 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomerisation and controlled condensation in an aqueous medium of allyl alcohol catalysed by new water-soluble rhodium complexes with 1,3,5-triaza-7-phosphaadamantane (PTA)†
Piotr
Smoleński
*ab,
Marina V.
Kirillova
b,
M. Fátima C.
Guedes da Silva
bc and
Armando J. L.
Pombeiro
*b
aFaculty of Chemistry, University of Wrocław, 50-383, ul. F. Joliot-Curie 14, Wrocław, Poland. E-mail: piotr.smolenski@chem.uni.wroc.pl
bCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, Technical University of Lisbon, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: pombeiro@ist.utl.pt
cUniversidade Lusófona de Humanidades e Tecnologias, ULHT Lisbon, Av. do Campo Grande, 376, 1749-024, Lisbon, Portugal
First published on 29th May 2013
Abstract
New aqua-soluble rhodium(I) [Rh(CO)(PTA)4]Cl (1) (PTA = 1,3,5-triaza-7-phosphaadamantane) and rhodium(III) [RhCl2(PTA)4]Cl (2) complexes have been synthesized via the reaction of [{Rh(CO)2(μ-Cl)}2] or RhCl3·3H2O, respectively, with stoichiometric amounts of PTA in ethanol. Compound 1 is also obtained upon reduction of 2 in an H2/CO atmosphere. They have been characterized by IR, 1H and 31P{H} NMR spectroscopies, elemental and single crystal X-ray diffraction analyses. While compound 1 shows distorted square-pyramid geometry (τ5 = 0.09) with a P3C-type basal plane, compound 2 is octahedral with the chloro ligands in the cis position. The hydride rhodium(I) complex [RhH(PTA)4] (3) is formed upon the addition of NaBH4 to an aqueous solution of 1 or 2. Compounds 1–3 (in the case of 2 upon reduction by H2) act as homogeneous catalysts, or catalyst precursors, in the isomerisation and condensation of allyl alcohol at room temperature and in an aqueous medium. The product selectivity is easily controlled by changing the concentration of the base in the reaction mixture, thus resulting in the exclusive formation of either 3-hydroxy-2-methylpentanal (HP) or 2-methyl-2-pentenal (MP) in quantitative yields.
Introduction
The application of the aminophosphine 1,3,5-triaza-7-phosphaadamantane (PTA)1 as a water-soluble ligand for the synthesis of different metal complexes is a growing research area, since many coordination compounds of PTA show interesting catalytic,1,2 biological1,3 or photoluminescent properties.1,4 In particular, Rh and Ru complexes with PTA and its N-alkylated derivatives have been extensively studied, finding their wide application as efficient and versatile catalysts for the hydrogenation, hydroformylation and isomerisation of unsaturated organic substrates in aqueous media.5 Besides, in the case of hydrogenation reactions, it was shown that rhodium complexes bearing more than three PTA ligands in the coordination sphere are much less active than those having only two PTA moieties.6 For example, complex [RhCl(CO)(PTA)2]7 is a catalyst in water gas shift reaction (WGSR), while [RhCl(PTA-H)(PTA)2] (PTA-H = N-protonated PTA)6 catalyzes the hydrogenation of unsaturated aldehydes. Interestingly, in the presence of an excess of PTA, the latter complex is completely inactive due to the formation of the coordinatively saturated Rh-compound [RhCl(PTA-H)3(PTA)]Cl3.6 On the other hand, rhodium(I) complexes of the type [RhH(PTA-R)4]I4 (R = Me, Et)8 that bear the N-alkylated PTA cationic derivatives (PTA-R)+ instead of neutral PTA are efficient catalysts for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond reduction.
C bond reduction.
Rhodium(I) acetylacetonato complexes with PTA, P(CH2CH2CN)3 and P(m-C6H4SO3Na)3 ligands have also been studied in the hydrogenation of allyl alcohol to n-propanol (Scheme 1). Isomerisation to propanal is also observed, although as a minor product. In the presence of these catalysts, the alcohol undergoes isomerisation to enol before its hydrogenation to n-propanol.9
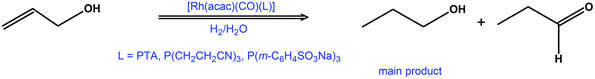 | ||
| Scheme 1 Hydrogenation and isomerisation of allyl alcohol. | ||
Since the rhodium complexes with a higher number of coordinated PTA are less active in the CC bond reduction reactions, it would be interesting to examine them towards the secondary reaction of alcohol isomerisation to aldehyde. A catalytic system generated in situ from [Rh(COD)(MeCN)2](BF4) and PTA was previously reported10a,b to catalyse the isomerisation of allylic alcohols into carbonyl compounds in aqueous media, but no aldol condensation products were observed, even in the presence of NaOH. Aldol condensation, which could follow the isomerisation, is one of the important reactions of aldehydes in synthetic organic chemistry, especially because of C–C bond formation.10c Products of aldol condensation find wide applications in the pharmaceutical field, as plasticizers, detergents, fragrances and cosmetics.11 One of them, 2-methylpentenal, is an industrially important chemical used as an intermediate for the synthesis of various pharmacologically active compounds.12 Commercially, condensation reactions are carried out in the presence of strong bases or acids (sodium hydroxide or sulphuric acid) and require high temperatures, thus displaying many disadvantages such as corrosion and environmental problems, and complex workup.11,12
Therefore, the search for catalytic systems without some of those disadvantages deserves to be explored and water soluble rhodium complexes are promising candidates.
