 12.6 & 9.4 Hz, JHH = 2.5 Hz, 2H, C6H2F2-H3); 5.77 (dd, JHF = 8.8 Hz, JHH = 2.4 Hz, 2H, C6H2F2-H5); 5.59 (s, 4H, CH2). 13C NMR (CD3CN) δ 56.0 (s, CH2), 99.1 (t, 2JCF = 27.3 Hz, C3), 114.5 (dd, 2JCF = 17.8 Hz, 4JCF = 3.0 Hz, C5), 123.9 (s, Cd), 124.1 (s, Cb), 124.4 (s, CHN3), 128.8 (s, Ph), 129.0 (dd, 2JCF = 5.7 Hz, 4JCF = 2.6 Hz, C1), 129.6 (s, Ph), 129.7 (s, Ph), 134.1 (s, Ci), 140.1 (s, Cc), 140.6 (s, CN3), 150.5 (s, Ca), 151.2 (d, 3JCF = 7.0 Hz, C6), 161.1 (dd, 1JCF = 206.2 Hz, 3JCF = 12.7 Hz, C2), 163.5 (dd, 1JCF = 202.2 Hz, 3JCF = 13.1 Hz, C4), 164.3 (dd, 3JCF = 7.0 Hz, 5JCF = 1.0 Hz, Ce). 19F{1H} NMR (CD3CN) −72.8 (d, JPF = 706 Hz, PF6); −109.1 (d, JFF = 10.2 Hz, C6H2F2-F4); −111.0 (d, JFF = 10.1 Hz, C6H2F2-F2). MS (ESI) m/z 889.2 ([IrC40F4N28H28]+); HRMS (ESI) calcd 889.199681, found 889.200748.
X-ray crystallography
Single crystal X-ray diffraction data were collected on a Bruker Apex Duo diffractometer equipped with a graphite monochromated Mo(Kα) radiation source (0.071073 nm) and a cold stream of N2 gas. Summarised crystal and refinement data are presented in Table 6. Preliminary scans were employed to assess crystal quality, lattice symmetry, ideal exposure time etc. prior to collecting a full sphere of diffraction intensity data using SMART operating software.54 Intensities were then integrated from several series of exposures, merged and corrected for Lorentz and polarisation effects using SAINT software.55 Solutions were generated by conventional heavy atom Patterson or direct methods and refined by full-matrix non-linear least squares on all F2 data, using SHELXS-97 and SHELXL software respectively (as implemented in the SHELXTL suite of programs).56 Empirical absorption corrections were applied based on multiple and symmetry-equivalent measurements using SADABS.57 All structures were refined until convergence (max shift/esd < 0.01) and in each case, the final Fourier difference map showed no chemically sensible features.| 2 | 3 | |
|---|---|---|
| Formula | C40H32F6IrN8P | C40H28F10IrN8P |
| Mr/g mol−1 | 961.91 | 1033.87 |
| Temperature/K | 150 | 150 |
| Space group | P | C2/c |
| a/Å | 13.0144(7) | 16.7186(6) |
| b/Å | 13.3032(8) | 26.4926(9) |
| c/Å | 24.2891(15) | 9.3340(3) |
| α/° | 78.746(1) | 90 |
| β/° | 87.429(1) | 109.714(1) |
| γ/° | 64.527(1) | 90 |
| V/Å3 | 3719.8(4) | 3891.9(2) |
| Dc/g cm−3 | 1.718 | 1.764 |
| Z | 4 | 4 |
| μMo/mm−1 | 3.705 | 3.562 |
| ‘T’min, max | 0.357, 0.525 | 0.580, 0.720 |
| 2θmax | 61.02 | 71.26 |
| Nref | 18570 | 8008 |
| R1 | 0.0250 | 0.0222 |
| wR2 | 0.0529 | 0.0524 |
| S | 1.007 | 1.040 |
Computational details
DFT calculations were carried out using the NWChem 6.0 and 6.1 software package.58 Calculations were carried out using the B3LYP hybrid functional (20% Hartree–Fock),59 Stuttgart relativistic small core ECP for iridium60 and 6-311G* basis sets61 for all other atoms. Molecular structures and molecular orbitals were visualized using the ccp1 graphical user interface. The ground state geometries of all complexes were first optimized and molecular orbital energies determined. TD-DFT calculations were then used at the ground state geometries to derive vertical excitation energies and hence simulated absorption spectra. Δ-SCF calculations were then carried out to obtain estimated luminescent emission energies; first, the geometries of the lowest lying triplet states were optimized using the constraint of a spin multiplicity of 3. The singlet ground state energies at these triplet excited state geometries were then determined in single point calculations. The difference in energy between the triplet excited states and the singlet states at the same geometry are therefore the theoretical emission energies.Acknowledgements
The authors thank the Leverhulme Trust for funding this research. As members of the HPC UK Materials Chemistry Consortium we also thank the EPRSC (grant no. EP/F067496) and the UK's national supercomputing service HECToR for computational resources used in this work. We also acknowledge the University of Huddersfield Centre for High Performance Computing. We also thank Dr Marcus Chadha, Innovative Physical Organic Solutions, University of Huddersfield, for mass spectrometry assistance.Notes and references
- F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464–3471 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS.
- L. Chassot, E. Muller and A. Vonzelewsky, Inorg. Chem., 1984, 23, 4249–4253 CrossRef CAS.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265–287 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- A. Y.-Y. Tam, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, Chem. Commun., 2011, 47, 3383–3385 RSC.
