 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferracyclic carbamoyl complexes related to the active site of [Fe]-hydrogenase†
Peter J. Turrell, Amanda D. Hill, Saad K. Ibrahim, Joseph. A. Wright and Christopher J. Pickett*
Energy Materials Laboratory, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: c.pickett@uea.ac.uk
First published on 12th April 2013
Abstract
The active site of the [Fe]-hydrogenase features an iron(II) centre bearing cis carbonyl groups and a chelating pyridine–acyl ligand. Reproducing these unusual features in synthetic models is an intriguing challenge, which will allow both better understanding of the enzymatic system and more fundamental insight into the coordination modes of iron. By using the carbamoyl group as a surrogate for acyl, we have been able to synthesize a range of ferracyclic complexes. Initial reaction of Fe(CO)4Br2 with 2-aminopyridine yields a complex bearing a labile solvent molecule, which can be replaced by stronger donors bearing phosphorus atoms to produce a number of derivatives. Introduction of a hydroxy group using this method is unsuccessful both with a free OH group and when this is silyl-protected. In contrast, the analogous reactions starting from 2,6-diaminopyridine does allow synthesis of complexes bearing a pendant basic group.
Introduction
Biologically, activation of dihydrogen occurs by three phylogenetically-distinct types of hydrogenase enzyme: [NiFe], [FeFe]- and [Fe].1,2 The redox-active [NiFe]- and [FeFe]-hydrogenases have been the focus of studies both at the biological and synthetic levels, with a very large number of models reported for the di-iron system. In contrast, the redox-inactive [Fe]-hydrogenase has to date received less attention. This enzyme catalyses the heterolytic cleavage of dihydrogen into a proton and a hydride, delivering the latter to the substrate methylenetetrahydromethanopterin.3,4 The crystal structure of the [Fe]-hydrogenase revealed that the iron centre is ligated by an unusual acyl group (Fig. 1).5,6![Active site of the [Fe]-hydrogenase (X = solvent).6](/image/article/2013/DT/c3dt50642h/c3dt50642h-f1.gif) | ||
| Fig. 1 Active site of the [Fe]-hydrogenase (X = solvent).6 | ||
Prior to the confirmation of the structure of the [Fe]-hydrogenase by crystallography, a small number of model compounds had been synthesized based on the available spectroscopic evidence.7 The design of the majority of these focused on reproduction of a cis carbonyl arrangement at iron, and supported the formulation of an iron(II) centre in the enzyme,8–11 while others introduced pyridine and sulfur ligands around the metal centre.12,13 Following the publication of the revised crystal structure of the [Fe]-hydrogenase, Hu and coworkers have described the synthesis of five- and six co-ordinate acyl models (Fig. 2)14–17 and related dimeric sulfur-bridged structures.18 We have communicated a facile route to ferracyclic analogues possessing an intramolecularly-generated carbamoyl group,19 and described the formation of a thiolate-containing structure which closely reproduces the geometry of the enzyme active site (Fig. 2).
![Monomeric thiolate-containing models of [Fe]-hydrogenase active site reported by Hu and coworkers (left)15 and Pickett and coworkers (right).19](/image/article/2013/DT/c3dt50642h/c3dt50642h-f2.gif) | ||
| Fig. 2 Monomeric thiolate-containing models of [Fe]-hydrogenase active site reported by Hu and coworkers (left)15 and Pickett and coworkers (right).19 | ||
Herein, we now report synthetic and structural details of some new ferracyclic carbamoyl systems, including the identification of a key intermediate in the synthetic pathway, and the first example of a structural model of [Fe]-hydrogenase bearing a basic group at the 6-position on the pyridine ring.