Hence, we report herein the synthesis, structural analysis and catalytic properties of new water-soluble and water-stable rhodium complexes bearing the aminophosphine PTA, [Rh(CO)(PTA)4]Cl (1) and [RhCl2(PTA)4]Cl (2). Besides, formation of the hydride rhodium(I) complex [RhH(PTA)4] (3) upon addition of NaBH4 to an aqueous solution of 1 or 2 has been monitored by 31P{1H} NMR. The compounds have been characterized by IR, 1H and 31P{1H} spectroscopies, elemental and single crystal X-ray diffraction analyses (for 1 and 2). Complexes 1–3 were found to efficiently catalyze condensation of allyl alcohol under ambient conditions in water as a solvent.
Experimental
All syntheses were performed under an inert atmosphere of dry oxygen-free dinitrogen, using standard Schlenk techniques. Solvents were dried and distilled prior to use. [{Rh(CO)2(μ-Cl)}2], RhCl3·3H2O, NaBH4, allyl alcohol (Aldrich) and standards for GC (Aldrich and POCh) were used as received, while 1,3,5-triaza-7-phosphaadamantane (PTA) was synthesized in accordance with modified literature methods.13 Elemental analyses were performed on a Vario EL III apparatus. Infrared spectra (4000–400 cm−1) were recorded with a Bruker IFS 1113v instrument in KBr pellets. NMR spectra were measured using a Bruker 300 AMX spectrometer. 1H and 13C chemical shifts (δ) are given in ppm relative to Si(Me)4, and δ(31P) relative to 85% H3PO4. Coupling constants are in Hz; abbreviations: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad.Catalytic reactions were carried out in an autoclave (150 mL of capacity) (Berghof), and at atmospheric pressure in glass vessels. An aqueous solution (10 mL) containing one of the complexes 1–3 (0.01 mmol), allyl alcohol (10 mmol) and the appropriate amount of NaOH was stirred at room temperature for 30 min. During this time, the formation of 2-methyl-2-pentenal (MP) as a colourless organic phase was observed. A second product, 3-hydroxy-2-methylpentanal (HP), that is partially soluble in water, is formed at a low concentration of NaOH. Therefore, immediately after the reaction, the liquid mixture was separated from the solid residue by the vacuum transfer technique and the organic/aqueous phases were analyzed using gas chromatographs (HP model 5890 and 6890), with FID and MS detectors, with capillary columns HP5 (30 m × 0.32 mm × 0.25 μm) and HP-INNOWax (30 m × 0.5 mm × 0.25 μm). The solid residue, as well as the liquid phase, were additionally analyzed by NMR and/or IR spectroscopy. Blank tests have shown no product formation in the absence of Rh catalysts. The catalyst recycling was performed in the presence of each complex in 0.20 M NaOH as follows: after each run the organic product was removed in a phase separator, followed by the introduction of a new amount of allyl alcohol substrate.
[Rh(CO)(PTA)4]Cl (1)
Method A. To an ethanolic solution (20 mL) of [{Rh(CO)2(μ-Cl)}2] (97.2 mg, 0.25 mmol) PTA was added (314 mg, 2.00 mmol) and the mixture was stirred at room temperature for 1 h. A microcrystalline yellow product was collected by filtration, washed with cold ethanol (2 × 10 mL) and dried in vacuo to give 1. Yield: 65% (258.4 mg) based on [{Rh(CO)2(μ-Cl)}2]. Complex 1 is soluble in H2O (S25 °C ≈ 30 mg mL−1), DMSO and MeOH, less soluble in EtOH, and insoluble in C6H6 and alkanes. C25H48ClN12OP4Rh (795.0): calcd C 37.77; H 6.09; N 21.14; found C 37.85, H 6.01, N 21.07. IR (KBr): 2938 (w), 2929 (w), ν(CH), 1992 (s), ν(CO), 1704 (m), 1590 (m), 1426 (s), 1280 (m), 1245 (s), 1072 (s), 1015 (s), 1001 (s), 975 (s), 949 (s), 810 (m), 728 (m), 610 (m) and 560 (s) (PTA bands). 1H NMR (300 MHz, D2O): δ 5.15 and 5.12 (2d, J(AB) = 12 Hz, 24H, NCH2N, PTA), 4.16 (s, 24H, PCH2N, PTA). 31P{1H} NMR (121.4 MHz, D2O): δ −56.6 (br s, PTA).Method B. An aqueous solution (10 mL) of 2 (221.0 mg, 0.25 mmol) was transferred into the autoclave and the mixture was stirred at room temperature for 1 h under a CO/H2 atmosphere [p(CO) = p(H2) = 1.0 MPa]. After depressurization of the mixture, i-propanol (10 mL) was added, and the yellow microcrystalline solid was filtered off, washed with cold i-propanol (2 × 5 mL) and dried in vacuo to give 1 in 70% yield.