- P. Coppo, E. A. Plummer and L. De Cola, Chem. Commun., 2004, 1774–1775 RSC.
- D. L. Davies, M. P. Lowe, K. S. Ryder, K. Singh and S. Singh, Dalton Trans., 2011, 40, 1028–1030 RSC.
- W. Mroz, C. Botta, U. Giovanella, E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore and J. A. G. Williams, J. Mater. Chem., 2011, 21, 8653–8661 RSC.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2569 CrossRef.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC.
- M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. Van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13 CrossRef CAS.
- J. D. Crowley and D. A. McMorran, Top. Heterocycl. Chem., 2012, 28, 31 CrossRef CAS.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- T. Romero, R. A. Orenes, A. Espinosa, A. Tarraga and P. Molina, Inorg. Chem., 2011, 50, 8214 CrossRef CAS.
- B. Schulze, C. Friebe, M. D. Hager, A. Winter, R. Hoogenboom, H. Goerls and U. S. Schubert, Dalton Trans., 2009, 787–794 RSC.
- J. T. Fletcher, B. J. Bumgarner, N. D. Engels and D. A. Skoglund, Organometallics, 2008, 27, 5430–5433 CrossRef CAS.
- B. Happ, C. Friebe, A. Winter, M. D. Hager, R. Hoogenboom and U. S. Schubert, Chem.–Asian. J., 2009, 4, 154–163 CrossRef CAS.
- M. Obata, A. Kitamura, A. Mori, C. Kameyama, J. A. Czaplewska, R. Tanaka, I. Kinoshita, T. Kusumoto, H. Hashimoto, M. Harada, Y. Mikata, T. Funabiki and S. Yano, Dalton Trans., 2008, 3292–3300 RSC.
- M. Felici, P. Contreras-Carballada, Y. Vida, J. M. M. Smits, R. J. M. Nolte, L. De Cola, R. M. Williams and M. C. Feiters, Chem.–Eur. J., 2009, 15, 13124–13134 CrossRef CAS.
- M. Juriceck, M. Felici, P. Contreras-Carballada, J. Lauko, S. R. Bou, P. H. J. Kouwer, A. M. Brouwer and A. E. Rowan, J. Mater. Chem., 2011, 21, 2104 RSC.
- S. Zanarini, M. Felici, G. Valenti, M. Maracaccio, L. Prodi, S. Bonacchi, P. Contreras-Carballada, R. M. Williams, M. C. Feiters, R. J. M. Nolte, L. De Cola and F. Paolucci, Chem.–Eur. J., 2011, 17, 4640 CrossRef CAS.
- J. M. Fernandez-Hernandez, C.-H. Yang, J. I. Beltran, V. Lemaur, F. Polo, R. Frohlich, J. Cornil and L. De Cola, J. Am. Chem. Soc., 2011, 133, 10543 CrossRef CAS.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfet, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. Gonzalez and U. S. Schubert, Organometallics, 2009, 28, 5478–5488 CrossRef CAS.
- M. Felici, P. Contreras-Carballada, J. M. M. Smits, R. J. M. Nolte, R. M. Williams, L. De Cola and M. C. Feiters, Molecules, 2010, 15, 2039–2059 CrossRef CAS.
- J. M. Fernandez-Hernandez, J. I. Beltran, V. Lemaur, M.-D. Galvez-Lopez, C.-H. Chien, F. Polo, E. Orselli, R. Frohlich, J. Cornil and L. De Cola, Inorg. Chem., 2013, 52, 1812 CrossRef CAS.
- K. Hasan and E. Zysman-Colman, Inorg. Chem., 2012, 51, 12560 CrossRef CAS.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514 CrossRef CAS.
- K. N. Swanick, S. Ladouceur, E. Zysman-Colman and Z. Ding, Chem. Commun., 2012, 48, 3179–3181 RSC.
- K. N. Swanick, S. Ladouceur, E. Zyzman-Colman and Z. Ding, Angew. Chem., Int. Ed., 2012, 51, 11079–11082 CrossRef CAS.
- U. Monkowius, S. Ritter, B. Konig, M. Zabel and H. Yersin, Eur. J. Inorg. Chem., 2007, 4597–4606 CrossRef CAS.
- C. E. Welby, S. Grkinic, A. Zahid, B. S. Uppal, E. A. Gibson, C. R. Rice and P. I. P. Elliott, Dalton Trans., 2012, 41, 7637–7646 RSC.
- A. Mattiuzzi, I. Jabin, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2011, 40, 7395–7402 RSC.
- L. Donato, P. Abel and E. Zysman-Colman, Dalton Trans., 2013, 42, 8402–8412 RSC.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- S. Bettington, A. L. Thompson, A. Beeby and A. E. Goeta, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, M827–M829 CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
- E. Baranoff, B. F. E. Curchod, F. Monti, F. Steimer, G. Accorsi, I. Tavernelli, U. Rothlisberger, R. Scopelliti, M. Graetzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 799–811 CrossRef CAS.
- Y. Ohsawa, S. Sprouse, K. A. King, M. K. Dearmond, K. W. Hanck and R. J. Watts, J. Phys. Chem., 1987, 91, 1047–1054 CrossRef CAS.
- SMART Diffractometer Control Software, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- SAINT Integration Software, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1994 Search PubMed.
- SHELXTL Program System, Vers 5.1, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: A Program for Absorption Correction with Siemens SMART System, University of Gottingen, Germany, 1996 Search PubMed.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary DFT data and CIF files. CCDC 938365 and 938366. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51284c |
| This journal is © The Royal Society of Chemistry 2013 |