DOI:
10.1039/C3DT50617G
(Paper)
Dalton Trans., 2013,
42, 9921-9929
Tuning affinity and reversibility for O2 binding in dinuclear Co(II) complexes†
Received
6th March 2013
, Accepted 4th May 2013
First published on 9th May 2013
Abstract
The O2 binding affinity of a series of dicobalt(II) complexes can be tuned between p(O2)50% = 2.3 × 10−3 and 700 × 10−3 atm at 40 °C by varying the number of H and Cl atoms in the bridging acetato ligands of [Co2(bpbp)(CH(3−n)ClnCO2)(CH3CN)2]2+, where bpbp− = 2,6-bis(N,N-bis(2-pyridylmethyl)aminomethyl)-4-tert-butylphenolate and n = {0, 1, 2, 3}. O2 binds most strongly to the deoxy complex containing the acetato bridge and the O2 affinity decreases linearly as the number of Cl atoms is increased from 0 to 3 in [Co2(bpbp)(O2)(CH3CO2)]2+, [Co2(bpbp)(O2)(CH2ClCO2)]2+, [Co2(bpbp)(O2)(CHCl2CO2)]2+ and [Co2(bpbp)(O2)(CCl3CO2)]2+. The O2 affinities can be qualitatively correlated with both the pKa value of the parent acetic or chloroacetic acid and the redox potential of the O22−/O2˙− couple measured for the peroxide-bridged complexes. The redox potential varies between 510 mV (vs. Fc0/+) for the acetato-bridged complex to 696 mV for the trichloroacetato-bridged system. Despite the clear difference in reactivity in solution, there are no clear trends which can be correlated to O2 affinity in the O–O bond lengths in the X-ray crystal structures at 180 K (1.415(4)–1.424(2) Å) or in the frequencies of the peroxido O–O stretch in the solid-state resonance Raman spectra at 298 K (830–836 cm−1). Using density functional theory calculations, we conclude that the Co(II) atoms of the deoxy complexes coordinate solvent molecules as auxiliary ligands and that a conformation change of the ligand is involved in the reversible O2 binding process. The alternative of five coordination in the deoxy Co(II) complexes is therefore seen as less likely. The crystal structure and p(O2)50% are also reported for the 1-naphthoato-bridged oxy complex [Co2(bpbp)(O2)(C10H7O2)]2+, and the O2 binding affinity in that case is also qualitatively consistent with the expectation from the pKa of the parent 1-naphthoic acid.
Introduction
Reversible dioxygen binding is a life-supporting process for respiring organisms carried out by three classes of metalloproteins: hemoglobin, hemerythrin, and hemocyanin, the latter two having dimetallic active sites. Although cobalt has not been found in any naturally occurring dioxygen carrier, it was shown over four decades ago that cobalt-substituted hemoglobin (coboglobin), is able to bind O2 reversibly, albeit with lower affinity than the native iron protein. Oxycoboglobin is formulated as Co(III) with a coordinated superoxide.1 Mononuclear cobalt complexes based on salen2 and corrole3ligands can also achieve reversible O2 binding, although this property can be quenched by formation of peroxido-bridged dimers.2,4 Dimer formation in hemoglobin is prevented by the protein, and the similar prevention of dimerisation has been achieved for model systems with the sterically hindered “picket fence” porphyrin type ligands5 or by incorporating Co–salen complexes into zeolites6 and polymers.7–9 Suzuki and co-workers10–12 have reported that reversible O2 binding can also occur for dicobalt(II) systems based on alkoxido and phenolato-hinged dinucleating ligands. In these, both cobalt atoms are oxidised from Co(II) to Co(III) by the oxidative addition of dioxygen, thus facilitating the reduction of O2 to peroxide rather than the highly reactive superoxide as is the case for the mononuclear systems and which are prone to irreversible self-annihilation reactions.