[RhCl2(PTA)4]Cl·EtOH (2·EtOH)
To an ethanolic solution (20 mL) of RhCl3 (131.65 mg, 0.50 mmol) PTA was added (314 mg, 2.00 mmol) and the resulting mixture was stirred at room temperature for 3 h. The microcrystalline dark yellow product was collected by filtration, washed with cold ethanol (2 × 5 mL) and dried in vacuo to give 2·EtOH. Yield: 75% (331.5 mg) based on RhCl3. Complex 2 is soluble in H2O (S25 °C ≈ 10 mg mL−1) and DMSO, less soluble in EtOH, and insoluble in C6H6 and alkanes. C26H54Cl3N12OP4Rh (883.9): calcd C 35.33; H 6.16; N 19.01; found C 35.35, H 6.10, N 19.10. IR: 3359, (s br) ν(OH), 2945, 2930, (2s br) ν(CH), 1700 (m), 1595 (m), 1426 (s), 1280 (m), 1245 (s), 1072 (s), 1050 (m), 1015 (s), 1001 (s), 975 (s), 949 (s), 880 (m), 810 (m), 728 (m), 610 (m) and 563 (s) (PTA and EtOH bands). 1H NMR (300 MHz, D2O): δ 5.17 and 5.14 (2d, J(AB) = 13 Hz, 24H, NCH2N, PTA), 4.16 (s, 24H, PCH2N, PTA), 3.67 (q, 2H, EtOH), 1.16 (t, 3H, EtOH). 31P{1H} NMR (121.4 MHz, D2O): δ −46.0 (br s, PTA).[RhH(PTA)4] (3)
To an aqueous solution (5 mL) of 1 or 2 [198.7 mg (1), 221.0 mg (2), 0.25 mmol] NaBH4 was added (94.6 mg, 2.50 mmol) and the mixture was stirred at room temperature for 5 min. Addition of cold iso-propanol (10 mL) caused the precipitation of the microcrystalline off-white product, which was collected by filtration, washed with diethyl ether (2 × 5 mL) and dried in vacuo to give 3. Yield: 55 and 65% (based on 1 and 2, respectively). Complex 3 is soluble in H2O (S25 °C ≈ 20 mg mL−1), less soluble in EtOH, and insoluble in C6H6. C24H49N12P4Rh (732.53): calcd C 39.35; H 6.74; N 22.95; found C 38.25, H 6.19, N 21.80. IR (KBr): 2930 (w), 2901 (w), ν(CH), 1942 (m), ν(Rh-H), 1242 (s), 1103 (m), 1014 (s), 1002 (s), 974 (s), 825 (s), 747 (s), 744 (m), 588 (m), 562 and 562 (s) (PTA bands). 1H NMR (300 MHz, D2O): δ 4.69 and 4.61 (2d, J(AB) = 13 Hz, 12H, NCH2N, PTA), 4.63 and 4.55 (2d, J(AB) = 13 Hz, 12H, NCH2N, PTA), 4.17 (br s, 12H, PCH2N, PTA), 4.08 (br s, 12H, PCH2N, PTA), −11.2 (ddtd, 2J(H-Pa) = 123.0 Hz, 2J(H-Pb) = 2J(H-Pc) = 25.6 Hz, 1J(H-Rh) = 13.6 Hz). 1H{31P} NMR (300 MHz, D2O): δ 4.69 and 4.61 (2d, J(AB) = 13 Hz, 12H, NCH2N, PTA), 4.63 and 4.55 (2d, J(AB) = 13 Hz, 12H, NCH2N, PTA), 4.17 (s, 12H, PCH2N, PTA), 4.08 (s, 12H, PCH2N, PTA), −11.2, (d, 1J(H-Rh) = 13.6 Hz). 31P NMR (121.4 MHz, D2O): δ −39.5 (Pb, dtd, 1J(Pb-Rh) = 94.1 Hz, 2J(Pb-Pa) = 2J(Pb-Pc) = 23.7 Hz, 2J(H-Pb) = 25.6 Hz), −53.55 (Pa, dtd, 1J(Pa-Rh) = 85.2 Hz, 2J(Pc-H) = 123.0 Hz), −53.65 (Pc, dtd, 1J(Pc-Rh) = 85.2 Hz, 2J(Pc-H) = 123.0 Hz). 31P{1H} NMR (121.4 MHz, D2O): δ −39.5 (Pb, dtd, 1J(Pb-Rh) = 94.1 Hz, 2J(Pb-Pa) = 2J(Pb-Pc) = 23.7 Hz), −53.55 (Pa, dt, 1J(Pa-Rh) = 85.2 Hz), −53.65 (Pc, dt, 1J(Pc-Rh) = 85.2 Hz).Refinement details for the X-ray crystal structure analysis of 1–2
Single crystals suitable for X-ray-analyses were grown from the reaction filtrate at 4 °C. Intensity data were collected using a Bruker AXS-KAPPA APEX II diffractometer with graphite monochromated Mo-Kα radiation. Data were collected at 150 K using omega scans of 0.5° per frame and a full sphere of data was obtained. Cell parameters were retrieved using Bruker SMART software and refined using Bruker SAINT14 on all the observed reflections. Absorption corrections were applied using SADABS.14 Structures were solved by direct methods by using the SHELXS-97 package15a and refined with SHELXL-9715a with the WinGX graphical user interface.15b All hydrogen atoms were inserted in calculated positions. There were disordered molecules present in the structure of complex 2. Since no obvious major site occupations were found for those molecules, it was not possible to model them. PLATON/SQUEEZE16 was used to correct the data and a potential volume of 1381.7 Å3 was found with ca. 209 electrons per unit cell worth of scattering. The electron count suggests the presence of one molecule of ethanol per unit cell, which was confirmed by 1H NMR (see Experimental). Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms and isotropic for most of the remaining atoms were employed. The selected bond distances (Å) and angles (°) are given in the footnotes of Fig. 1 and 2, and the crystallographic data and refinement parameters are summarized in Table 1. CCDC no. 896276 and 896277 contain the supplementary crystallographic data of 1 and 2 for this paper.![An ORTEP view of [Rh(CO)(PTA)4]Cl (1). The ellipsoids are drawn at the 50% probability level and the H atoms are omitted for clarity. Selected bond distances and angles (Å, °): P1–Rh1 2.334(3), P2–Rh1 2.339(5), P3–Rh1 2.401(5), C1–Rh1 1.92(2), C1–O1 1.08(2); O1–C1–Rh1 177(2), C1–Rh1–P1 84.47(18), C1–Rh1–P2 159.1(7), P1–Rh1–P2 90.97(10), C1–Rh1–P3 103.7(7), P1–Rh1–P3 102.83(9), P2–Rh1–P3 97.18(18). Symmetry operation to generate equivalent atoms: x, 1/2 − y, z.](/image/article/2013/DT/c3dt50992c/c3dt50992c-f1.gif) | ||
| Fig. 1 An ORTEP view of [Rh(CO)(PTA)4]Cl (1). The ellipsoids are drawn at the 50% probability level and the H atoms are omitted for clarity. Selected bond distances and angles (Å, °): P1–Rh1 2.334(3), P2–Rh1 2.339(5), P3–Rh1 2.401(5), C1–Rh1 1.92(2), C1–O1 1.08(2); O1–C1–Rh1 177(2), C1–Rh1–P1 84.47(18), C1–Rh1–P2 159.1(7), P1–Rh1–P2 90.97(10), C1–Rh1–P3 103.7(7), P1–Rh1–P3 102.83(9), P2–Rh1–P3 97.18(18). Symmetry operation to generate equivalent atoms: x, 1/2 − y, z. | ||
![An ORTEP view of [RhCl2(PTA)4]Cl (2). The ellipsoids are drawn at the 50% probability level. The crystallization ethanol molecule and H atoms are omitted for clarity. Selected bond distances and angles (Å, °): P1–Rh1 2.3756(9), P2–Rh1 2.2872(9), P3–Rh1 2.3204(8), P4–Rh1 2.3997(9), Cl1–Rh1 2.4192(9), Cl1–Rh1 2.4192(9), Cl2–Rh1 2.4200(8); P2–Rh1–P1 92.06(3), P2–Rh1–P3 95.59(3), P2–Rh1–P4 93.55(3), P3–Rh1–P1 95.83(3), P3–Rh1–P4 99.24(3), Cl1–Rh1–Cl2 85.69(3).](/image/article/2013/DT/c3dt50992c/c3dt50992c-f2.gif) | ||
| Fig. 2 An ORTEP view of [RhCl2(PTA)4]Cl (2). The ellipsoids are drawn at the 50% probability level. The crystallization ethanol molecule and H atoms are omitted for clarity. Selected bond distances and angles (Å, °): P1–Rh1 2.3756(9), P2–Rh1 2.2872(9), P3–Rh1 2.3204(8), P4–Rh1 2.3997(9), Cl1–Rh1 2.4192(9), Cl1–Rh1 2.4192(9), Cl2–Rh1 2.4200(8); P2–Rh1–P1 92.06(3), P2–Rh1–P3 95.59(3), P2–Rh1–P4 93.55(3), P3–Rh1–P1 95.83(3), P3–Rh1–P4 99.24(3), Cl1–Rh1–Cl2 85.69(3). | ||
| 1 | 2 | |
|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|. b wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. | ||
| Empirical formula | C25H48ClN12OP4Rh | C24H48Cl3N12P4Rh |
| Molecular weight | 794.99 | 837.88 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/m (No. 11) | C2/c (No. 15) |
| a (Å) | 9.931(5) | 27.2415(10) |
| b (Å) | 14.531(7) | 11.3351(5) |
| c (Å) | 10.807(5) | 25.7238(11) |
| β (°) | 97.59(2) | 108.5830(10) |
| V (Å3) | 1545.9(12) | 7529.0(5) |
| Z | 2 | 8 |
| ρ calc (Mg m−3) | 1.708 | 1.478 |
| μ (Mo Kα) (mm−1) | 0.892 | 0.872 |
| F (000) | 824 | 3456 |
| No. reflections collected | 9018 | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 989 989 |
| No. reflections unique/observed | 2823/1296 | 8182/5810 |
| R int. | 0.2330 | 0.0540 |
| Final R1a, wR2b (I ≥ 2σ) |
0.0734, 0.1724 | 0.0636, 0.0885 |
| Goodness-of-fit on F2 | 0.948 | 0.965 |
Results and discussion
Syntheses and characterization
Treatment of an ethanolic solution of [{Rh(CO)2(μ-Cl)}2] with a stoichiometric quantity of PTA affords (Scheme 2) [Rh(CO)(PTA)4]Cl (1), while [RhCl2(PTA)4]Cl (2) is formed in the presence of RhCl3·3H2O instead of [{Rh(CO)2(μ-Cl)}2] (Scheme 2). | ||
| Scheme 2 Syntheses of 1–3 with a numbering scheme for the PTA ligands in 3 (see Experimental). | ||