Results and discussion
Formation of carbamoyl ligands has been previously reported following reaction of rhenium and ruthenium carbonyl complexes with amine-substituted nitrogen-containing heterocycles.20–22 Evidently coordination of the heterocyclic nitrogen atom to the metal centre allows intramolecular attack at an electrophilic CO (Scheme 1). This approach was adapted to synthesise the first ferracyclic carbamoyl structures.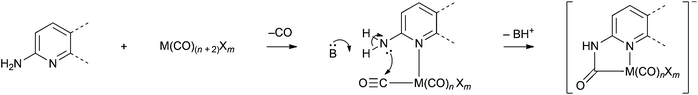 | ||
| Scheme 1 Generalised mechanism for metallocyclic carbamoyl formation. | ||
Primary ring-forming reaction
Reaction of Fe(CO)4Br2 with 2-aminopyridine in CH2Cl2 results in the formation of a yellow precipitate 1 with evolution of carbon monoxide (Scheme 2). This material is insoluble in non-coordinating solvents (see below) and was not previously identified.19 In the coordinating solvent MeCN, 1 reacted to give complex 2 which was fully characterised by X-ray crystallography. Compound 2 provided a starting point for the synthesis of thiolate derivatives related to the active site of [Fe]-hydrogenase (Scheme 2). We have now found that by slow diffusion of a solution of Fe2(CO)4Br2 in CH2Cl2 into 2-aminopyridine in hexanes we can obtain well-formed crystals of 1 suitable for X-ray analysis. This reveals that 1 is a salt with the anionic component bearing the anticipated carbamoyl functionality and cis carbonyl groups: the cationic fragment is a protonated 2-aminopyridine (Fig. 3). The Br–Fe–Br angle in 1 [93.48(3)°] is very similar to that in Fe(CO)4Br2, [average 92.75(17)°, see ESI† for details], suggesting that the steric requirement of the pyridinyl–carbamoyl ligand is small. The two Fe–Br distances in 1 are significantly different, with the bromide trans to the carbamoyl [2.5185(8) Å] longer than that trans to a carbonyl [2.4578(9) Å]; the latter is in line with the average Fe–Br distance in Fe(CO)4Br2 [2.427(3) Å]. High-resolution mass spectroscopy of bulk1 (formed as the yellow insoluble powder) confirmed the presence of an M− species at m/z 392.7999, with the appropriate isotope pattern for the presence of two bromide groups. The attenuated total reflectance (ATR) infrared spectrum of bulk 1 shows two CO stretches at 1982 cm−1 and 2040 cm−1 along with a carbamoyl peak at 1660 cm−1.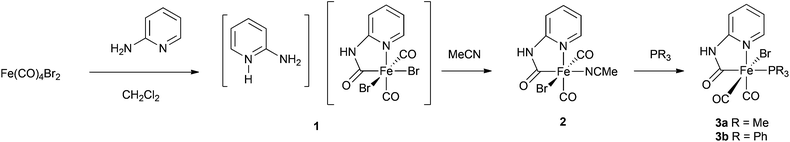 | ||
| Scheme 2 Synthesis of 3. | ||
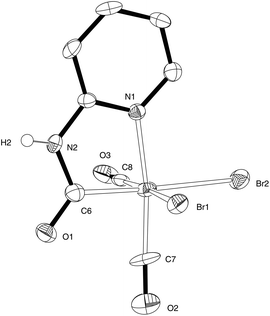 | ||
| Fig. 3 ORTEP representation of the anion of 1 showing 50% probability ellipsoids; hydrogen atoms except for H(2) have been omitted for clarity. Selected average bond lengths (Å) and angles (°) with estimated standard deviations: Fe(1)–Br(1) 2.4578(9), Fe(1)–Br(2) 2.5185(8), Fe(1)–C(6) 1.921(5), Fe(1)–C(7) 1.769(5), Fe(1)–C(8) 1.844(6), Fe(1)–N(1) 2.004(4); Br(1)–Fe(1)–Br(2) 93.48(3), C(6)–Fe(1)–N(1) 81.78(19), C(7)–Fe(1)–C(8) 89.1(2). | ||
As reported earlier, dissolution of 1 in MeCN followed by concentration and cooling yielded 2·MeCN as a crystalline solid (Scheme 2), with X-ray diffraction confirming loss of hydrogen bromide from 1 and retention of the carbamoyl and cis carbonyl groups.19 Here we note that the X-ray structure of 2 shows that the carbamoyl group retains significant double bond character, with the CO bond distance [1.229(3) Å] significantly longer than that for the two carbonyls [1.136(3) Å and 1.086(3) Å] but shorter than would be anticipated for a C–O single bond. The C–N distance [1.383(3) Å] also confirms the partial double-bond character of this unit. Examination of the C–Fe and N–Fe bond lengths in 1 and 2 shows that they are not significantly different, whilst the C–Fe–N angles in the two compounds are very similar [81.84(18)° and 82.68(9)°, respectively]. The carbamoyl ligand unit is therefore insensitive to the wider metal environment: with the exception of compound 3a (vide infra), this pattern is seen for all of the monomeric carbamoyl complexes we have examined.