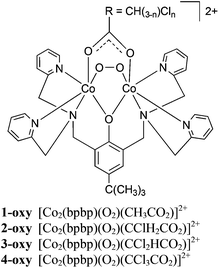
Materials displaying fully reversible O2 binding properties have potential industrial and medical uses where storage, concentration gradients akin to that shown by the life-sustaining complementary functions of hemoglobin and myoglobin, and release of oxygen need to be finely controlled.13,14 For example, incorporation of complexes able to bind oxygen reversibly into membranes has been used to increase oxygen permeability, thereby facilitating concentration of oxygen from air.15,16 We report here a systematic study of the electronic effects of the supporting bridging carboxylato co-ligand on the O2 affinity of the deoxy forms of the series of complexes which include those shown in Scheme 1. This work demonstrates a relatively facile method for tuning O2 affinity, a property that could be used in the design of metal–organic materials for applications in creating gradients of O2 concentration.
 |
| | Scheme 1 | |
Results and discussion
Synthesis
The peroxido bridged dicobalt(III) complexes (Scheme 1, n = 0, 1, 2, 3) or naphthalene-1-carboxylato, were synthesized by reacting 2,6-bis(N,N-bis(2-pyridylmethyl)aminomethyl)-4-tert-butylphenol (Hbpbp) with two equivalents Co(BF4)2·6H2O or Co(ClO4)2·6H2O and one equivalent of the co-ligand precursor in the presence of air or pure O2 at room temperature or below. At ambient temperatures, [Co2(bpbp)(O2)(CH3CO2)]2+ (1-oxy) and [Co2(bpbp)(O2)(CClH2CO2)]2+ (2-oxy) were formed in air, while [Co2(bpbp)(O2)(CCl2HCO2)]2+ (3-oxy) was formed in an atmosphere of pure oxygen at elevated pH. [Co2(bpbp)(O2)(CCl3CO2)]2+ (4-oxy) was formed under an atmosphere of pure oxygen at elevated pH and at −20 °C. Thus with increasing electron-withdrawing character in the bridging acetato ligand, more forcing conditions are required to ensure formation of the peroxido complexes. The related complex [Co2(bpbp)(O2)(O2C-naphth)]2+ (5-oxy) which contains the co-bridging naphthalene-1-carboxylato group was prepared analogously to 1-oxy and 2-oxy. The dicobalt(III) oxy complexes are dark brown and the deoxy complexes are pink. The deoxy complexes of the Cl-substituted carboxylates were on some occasions obtained as very air-sensitive pink powders by working under an inert atmosphere. However, single crystals suitable for X-ray structural analysis were not forthcoming. These powders turn brown rapidly on exposure to air. The reversible binding process is observed easily in methanol or acetonitrile solution by warming the oxy complexes in order to effect the dark brown to pink colour change. On cooling, O2 is absorbed and bound again and the dark brown colour returns. This process can be repeated many times in organic solvent solution. Evacuation and purging with N2 also removes O2.
In accordance with expected ligand-field stabilization effects, the auxiliary bridging groups will be relatively labile in the deoxy dicobalt(II) forms. In this situation, solvent coordination can compete with the carboxylatoligands, especially those that are less basic. In water, the consequence is the scrambling reaction depicted in eqn (1). We have shown previously that the more acidic carboxylic acids favour formation of the dicarboxylato-bridged complexes which do not bind O2.17 The generation of [Co2(bpbp)(RCOO)2]+ species was substantiated by ESI mass spectrometry and was seen to be promoted if a significant amount of water was present. When waterligands fill all of the auxiliary coordination sites, an outer-sphere oxidation of the complex in the presence of air produces the “met” hydroxido-aqua complexes irreversibly,18 according to eqn (2). Both of these reactions will compete with reversible O2 binding and in the presence of large concentrations of water and/or when the parent carboxylic acid is relatively acidic, water inhibits O2 binding.
| | | 2[Co2(bpbp)(O2)(RCOO)]2+ + 4H2O → 2O2 + [Co2(bpbp)(RCOO)2]+ + [Co2(bpbp)(H2O)4]3+ | (1) |
| | | [Co2(bpbp)(H2O)4]3+ → [Co2(bpbp)(OH)2(H2O)2]3+ + 2H+ + 2e− | (2) |
Resonance Raman spectra performed in the solid state show clearly the band due to ν(O–O) which are observed at 830.63, 837.76, 836.41 and 837.76 cm−1 for 1-oxy, 2-oxy, 3-oxy, and 4-oxy respectively (measurements averaged over several crystal orientations, ESI Fig. S2†). These values are consistent with the peroxido formulation.19,20 No correlation is evident between the positions of the bands and the electron-withdrawing effect of the carboxylatoligand. For all four complexes, the intensity of the ν(O–O) band decreased over time as the spectra were recorded, and as the colour of the sample changed from dark brown to light pink. This indicated that deoxygenation of the complexes was caused by the laser, either directly by photoactivation or by local heating of the sample.