Additionally, compound 2 can be transformed to 1 by reduction of the Rh(III) to the Rh(I) complex in a CO/H2 atmosphere, while the hydride Rh(I) complex [RhH(PTA)4] 3 forms directly from an aqueous solution of 1 or 2 in the presence of NaBH4 (Scheme 2). The complexes were isolated as yellow (1–2) or off-white (3) microcrystalline solids in 55–75% yields, and characterized by IR, 1H and 31P NMR spectroscopies, elemental and (for 1 and 2) single crystal X-ray diffraction analyses. 1 and 2 are relatively air stable in the solid state and in aqueous solutions, while compound 3 is stable in solid state under inert conditions. The complexes are soluble in polar solvents, such as H2O and Me2SO, less soluble in medium polarity solvents such as EtOH, n-PrOH, and insoluble in non-polar ones such as toluene and hexane.
Spectroscopy
The CO (in 1) and H (in 3) ligands are easily identified by their characteristic IR ν(CO) and ν(Rh–H) bands at 1992 and 1942 cm−1, respectively, which are comparable to those observed in related complexes.5f,7,8 The IR spectra of 1–3 show related features with typical vibrations due to the PTA ligand, including some characteristic bands (1100–900 cm−1) associated with the ν(C–X) (X = N, P) vibrations. Besides, several bands due to νas and νs(CH) are also detected in the 2945–2901 cm−1 range.1The 1H NMR spectra of 1–3 in D2O show two characteristic types of methylene protons for the coordinated PTA. One of them, assigned to the P–CH2–N moiety, occurs as a singlet at δ 4.08–4.17 whereas the other one, corresponding to the N–CH2–N group, displays an AB spin system centred at 4.65–5.15 ppm, attributed to the N–CHax–N and N–CHeq–N protons, as previously reported.17 Additionally, in the 1H NMR spectrum of 3, a double set of methylene protons centred at δ 4.65 and 4.59 (for NCH2N), as well as δ 4.17 and 4.08 (for PCH2N), assigned as two Pb and Pa,c, were observed due to the nonequivalence of the PTA ligands (see Scheme 2). The multiplicity of the hydride resonance at δ −11.2 confirms the nonequivalence of the coordinated ligands in 3. In contrast to the previously described analogue [RhH(PTA-Me)4]I4 (PTA-Me = N-methyl-1,3,5-triaza-7-phosphaadamantane cation), the 31P{1H} and 31P NMR spectra of 3 reveal the presence of three types of nonequivalent PTA ligands (three resonances at δ −39.5, −53.55 and −53.65 corresponding to the Pb, Pa and Pc ligands, respectively) in the coordination sphere of the rhodium(I) centre. For 1 and 2, in the 31P{1H} NMR spectra, only a broad singlet is observed at δ −56.6 and −46.0, respectively, due to an average effect of a dynamic process.
X-ray crystal structures of 1 and 2
The crystal structure of compound 1 indicates that the molecule exhibits a crystallographically imposed mirror plane containing Rh1, C1, O1, P2, N22, C22, C25, P3, N31, C31 and C35 atoms, as shown in Fig. 1. The geometry of the Rh atom in 1 is square pyramid (τ5 = 0.09) with three of the PTA ligands and the carbonyl group in equatorial sites therefore defining a P3C type basal plane. The metal is located 0.4475(15) Å above this basal plane deviating towards the P3-phosphine which is in the axial position. The Rh–C bond distance of 1.92(2) Å is longer than that found in [Rh{P(3-py)3}2(CO)Cl] [1.811(2) Å]18 or in [Rh(Tpms)(CO)(PTA)] [Tpms = tris(1-pyrazolyl)methanesulfonate; 1.813(4) Å]19 while that of C–O is shorter [1.08(2) Å against 1.138(2) or 1.139(6) Å, respectively].In the structure of 2 (Fig. 2) the metal exhibits a rather distorted octahedral environment around the Rh centre, filled by four neutral PTA moieties and two chloride ligands in the cis-configuration. The P2–Rh–P3 angle is 95.59(3)° and that of P1–Rh–P4 is 163.31(3)°. The Rh–P bond distances for the phosphine groups trans to chloride (2.2871 and 2.3203 Å) are shorter than those for the mutually trans Rh–P ones (2.3757 and 2.3997 Å) indicating a greater trans effect of the neutral phosphine ligand, relative to the chloride anion.