Substitution chemistry of complex 2
Complex 2 possesses a potential labile solvato ligand, MeCN. We have shown that this can be replaced by THF and reversibly by CO:19 here we further explore the substitution of the solvato ligands MeCN and THF with phosphines.Slow addition of a dilute solution of PMe3 to 2 in THF enabled the isolation of 3a in which the ligating solvent is replaced by the phosphine ligand (Scheme 2, Fig. 4). Compared to complexes 1 and 2, the carbamoyl ligand in 3a is ‘twisted’ such that the pyridine ring does not lie directly above the Fe–P bond, reducing steric clash between the phosphine and pyridine groups. In solution, the IR spectrum of 3a shows maxima at 1934 cm−1, 1967 cm−1 and 2035 cm−1, absorptions which are shifted to lower wavenumbers with respect to the parent complex and indicative of better electron-donating ability of the PMe3 ligand compared with MeCN at the Fe(II) centre.
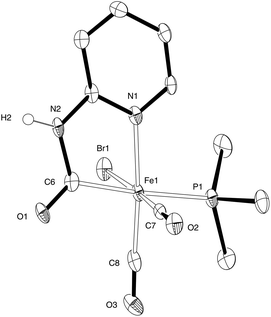 | ||
| Fig. 4 ORETP representation of the structure of 3a showing 50% probability ellipsoids; hydrogen atoms except for H(2) have been omitted for clarity. Selected average bond lengths (Å) and angles (°) with estimated standard deviations: Fe(1)–Br(1) 2.4914(6), Fe(1)–C(6) 1.981(3), Fe(1)–C(7) 1.760(4), Fe(1)–C(8) 1.779(4), Fe(1)–N(1) 2.006(3), Fe(1)–P(1) 2.3152(10); C(7)–Fe(1)–C(8) 90.19(16), C(6)–Fe(1)–N(1) 82.10(13), Br(1)–Fe(1)–P(1) 87.86(3). | ||
Reaction of 2 with PMe3 in THF is evidently sensitive to moisture and/or oxygen, as a by-product which co-crystallised with 3a, [Fe(MeCN)4(PMe3)2][(Br3Fe)2O], has been identified, in which the carbamoyl ligand has been lost entirely from the metal centre (see ESI†).
The reaction of 2 in THF with one equivalent of PPh3 gave a mixture of mono- and di-substituted compounds. Modifying the reaction procedure by carrying out and working up under an atmosphere of CO allowed isolation of 3b, which exhibits solution IR stretches at 2040 cm−1 and 1990 cm−1 in the carbonyl region. Compound 3b gave satisfactory elemental analysis to support formulation as the monophosphine. The di-substituted complex Fe(CO)(PPh3)2(Br)(C6H5N2CO)·THF was isolated as a minor product which was characterized by crystallography (see ESI†).