The deoxygenated complexes could not be structurally characterized in the solid state. Thus it remains unclear if the cobalt ions are five-coordinated or, for example, solvent or counteranion-derived ligands occupy the sixth coordination site in the absence of peroxide. Suzuki and co-workers proposed that closely related and structurally uncharacterized deoxy dicobalt(II) complexes are five coordinated on the basis of solution electronic spectra and solid-state magnetic susceptibility data.12,21–24
X-ray crystal structures
The coordination mode of bpbp− is identical in all of the oxy complexes (Fig. 1). The phenolato O atom bridges between the two Co atoms and each dipyridylmethylamine groups are fac coordinated to one Co(II) metal centre. The coordination spheres are completed by the RCOO− and O22− groups bridging between the Co(II) metal centres. One O atom of the peroxido ligand (O2) lies trans to an amine donor (N1) and the other (O3) lies trans to a pyridyl donor (N5) of bpbp−. The same applies to the bridging carboxylato O donors (O4 trans to N2 and O5 trans to N4). The bond lengths around the Co atoms are listed in Table 1. The Co–ligand and O–O bond lengths might be expected to decrease stepwise from 1-oxy to 4-oxy as the bridging carboxylato ligand becomes more electron withdrawing. However, there are very few significant differences in the Co–ligand bonds. Likewise, there are no significant differences in the O–O bond lengths. Complex 5-oxy, which has the relatively bulky naphthoato ligand bridging between the two Co atoms, also has an essentially identical coordination sphere. The O–O bond length does appear to be significantly shorter in 5-oxy compared to 1-oxy and 2-oxy, but this does not provide any consistent indication of the p(O2)50% values, as discussed below.
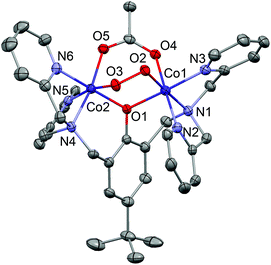 |
| | Fig. 1 Complex 1-oxy drawn with 50% displacement ellipsoids (H atoms omitted). The geometry is essentially identical in all other complexes. | |
Table 1 Selected distances and angles in 1–5 at 180 K (Å, °)
| |
1
|
2
|
3
|
4
|
5
|
|
Two complexes in the asymmetric crystallographic unit.
|
| Co1–O1 |
1.8943(16) |
1.8975(17) |
1.898(3) |
1.904(3) |
1.890(2) |
1.903(3) |
| Co1–O2 |
1.8640(17) |
1.8670(17) |
1.865(3) |
1.870(3) |
1.863(3) |
1.859(2) |
| Co1–O4 |
1.9141(17) |
1.9306(18) |
1.939(3) |
1.939(3) |
1.936(3) |
1.942(3) |
| Co1–N1 |
2.006(2) |
2.004(2) |
2.019(4) |
1.995(4) |
2.017(3) |
2.021(3) |
| Co1–N2 |
1.928(2) |
1.915(2) |
1.916(4) |
1.903(4) |
1.915(3) |
1.914(3) |
| Co1–N3 |
1.919(2) |
1.896(2) |
1.907(4) |
1.921(4) |
1.917(3) |
1.912(3) |
|
|
|
|
|
|
|
|
| Co2–O1 |
1.8898(15) |
1.8997(17) |
1.912(3) |
1.908(3) |
1.905(2) |
1.902(2) |
| Co2–O3 |
1.8600(16) |
1.8610(17) |
1.867(3) |
1.863(3) |
1.865(3) |
1.867(3) |
| Co2–O5 |
1.9160(17) |
1.9121(18) |
1.931(3) |
1.942(3) |
1.920(3) |
1.906(2) |
| Co2–N4 |
1.962(2) |