The lengths of the Rh–P bonds in both complexes [in the 2.2872(9)–2.401(5) Å range], the Rh–Cl in 2 [av. 2.4196(9) Å] and the bonding parameters within the PTA cages are comparable to those found in related Rh–PTA derivatives.6,20
Catalysis
Complexes 1–3 were tested as homogeneous catalysts, or catalyst precursors, for the isomerisation and controlled condensation of allyl alcohol to 3-hydroxy-2-methylpentanal (HP) and 2-methyl-2-pentenal (MP) under ambient conditions and in aqueous media (Table 2, Scheme 3a), which proceeds in three main steps. | ||
| Scheme 3 Pathway of allyl alcohol condensation in the presence (a) or in the absence (b) of 1–3. P = n-propanal; HP = 3-hydroxy-2-methylpentanal; MP = 2-methyl-2-pentenal. | ||
| Complex/substrate | Products | Yieldb [%] | |||||
|---|---|---|---|---|---|---|---|
| CNaOH [M] | |||||||
| 0 | 0.02 | 0.05 | 0.10 | 0.15 | 0.20 | ||
| a Reaction conditions: complex 1–3 (0.01 mmol); H2O (10 mL); substrate (10 mmol); 293 K; 30 min. b Determined by GC. c Reaction in an autoclave at p(H2) = 1.0 MPa. d Condensation of n-propanal under similar conditions in the absence of 1–3. | |||||||
| Entry | 1 | 2 | 3 | 4 | 5 | 6 | |
| 1/Allyl alcohol | n-Propanal (P) | 8 | 1 | <1 | 0 | 0 | 0 |
| 3-Hydroxy-2-methylpentanal (HP) | 0 | 50 | 95 | 29 | 1 | <1 | |
| 2-Methyl-2-pentenal (MP) | 0 | 1 | 3 | 70 | 99 | >99 | |
| Conversion of allyl alcohol [%] | 9 | 52 | 100 | 100 | 100 | 100 | |
| Entry | 7 | 8 | 9 | 10 | 11 | 12 | |
| 2/Allyl alcoholc | n-Propanal (P) | 5 | 2 | <1 | <1 | 0 | 0 |
| 3-Hydroxy-2-methylpentanal (HP) | 0 | 50 | 95 | 40 | 1 | 1 | |
| 2-Methyl-2-pentenal (MP) | 0 | 1 | 2 | 56 | 99 | 99 | |
| Conversion of allyl alcohol [%] | 7 | 60 | 100 | 100 | 100 | 100 | |
| Entry | 13 | 14 | 15 | 16 | 17 | 18 | |
| 3/Allyl alcohol | n-Propanal (P) | 11 | 5 | 1 | <1 | 0 | 0 |
| 3-Hydroxy-2-methylpentanal (HP) | 0 | 50 | 87 | 40 | 1 | <1 | |
| 2-Methyl-2-pentenal (MP) | 1 | 2 | 7 | 52 | 99 | >99 | |
| Conversion of allyl alcohol [%] | 14 | 60 | 95 | 96 | 100 | 100 | |
| Entry | 19 | 20 | 21 | 22 | 23 | 24 | |
| n-Propanal (P)d | 3-Hydroxy-2-methylpentanal (HP) | 0 | 0 | 0 | 0 | 0 | 0 |
| 2-Methyl-2-pentenal (MP) | 0 | <1 | <1 | <1 | <3 | <3 | |
| Conversion of n-propanal [%] | 0 | <1 | <1 | <1 | <4 | <4 | |
The first step (I) includes the isomerisation of allyl alcohol to n-propanal (P) and does not require the presence of NaOH (Table 2, Fig. 3a–c). At a low concentration of base (0.02–0.05 M) two molecules of n-propanal couple to 3-hydroxy-2-methylpentanal (HP, step II, Scheme 3a). A concentration of the base above 0.05 M allows dehydration. At 0.10 M of base in the reaction mixture, both 3-hydroxy-2-methylpentanal (HP) and 2-methyl-2-pentenal (MP, step III) are formed. A higher base concentration, in the 0.15–0.20 M range, results in the formation of 2-methyl-2-pentenal (MP) as the sole product in quantitative yield. Thus, upon variation of the base concentration in the reaction mixture, it is possible to control the formation of the appropriate product (HP or MP). The presence of the rhodium complex is essential for the steps I and III, since none of the products was observed in the absence of the Rh catalyst. Although we detected the formation of small amounts of 2-methyl-2-pentenal (MP) in the absence of 1–3 (Scheme 3b, Fig. 3d) when using n-propanal as the substrate, the presence of rhodium complexes considerably increases the rate of this process, and additionally allows the formation of 3-hydroxy-2-methylpentanal (HP), at an appropriate concentration of the base.