Basic functionality at the 6-position of the pyridine ring
With a secure route to models replicating the first coordination sphere of the active site of the [Fe]-hydrogenase, the next target is replicating the wider environment around the iron. The native enzyme contains a hydroxyl group pendant to the putative hydrogen bind site,6 and the involvement of this group in catalysis has been proposed.4 Theoretical studies suggest that the lone pair of the oxygen sufficiently basic enough to able to polarize the hydrogen–hydrogen bond, facilitating hydrogen activation.23,24Based on the formation of 1 by reaction of 2-aminopyridine with Fe(CO)4Br2, we sought to introduce a hydroxy group using 4a as the ligand precursor. Compound 4a (Fig. 5) has previously been synthesized from 2,6-diaminopyridine by reaction with HCl25 or H2SO4,26 but in neither case is spectroscopic data available. Modification of the procedure of Tisza and Joos25 allowed us to reliably access 4a in good yield. We have established by an X-ray diffraction study that 4a exists as the keto form 4a′ in the solid state (see ESI†), but this does not interfere with the reactivity of the hydroxy group in solution (see below). Reaction of 4a with Fe(CO)4Br2 led to a very rapid reaction with significant effervescence; IR indicated that all bound CO had been lost in this process. Attempts to moderate the reaction by cooling and dilution were unsuccessful. To exclude the possibility that this failure was due to reaction at two competing sites, we moved to protection of the hydroxyl group in 4a. Silylation using (i-Pr)3SiCl under standard conditions to yield 4b proceeded smoothly. However, reaction of 4b with Fe(CO)4Br2 gave similar results to that with 4a: loss of carbonyl signals in the infrared. It is possible that HBr liberated in the formation of the carbamoyl is able to attack the silyl group, deprotecting in situ and thus leading to the same reactivity seen with 4a.
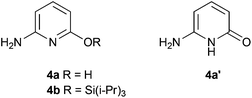 | ||
| Fig. 5 Oxygen-functionalised aminopyridines. | ||
We therefore turned to the introduction of an alternative basic group in the form of the second amine group in 2,6-aminopyridine. Carbamoyl formation on one side of this molecule would be expected to leave the second amine group free to participate in further chemistry. Following a similar procedure to that which produces compound 2, the diamine was added to a solution of Fe(CO)4Br2 (Scheme 3). Effervescence due to the liberation of carbon monoxide was observed and a pale yellow precipitate was rapidly formed. Dissolution of the yellow powder in MeCN gave a solution showing CO stretching frequencies at 1999 cm−1 and 2055 cm−1; this was tentatively assigned as 5. Even when stored in the dark at −20 °C, solutions of 5 were unstable whilst evaporation of the solvent left only intractable oils, precluding the acquisition of further data. We therefore added PMe3 directly to a crude preparation of 5 in THF, leading to the appearance of new IR bands at 1963 cm−1, 1985 cm−1 and 2042 cm−1. When crystallized from concentrated solutions,‡ compound 6 (Fig. 6) was obtained as orange blocks. A diffraction study confirmed the presence of a free amino group in this material, along with retention of the cis carbonyl arrangement as required. The geometry at iron is similar to the other carbamoyl complexes reported here, although it is notable that the bromide occupies the position trans to the carbamoyl here, in contrast to the arrangement in 3. This arrangement permits intramolecular hydrogen bonding to take place, with an H(3a)⋯Br(1) distance of 2.36 Å. The other protic hydrogen atoms in 6 are involved in intermolecular interactions as would be expected.
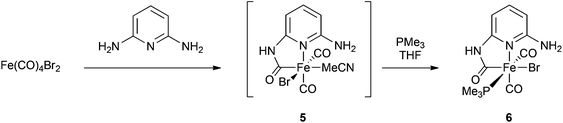 | ||