1.963(2) |
1.958(4) |
1.953(4) |
1.956(3) |
1.959(3) |
| Co2–N5 |
1.9994(19) |
2.001(2) |
2.000(4) |
2.002(4) |
1.988(3) |
1.984(3) |
| Co2–N6 |
1.915(2) |
1.920(2) |
1.923(4) |
1.920(4) |
1.912(3) |
1.921(3) |
|
|
|
|
|
|
|
|
| O2–O3 |
1.423(2) |
1.424(2) |
1.415(4) |
1.417(4) |
1.415(3) |
1.412(3) |
| Co1⋯Co2 |
3.1558(4) |
3.1645(6) |
3.1718(10) |
3.1642(10) |
3.1609(7) |
3.1601(7) |
| Co1–O1–Co2 |
113.02(8) |
112.91(8) |
112.73(15) |
112.20(16) |
112.79(12) |
112.30(12) |
Oxygen affinity in solution
The oxygen affinities of the deoxy complexes were evaluated in solution using UV-visspectroscopy. The oxygenation–deoxygenation process is fully reversible and can be repeated several times in acetonitrile, acetone, ethanol and methanol solutions. Following the method described by Drago and co-workers,25,26 the equilibrium constant for the uptake of O2 was extracted from a plot of the partial pressure p(O2) against p(O2)/(A − A0), where A is the absorbance at the given partial pressure of O2 and A0 is the absorbance for p(O2) = 0. A linear least-squares fit through the data points crosses the secondary axis at −1/K, where K is the equilibrium constant of eqn (3) (K = 1/p(O2)50%). The equilibrium constants at different temperatures were used to construct van't Hoff plots (Fig. 2), from which the thermodynamic data were derived.| |  | (3) |
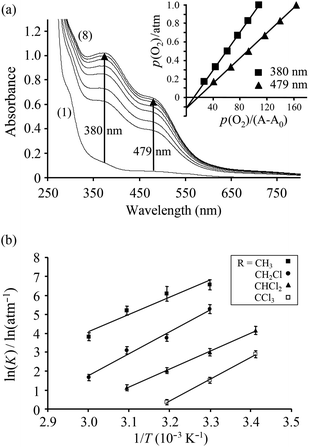 |
| | Fig. 2 (a) Spectral changes of 3 at various O2 partial pressures in CH3CN at 40 °C: (1) 0 atm, (2) 0.08 atm, (3) 0.17 atm, (4) 0.33 atm, (5) 0.50 atm, (6) 0.67 atm, (7) 0.83 atm, (8) 1.0 atm. The insert shows a plot of p(O2) vs. p(O2)/(A − A0) at 380 and 479 nm. (b) van't Hoff plots for oxygenation of the complexes 1-deoxy to 4-deoxy. | |
It is evident that the increasing number of electron-withdrawing Cl atoms on the auxiliary bridging carboxylatoligand serve to decrease the oxygen affinity in the complexes in going from 1-deoxy to 4-deoxy and at 40 °C, p(O2)50% = 2.3 ± 0.9 × 10−3 atm, 23 ± 4 × 10−3 atm, 132 ± 21 × 10−3 atm and 700 ± 90 × 10−3 atm for 1-deoxy, 2-deoxy, 3-deoxy and 4-deoxy, respectively. For comparison, p(O2)50% for hemoglobin under physiological conditions has been found to be 35.0 × 10−3 atm, and for myoglobin at 20 °C, p(O2)50% = 0.76 × 10−3 atm.27 Modification of the bridging carboxylato ligand provides a facile approach to tailor the oxygen affinity of dicobalt complexes of this type. A good measure for the electron-withdrawing ability of the carboxylatoligand is the pKa value of the parent acid, CH(3−n)ClnCO2−: pKa = 4.76, 2.86, 1.35, 0.66 for n = 0–3, respectively. Thus, the O2 binding affinity increases as the pKa increases. The pKa value of 1-naphthoic acid (3.69) is between that of acetic and chloroacetic acid. Accordingly, p(O2)50% for complex 5-deoxy was measured to be 8 × 10−3 atm in acetonitrile at 40 °C, between that of complex 1-deoxy and 2-deoxy.