 | ||
| Fig. 3 (a–c) Correlation between the NaOH concentration and the activity of the corresponding catalyst or catalyst precursors 1–3 in condensation of allyl alcohol (P = n-propanal; HP = 3-hydroxy-2-methylpentanal; MP = 2-methyl-2-pentenal). (d) Correlation between the NaOH concentration in condensation of n-propanal in the absence of 1–3. Reaction conditions are those of Table 2. | ||
The turnover frequencies (TOFs) achieve values as high as 2000 h−1 for all the complexes 1–3, at a NaOH concentration in the 0.05–0.2 M range. For comparison, the optimized system of [Rh(COD)(MeCN)2](BF4) and PTA catalyses the isomerisation of allylic alcohols into carbonyl compounds with a comparable rate (2400 h−1), but at a higher temperature.10a,b Separately, the catalytic condensation of n-propanal to the 3-hydroxy-2-methylpentanal (HP) and 2-methyl-2-pentenal (MP) mixture, with a TOF of 42 mmol of products per g catalyst per h using chitosan as a solid base catalyst at 100 °C, was previously reported.11
It should also be noted that the Rh(III) complex 2 is active only in the presence of H2, thus suggesting reduction, by this species, to a catalytically more active Rh(I) form. Expectedly, the presence of H2 (1.0–2.0 MPa) does not lead to hydrogenation of the allyl alcohol. Since no condensation product was observed in the absence of a base, the involvement of an Rh–monohydride complex in the reaction mechanism is suggested.21 Indeed, 1H NMR spectra of the complexes separated from the final reaction solution exhibit resonances centred at δ ca. −11.0, when the base was added. Moreover, the loss of CO from the coordination sphere of 1 (in the presence and in the absence of allyl alcohol) was confirmed by IR spectroscopy. Catalysts 1–3 were isolated at the end of the reaction in the presence of NaOH (0.20 M) and tested in two subsequent runs; the same level of activities towards the 2-methyl-2-pentenal (MP) formation was revealed.
Although some examples of isomerisation reactions for allyl and allylic alcohols10a,22 or condensation of various aldehydes10–12,23 have been described in the literature, to our knowledge, the complexes 1–3 appear to be the first examples of catalysts which allow not only the selective isomerisation of allyl alcohol to n-propanal (step I), but also (depending on the NaOH concentration) the reaction leading to the selective formation of the condensation products: 3-hydroxy-2-methylpentanal (HP, step II), and after dehydration only 2-methyl-2-pentenal (MP, step III) at a higher concentration of NaOH. Although the catalytic behaviours of other rhodium complexes with PTA and other water-soluble phosphines9,23 towards the transformation of allyl and allylic alcohols have been described previously, in such cases the isomerisation products have not been subsequently transformed into any condensation products.
Conclusions
The study reports a suitable way for the syntheses of new water-soluble rhodium complexes of the types [Rh(CO)(PTA)4]Cl (1), [RhCl2(PTA)4]Cl (2) and [RhH(PTA)4] (3), which is based on the reaction of [{Rh(CO)2(μ-Cl)}2] or RhCl3 with 1,3,5-triaza-7-phosphaadamantane (PTA). The solubility and stability in water of the obtained complexes encouraged their applications in aqueous catalysis. In fact, the work shows that they catalyze the isomerisation and controlled condensation of allyl alcohol, under ambient conditions and in aqueous media, thus displaying some advantages over the commercial methods. The selectivity can be controlled simply by changing the concentration of the base in the reaction mixture, thus resulting in the exclusive formation of either 3-hydroxy-2-methylpentanal (HP) or 2-methyl-2-pentenal (MP) in quantitative yields, at lower or higher concentrations of the base, respectively. The activity of the catalyst remains the same at least for the three following runs, thus allowing its efficient recycling. This study opens up the possibility of application of hydro-soluble PTA–Rh complexes in the controlled condensation of allyl alcohol, with an easy base-tuned selectivity in water and under mid conditions, broadening their application in catalysis under green conditions. It deserves to be further explored for other types of condensation catalyses.Acknowledgements
This work was supported by KBN (Grant no. N204 280438, Poland) and FCT (Grants BPD/20869/04, BPD/34926/2007 and project PEst-OE/QUI/UI0100/2011, Portugal). Special thanks to Mrs Magda Ligorowska for her contribution to this work.References
- For reviews, see: (a) A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955 CrossRef CAS; (b) J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555 CrossRef CAS.