| Scheme 3 Synthesis of 6. | ||
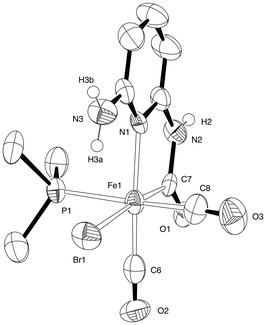 | ||
| Fig. 6 ORTEP representation of the structure of 6·MeCN showing 50% probability ellipsoids; hydrogen atoms except for H(2), H(3a) and H(3b) have been omitted for clarity. Selected average bond lengths (Å) and angles (°) with estimated standard deviations: Fe(1)–Br(1) 2.5488(18), Fe(1)–C(6) 1.917(11), Fe(1)–C(7) 1.789(13), Fe(1)–C(8) 1.826(14), Fe(1)–N(1) 2.077(8), Fe(1)–P(1) 2.270(3); C(6)–Fe(1)–N(1) 82.5(4), C(7)–Fe(1)–C(8) 92.8(5). | ||
Electrochemistry
Cyclic voltammetry of 2 in MeCN-0.1 M [NBu4][BF4] at vitreous carbon revealed irreversible waves for both oxidation and reduction. Oxidation gave a single wave at Ep,ox = +1.36 V versus Ag/AgCl, while reduction showed two waves at −1.17 V and −1.43 V versus Ag/AgCl. Notably the natural centre in the enzyme is redox inactive in the accessible physiological potential domain ca −1.0 to +1.0 V versus Ag/AgCl, and the observed oxidation and reduction potentials of the synthetic complex is in accord with this. The large HOMO–LUMO gap of some 2.5 V is indicative of the stability of the Fe(II) structure cf. Pearson's principle of maximum hardness.27 Cyclic voltammetry of 3a similarly revealed only irreversible oxidation and reduction processes at Eoxp = +1.26 V and Eredp = −1.36 V versus Ag/AgCl, respectively. Infrared spectroelectrochemistry of 3a in the MeCN electrolyte showed depletion bands at Eapplied = Ep but the absence of detectable growth bands in the CO region, indicative of oxidative decarbonylation on the seconds time-scale.Conclusions
Metallocyclic carbamoyl complexes are rare. We have now identified and structurally characterised a key precursor on a pathway which has provided access to a range substituted ferracyclic structures, including new phosphine derivatives (Scheme 2). In the [Fe]-hydrogenase structure nature deploys a hydroxyl-ketonic group at the 6-position on the pyridine ring in a ferracyclic structure and this is thought to be important in heterolytic cleavage of dihydrogen acting as proton acceptor. Although approaches to synthesise a synthetic analogue with a similar framework were unsuccessful, we have shown that the reaction of Fe(CO)4Br2 with 2,6-diaminopyridine affords a ferracyclic structure which has a basic amino group juxtaposed in the 6-position to the Fe centre, i.e. in the second coordination sphere, and this complex has been structurally characterised.Experimental
General
Unless otherwise stated, reactions were carried out under nitrogen using conventional air-sensitive techniques. Starting materials were purchased from Aldrich or Alfa Aesar and were used without further purification. Solvents were freshly distilled under an inert atmosphere of N2 from an appropriate drying agent. NMR spectra were recorded at 25 °C in 5 mm tubes using a Bruker Avance 300 spectrometer. FT-IR spectra were recorded on a Perkin-Elmer SpectrumBX instrument, fitted with a SensIR Technologies DuraSamplIR II ATR unit where appropriate. Elemental analyses were performed by London Metropolitan University. Cyclic voltammetric measurements were carried out using an Autolab PGSTAT 30 potentiostat. A conventional three-electrode arrangement was employed, consisting of a vitreous carbon working electrode, a platinum wire as the auxiliary electrode, and Ag/AgCl as a reference electrode. All measurements were done in dry degassed MeCN in the presence of 0.1 M [NBu4][BF4] as the supporting electrolyte at room temperature. Spectroelectrochemistry experiments were carried out using a Spectroelectrochemistry Partners SP-02 cell mounted on a Pike MIRacle ATR unit fitted to a Bruker Vertex 80 spectrometer.Fe(CO)4Br2