Cyclic voltammetry
Cyclic voltammograms were obtained for complexes 1-oxy–4-oxy in acetonitrile solution. At positive potentials versus Fc0/+, the bridging peroxido can be reversibly oxidised to a stable superoxido group.18 With a more electron-withdrawing bridging carboxylatoligand, the oxidation of the peroxido ligand becomes energetically more demanding and the reversible redox wave is shifted to higher potentials (Table 2). A linear correlation is observed between ΔrG for the oxygenation reaction and the oxidation potential of the oxy complexes which is associated with a O22−/O2˙− redox couple (Fig. 3). The trend illustrates that it becomes progressingly harder to remove an electron from the peroxido group of 4-oxy compared to that of 1-oxy.
![(a) Cyclic voltammograms of [Co2(bpbp)(O2)(RCO2)]2+ R = CH3, CH2Cl, CHCl2, CCl3 (1-oxy to 4-oxy) showing a shift of the reversible redox couple associated with the bridging O22−/O2•− when changing the electron-withdrawing ability of the bridging carboxylate ligand. Scan rate 100 mV s−1. (b) ln(K) for the oxygenation of 1-deoxy to 4-deoxy at 40 °C as a function of the redox potential.](/image/article/2013/DT/c3dt50617g/c3dt50617g-f3.gif) |
| | Fig. 3 (a) Cyclic voltammograms of [Co2(bpbp)(O2)(RCO2)]2+ R = CH3, CH2Cl, CHCl2, CCl3 (1-oxy to 4-oxy) showing a shift of the reversible redox couple associated with the bridging O22−/O2•− when changing the electron-withdrawing ability of the bridging carboxylate ligand. Scan rate 100 mV s−1. (b) ln(K) for the oxygenation of 1-deoxy to 4-deoxy at 40 °C as a function of the redox potential. | |
Table 2 Measured p(O2)50% values and reduction potentials of the O22−/O2˙− redox couple and pKa values of 1-oxy–4-oxy
| Complex |
p(O2)50% (atm) |
E (V) vs. Fc/Fc+ |
pKa |
|
1-oxy
|
2.3 × 10−3 |
0.510 |
4.76 |
|
2-oxy
|
2.3 × 10−2 |
0.568 |
2.86 |
|
3-oxy
|
1.3 × 10−1 |
0.634 |
1.35 |
|
4-oxy
|
7.0 × 10−1 |
0.696 |
0.66 |
Electronic structure analysis
The experimental data present a conundrum, in that the progressively weaker binding of O2 in going from 1-deoxy to 4-deoxy, is not reflected in any of the solid state properties of the oxy complexes. The O–O distances in 1-oxy–5-oxy are identical within experimental error, and there is no apparent trend in the resonance Raman O–O stretching frequencies. The oxygen affinity is, however, a function of both the oxy and deoxy forms, so we now consider possible structures for the latter. Attempts at in situ deoxygenation of single crystals invariably resulted in loss of crystallinity. In the crystal structure of complex 1-oxy at room temperature, the O–O bond length (1.409(4) Å) is shorter than at 180 K (the crystallographic difference compared to 1.423(2) Å being just significant), and this apparent increase in the O–O bond order is probably a precursor to the deoxygenation process. Heating above room temperature causes crystal decomposition, and we have not been able to obtain any definitive information about the structure of the deoxy form.‡ To address the question of the structural nature of the deoxygenated species, we turned to density functional theory.
The deoxygenated complexes could contain five-coordinate Co(II), as suggested by Suzuki,12,21–24 or six-coordinate Co(II) through solvent and/or counteranion coordination. The former seems likely in the solid state, while the latter seems more probable in the presence of a large excess of acetonitrile (the conditions under which we have measured p(O2)50%). The weakly coordinating ClO4− or BF4− counteranions should not compete strongly for the empty coordination sites, so this possibility was not considered for the calculations.