- F. Joó, Aqueous Organometallic Catalysis, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- (a) A. K. Renfrewa, L. Juillerat-Jeanneret and P. J. Dyson, J. Organomet. Chem., 2011, 696, 772 CrossRef; (b) C. Scolaro, T. J. Geldbach, S. Rochat, A. Dorcier, C. Gossens, A. Bergamo, M. Cocchietto, I. Tavernelli, G. Sava, U. Rothlisberger and P. J. Dyson, Organometallics, 2006, 25, 756 CrossRef CAS; (c) C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 12, 4161 CrossRef; (d) B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 3423 CrossRef CAS; (e) A. M. Kirillov, S. W. Wieczorek, A. Lis, M. F. C. Guedes da Silva, M. Florek, J. Król, Z. Staroniewicz, P. Smolenski and A. J. L. Pombeiro, Cryst. Growth Des., 2011, 11, 2711 CrossRef CAS.
- (a) F. Mohr, S. Sanz, E. R. T. Tiekink and M. Laguna, Organometallics, 2006, 25, 3084 CrossRef CAS; (b) F. Mohr, E. Cerrada and M. Laguna, Organometallics, 2006, 25, 644 CrossRef CAS; (c) A. M. Kirillov, P. Smoleński, Z. Ma, M. F. C. Guedes da Silva, M. Haukka and A. J. L. Pombeiro, Organometallics, 2009, 28, 6425 CrossRef CAS.
- (a) C. A. Mebi and B. J. Frost, Organometallics, 2005, 24, 2339 CrossRef CAS; (b) C. A. Mebi, R. P. Nair and B. J. Frost, Organometallics, 2007, 26, 429 CrossRef CAS; (c) P. J. Dyson, D. J. Ellis and G. Laurenczy, Adv. Synth. Catal., 2003, 1, 345 Search PubMed; (d) G. Laurenczy, F. Joó and L. Nádasdi, Inorg. Chem., 2000, 39, 5083 CrossRef CAS; (e) P. Smoleński, F. P. Pruchnik, Z. Ciunik and T. Lis, Inorg. Chem., 2003, 42, 3318 CrossRef; (f) F. P. Pruchnik, P. Smoleński, E. Gałdecka and Z. Gałdecki, New J. Chem., 1998, 1395 RSCP. Smoleński, J. Organomet. Chem., 2011, 696, 3867 CrossRef.
- D. J. Darensbourg, N. W. Stafford, F. Joó and J. H. Reibenspies, J. Organomet. Chem., 1995, 488, 99 CrossRef CAS.
- F. P. Pruchnik, P. Smoleński and I. Raksa, Pol. J. Chem., 1995, 69, 5 CAS.
- F. P. Pruchnik, P. Smoleński, E. Gałdecka and Z. Gałdecki, Inorg. Chim. Acta, 1999, 293, 110 CrossRef CAS.
- F. P. Pruchnik, P. Smoleński and K. Wajda-Hermanowicz, J. Organomet. Chem., 1998, 570, 63 CrossRef CAS.
- (a) N. Ahlsten, H. Lundberg and B. Martín-Matute, Green Chem., 2010, 12, 1628 RSC; (b) P. Lorenzo-Luis, A. Romerosa and M. Serrano-Ruiz, ACS Catal., 2012, 2, 1079 CrossRef CAS; (c) J. G. Stevens, R. A. Bourne and M. Poliakoff, Green Chem., 2009, 11, 409 RSC.
- T. Jose, N. Sudheesh and R. S. Shukla, J. Mol. Catal. A: Chem., 2010, 333 Search PubMed . 158.
- H. A. Patel, S. K. Sharma and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 286, 31 CrossRef CAS.
- (a) D. J. Daigle, A. B. Pepperman Jr. and S. L. Vail, J. Heterocycl. Chem., 1974, 11, 407 CrossRef CAS; (b) D. J. Daigle, Inorg. Synth., 1998, 32, 40 CrossRef CAS.
- Bruker, APEX2 & SAINT, AXS Inc., Madison, WI, 2004 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS; (b) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, C34 Search PubMed.
- (a) P. Smoleński and A. J. L. Pombeiro, Dalton Trans., 2008, 87 RSC; (b) A. M. Kirillov, P. Smoleński, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2007, 2686 CrossRef CAS.
- W. H. Meyer, R. J. Bowena and D. G. Billing, Z. Naturforsch., B, 2007, 62, 339 CAS.
- P. Smolenski, C. Dinoi, M. F. C. Guedes da Silva and A. J. L. Pombeiro, J. Organomet. Chem., 2008, 693, 2338 CrossRef CAS.
- P. Smoleński, A. M. Kirillov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Inorg. Chim. Acta, 2011, 378, 342 CrossRef.
- O. Pàmies and J.-E. Bäckvall, Chem.–Eur. J., 2001, 7, 5052 CrossRef.
- (a) R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27 CrossRef CAS; (b) B. Martín-Matute, K. Bogár, M. Edin, F. B. Kaynak and J.-E. Bäckvall, Chem.–Eur. J., 2005, 11, 5832 CrossRef.
- (a) S. K. Sharma, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 278, 135 CrossRef CAS; (b) A. Bartoszewicz, M. Livendahl and B. Martín-Matute, Chem.–Eur. J., 2008, 14, 10547 CrossRef CAS.
Footnote |
| † CCDC 896276 and 896277. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt50992c |
| This journal is © The Royal Society of Chemistry 2013 |