This was synthesized as previously reported.19 Crystals suitable for X-ray diffraction were grown by sublimation at reduced pressure.[C5H7N2][Fe(C6H5N2O)(CO)2Br2)] (1)
A solution of 2-aminopyridine (12 mg, 0.13 mmol) in hexane (5 cm3) was carefully layered onto a solution of Fe(CO)4Br2 (50 mg, 0.13 mmol) in CH2Cl2 (10 cm3). A small amount (ca 5 mg) of yellow crystalline [C5H7N2][Fe(C6H5N2O)(CO)2Br2)] was formed upon the slow diffusion of the solutions through each other over the course of five days and the isolated material was characterized by X-ray diffraction. Found C 30.11, H 2.31, N 11.01%; C13H12Br2FeN4O3·0.5(CH2Cl2) requires C 30.57, H 2.47, N 10.56%. m/z (orbitrap) calcd for anion C8H5N2Br2FeO3 392.8001, found 392.7999.FeBr(C6H5N2O)(CO)2(PMe3) (3a)
A solution of PMe3 (0.065 cm3, 0.063 mmol) dissolved in THF (10 cm3) was added drop wise to 2 (240 mg 0.607 mmol) in THF (10 cm3) over 30 minutes. During the addition, the solution turned from orange-red to a darker red-purple. The volume of the solution was reduced to 2 cm3 under vacuum before cooling to −20 °C. A white solid precipitated; the supernatant was decanted and the solvent was removed under reduced pressure yielding a sticky oil. This was taken in MeCN (ca 1 cm3) and evaporated in vacuum three times to co-evaporate the residual THF. The solid was recrystallized from a minimum of MeCN at −20 °C, give of red crystals of X-ray quality (8 mg, 3%). Found C 34.04, H 3.55, N 7.12%; C11H14BrFeN2O3P requires C 33.95, H 3.63, N 7.20%. νmax/cm−1 (MeCN) 2035, 1967, 1934. m/z (CI−) 361.8, 359.9 (M − H − CO). 31P NMR (121 MHz, CD2Cl2): δ −12.51.FeBr(C6H5N2O)(CO)2(PPh3) (3b)
Solid PPh3 (211 mg, 0.805 mmol) was added to 2 (316 mg 0.805 mmol) in THF (20 cm3). The solvent volume was reduced by sparging with CO, leading to the formation of a red-brown precipitate. The final brown paste was washed with cold THF (−20 °C, 5 cm3) to leave a red-brown powder (315 mg, 68%). Found C 54.19, H 3.60, N 4.89%; C26H20BrFeN2O3P requires C 54.29, H 3.50, N 4.87%. νmax/cm−1 (THF) 2040, 1990, 1666, 1620. 31P NMR (121 MHz, CD2Cl2): δ 37.47.6-Aminopyridin-2-ol (4a)
2,6-Diaminopyridine (12.5 g, 114 mmol) was refluxed in HCl (10%, 300 cm3) for three hours. The solvent was distilled out at atmospheric pressure to leave 50 cm3 of concentrated solution. On cooling, a pale crystalline material precipitated and was recovered by filtration. The crude material was redissolved in a minimum of hot water and the pH adjusted to 11 by addition of 40% NaOH solution. Cooling to 0 °C led to the formation of needle-like crystals of the product. After washing with CH2Cl2, this was recrystallized from hot water (4.0 g, 32%). Crystals suitable for X-ray diffraction were obtained from this second crystallization. 1H NMR (300 MHz, D2O): δ 5.54 (m, 1H), 5.57 (d, 1H, J = 3.9 Hz), 7.23 (t, 1H, J = 8.3 Hz). 13C NMR (75 MHz, D2O): 92.23, 101.81, 145.75, 151.59, 163.81. m/z (EI+) 110.0.6-{[Tris(propan-2-yl)silyl]oxy}pyridin-2-amine (4b)
6-Aminopyridin-2-ol 4a (618 mg, 5.62 mmol) was dissolved in THF (100 cm3) and cooled to −78 °C. Butyl lithium (1.6 M in THF, 3.51 cm3) was added slowly, and the reaction stirred for 2 h before warming to room temperature. Tris(isopropyl)silyl chloride (1.20 cm3, 5.62 mmol) was added, and reaction stirred for 48 h. The solvent was removed at reduced pressure and the product extracted from the residue with CH2Cl2. This was then filtered and the solvent removed at reduced pressure to leave a yellow/brown oil, which was used directly (910 mg, 61%). 1H NMR (300 MHz, CD2Cl2): δ 1.10 (d, 27H, J = 7.4 Hz), 1.35 (octet, 3H, J = 7.4 Hz), 6.04 (d, 1H, J = 1.7 Hz), 6.07 (d, 1H, J = 1.7 Hz), 7.32 (t, 1H, J = 7.8 Hz). m/z (orbitrap) calcd for C14H25N2OSi 267.1887, found 267.1892.FeBr(C6H6N3O)(CO)2(PMe3) (6)
Solid 2,6-diaminopyridine (186 mg, 1.71 mmol) was added to a solution of Fe(CO)4Br2 (560 mg, 1.71 mmol) in CH2Cl2 (25 cm3); gas was evolved and a precipitate formed. After two hours, the precipitate was recovered by filtration and dried in vacuo; this gave 557 mg of yellow solid. The solid was taken up in THF (10 cm3), and a solution of PMe3 (0.14 cm3, 1.35 mmol) in THF (15 cm3) was added. After one hour, the volume was reduced to approximately 5 cm3 and the solution cooled to −20 °C overnight. The solution was filtered to remove any white precipitate that had formed, then dried under vacuum to give a sticky orange oil. Recrystallization of this oil from a minimum of MeCN (∼3 cm3) at −20 °C gave a mass of homogeneous crystals of 8 (51 mg, 7.5%). Compound 6 was isolated as orange blocks suitable for X-ray diffraction study. Found C 32.46, H 3.89, N 10.21%; C11H16BrFeN3O3P requires C 32.62, H 3.98, N 10.38%. νmax/cm−1 (THF) 1963, 1985, 2042. m/z (CI−) 376.9, 374.9 (M − H − CO).Crystallography
Crystals were suspended in oil, and one was mounted on a glass fibre and fixed in the cold nitrogen stream of the diffractometer. Data were collected an Oxford Diffraction Xcalibur-3 CCD diffractometer equipped with Mo-Kα (λ = 0.71073 Å) radiation and graphite monochromator (1 and 6·MeCN), or a Bruker–Nonius FR591 molybdenum rotating anode and confocal mirrors [Fe(CO)4Br2 and 3a]. Data were processed using the CrysAlisPro program (1 and 6·MeCN)28 or the DENZO and COLLECT programs [Fe(CO)4Br2 and 3a].29 Structures were determined by the direct methods routines in the Superflip program30 (1 and 3a) or SHELXS program31 [Fe(CO)4Br2 and 6·MeCN], and were refined by full-matrix least-squares methods on F2 in SHELXL.31 Non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms bound to nitrogen were located in the Fourier difference map and freely refined where possible; in all other cases they were included in idealized positions and their Uiso values were set to ride on the Ueq values of the parent carbon atoms. Crystallographic data for previous-unpublished structures are summarized in Table 1 and S1† (ESI†). CCDC 922917–922924 contain the supplementary crystallographic data for Fe(CO)4Br2, 1, 3a and 6·MeCN and the supporting structures.| Fe(CO)4Br2 | 1 | 3a | 6·MeCN | |
|---|---|---|---|---|
| Formula | C4Br2FeO4 | C8H5Br2FeN2O3, C5H7N2 | C11H14BrFeN2O3P | C11H15BrFeN3O3P, C2H3N |
| M | 327.71 | 487.94 | 388.97 | 445.04 |
| Space group | C2 | P21/n | P21/a | P21/c |
| a/Å | 6.7608(4) | 11.2197(6) | 13.2378(3) | 9.8203(17) |
| b/Å | 10.2296(5) | 12.6286(7) | 7.5324(3) | 12.719(2) |
| c/Å | 12.2954(7) | 11.8538(6) | 15.4174(5) | 15.139(2) |
| β/° | 103.801(3) | 105.142(5) | 110.218(2) | 91.830(14) |
| V/Å3 | 825.80(8) | 1621.24(15) | 1442.58(8) | 1890.0(5) |
| T/K | 120(2) | 140(2) | 120(2) | 140(2) |
| Z | 4 | 4 | 4 | 4 |
| Rint | 0 | 0.074 | 0.056 | 0.165 |
| R1 [I > 2σI] | 0.068 | 0.042 | 0.040 | 0.091 |
| wR2 (all data) | 0.177 | 0.085 | 0.079 | 0.277 |
Acknowledgements
PJT and ADH thank the Engineering and Physical Sciences Research Council (EPSRC) for Departmental Training Award studentships. JAW thanks the Biotechnology and Biological Sciences Research Council (BBSRC) and EPRSC for funding (grant numbers BB/E023290/1 and EP/F047878/1). The authors thank the EPSRC National Mass Spectrometry Service, Swansea for mass spectroscopy data, and the EPSRC National Crystallography Service for collection of diffraction data.32References
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2009, 253, 1476–1494 CrossRef CAS.
- S. Shima and U. Ermler, Eur. J. Inorg. Chem., 2011, 963–972 CrossRef CAS.
- M. J. Corr and J. A. Murphy, Chem. Soc. Rev., 2011, 40, 2279–2292 RSC.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS.
- T. Hiromoto, K. Ataka, O. Pilak, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer, S. Shima and U. Ermler, FEBS Lett., 2009, 583, 585–590 CrossRef CAS.
- J. A. Wright, P. J. Turrell and C. J. Pickett, Organometallics, 2010, 29, 6146–6156 CrossRef CAS.
- Y. Guo, H. Wang, Y. Xiao, S. Vogt, R. K. Thauer, S. Shima, P. I. Volkers, T. B. Rauchfuss, V. Pelmenschikov, D. A. Case, E. E. Alp, W. Sturhahn, Y. Yoda and S. P. Cramer, Inorg. Chem., 2008, 47, 3969–3977 CrossRef CAS.
- B. V. Obrist, D. Chen, A. Ahrens, V. Schünemann, R. Scopelliti and X. Hu, Inorg. Chem., 2009, 48, 3514–3516 CrossRef CAS.
- X. Wang, Z. Li, X. Zeng, Q. Luo, D. J. Evans, C. J. Pickett and X. Liu, Chem. Commun., 2008, 3555–3557 RSC.
- T. Liu, B. Li, C. V. Popescu, A. Bilko, L. M. Pérez, M. B. Hall and M. Y. Darensbourg, Chem.–Eur. J., 2010, 16, 3083–3089 CrossRef CAS.
- A. M. Royer, M. Salome-Stagni, T. B. Rauchfuss and W. Meyer-Klaucke, J. Am. Chem. Soc., 2010, 132, 16997–17003 CrossRef CAS.
- D. Chen, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2010, 132, 928–929 CrossRef CAS.
- D. Chen, R. Scopelliti and X. Hu, Angew. Chem., Int. Ed., 2010, 49, 7512–7515 CrossRef CAS.
- D. Chen, R. Scopellitti and X. Hu, Angew. Chem., Int. Ed., 2011, 50, 5671–5673 CrossRef CAS.
- D. Chen, A. Ahrens-Botzong, V. Schünemann, R. Scopelliti and X. Hu, Inorg. Chem., 2011, 50, 5249–5257 CrossRef CAS.
- D. Chen, R. Scopelliti and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 1919–1921 CrossRef CAS.
- B. Hu, D. Chen and X. Hu, Chem.–Eur. J., 2012, 18, 11528–11530 CrossRef CAS.
- P. J. Turrell, J. A. Wright, J. N. T. Peck, V. S. Oganesyan and C. J. Pickett, Angew. Chem., Int. Ed., 2010, 49, 7508–7511 CrossRef CAS.
- J.-L. Zuo, W.-F. Fu, C.-M. Che and K.-K. Cheung, Eur. J. Inorg. Chem., 2003, 255–262 CrossRef CAS.
- P. Nombel, N. Lugan, B. Donnadie and G. Lavigne, Organometallics, 1998, 18, 187–196 CrossRef.
- Formation of a carbamoyl heterocycle on ruthenium has also been described by electrochemical reduction: T. Tomon, T.-A. Koizumi and K. Tanaka, Angew. Chem., Int. Ed., 2005, 44, 2229–2232 CrossRef CAS.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 10901–10908 CrossRef CAS.
- A. Dey, J. Am. Chem. Soc., 2010, 132, 13892–13901 CrossRef CAS.
- E. T. Tisza and B. Joos, US Patent, 1863676, 1932 Search PubMed.
- O. A. Seide and A. I. Titow, Ber. Dtsch. Chem. Ges., 1936, 69, 1884–1893 CrossRef.
- R. G. Pearson, Acc. Chem. Res., 1993, 26, 250–255 CrossRef CAS.
- CrysAlisPro, Oxford Diffraction Ltd., Abingdon, UK, 2010 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 922917–922924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt50642h |
| ‡ When crude 6 is crystallized slowly from dilute solution, partial decomposition takes place leading to the isolation of Fe(CO)(PMe3)2{C6H6N3CO(FeBr3)}(MeCN). This dinuclear material features a FeBr3− group bound to the carbamoyl oxygen. See ESI† for further details. |
| This journal is © The Royal Society of Chemistry 2013 |