We have considered the displacement reaction shown in eqn (4), where O2 replaces either 2, 1 or 0 CH3CN ligands in the coordination sphere. ΔrG values for the twelve reactions (n = 0–3) are collected in Table 3, where they are compared to the experimental values. Note that the difficulties associated with computing accurately the entropies of dissociation of CH3CN into bulk solvent preclude an accurate determination of the absolute value of ΔrG but we expect these errors to be constant across the series of chemically related compounds, 1-deoxy–4-deoxy, and so we expect to reproduce the observed trend towards stronger binding of O2 for the complexes with the less electron withdrawing bridging carboxylato ligands.
| | | [Co2(bpbp)(CH(3–n)ClnCO2)(CH3CN)m]2+ + O2 ⇌ [Co2(bpbp)(CH(3–n)ClnCO2)(O2)]2+ + mCH3CN | (4) |
Table 3 Calculated and experimental ΔrG values (kJ mol−1) for the binding of O2 to dicobalt(II) complexes of bpbp−. The number of acetonitrile molecules coordinated to Co in the optimised structures of 1-deoxy to 4-deoxy is given as m
| Complex |
m = 0 |
m = 1 |
m = 2 |
Exp. |
|
1
|
87.5 |
90.0 |
39.4 |
−15.8 |
|
2
|
84.6 |
85.9 |
44.4 |
−9.8 |
|
3
|
82.4 |
96.6 |
48.3 |
−5.3 |
|
4
|
89.9 |
100.7 |
53.7 |
−0.9 |
If solvent is assumed not to coordinate to the deoxy species (i.e. m = 0) then the structures obtained by removing the O2 unit and reoptimising with two high-spin (S = 3/2) Co(II) centres fail to reproduce the observed tendency towards stronger binding for 1-deoxy relative to 4-deoxy. The two limiting forms (n = 0 and n = 3) have very similar computed ΔrG while the middle two members are slightly more negative. The computed ΔrG values, for the optimised structures where one acetonitrile molecule is coordinating to a single Co(II) centre predict oxygen binding to become stepwise less favourable from 2-deoxy to 4-deoxy, but the trend does not include 1-deoxy. The structures where both Co(II) centres are coordinatively saturated by CH3CN placed in the binding pocket of O2 are quite strained as the removal of the O2 unit leaves a cavity that is a bit too small to accommodate two CH3CN molecules in close proximity to each other. Alternative minima that are 5–14 kJ mol−1 lower in energy are found if the tridentate dipyridylmethylamine moiety with its tertiary aminetrans to a carboxylato O is allowed to rearrange from a fac conformation in the oxy form to a mer conformation in the deoxy form. This opens up a coordination site for one acetonitrile molecule trans to the phenolato O atom, directed away from the second acetonitrileligand (Fig. 4). Whilst this configuration is not the most common for dinuclear complexes of bpbp− it has precedence in the X-ray crystal structures of [V2(bpbp)(O)3(H2O)]2+,28 [Mn2(bpbp)(ClO4)2(THF)]2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 and [FeCuF2(bpbp)(H2O)]2+.30 It is interesting to note that all of these structures contain different first coordination spheres at the two metal ions, in contrast to the majority of complexes of this type. Using the more stable conformation for the coordinatively saturated species we reproduce the trend in ΔrG well, predicting increases in steps of 5 kJ mol−1 per Cl atom substituted into the bridging carboxylato ligand similar to the effect observed in the experimental measurements (Table 3). Based on these results we suggest that 1-, 2-, 3- and 4-deoxy have two solvent molecules coordinated to complete an octahedral coordination environment at each cobalt atom. The O2 binding pathway could conceivably proceed via formation of the transient superoxido intermediate, [CoII(CH3CN)CoIII(O2)(bpbp)(CH3COO)]2+, formed through acetonitrile substitution by O2 at one Co site (that in which the acetonitrile is trans to the amine in Fig. 4). The distal O atom of the resultant superoxide then attacks the second Co site and the cispyridine on that Co moves around, thereby expelling the remaining acetonitrile.
29 and [FeCuF2(bpbp)(H2O)]2+.30 It is interesting to note that all of these structures contain different first coordination spheres at the two metal ions, in contrast to the majority of complexes of this type. Using the more stable conformation for the coordinatively saturated species we reproduce the trend in ΔrG well, predicting increases in steps of 5 kJ mol−1 per Cl atom substituted into the bridging carboxylato ligand similar to the effect observed in the experimental measurements (Table 3). Based on these results we suggest that 1-, 2-, 3- and 4-deoxy have two solvent molecules coordinated to complete an octahedral coordination environment at each cobalt atom. The O2 binding pathway could conceivably proceed via formation of the transient superoxido intermediate, [CoII(CH3CN)CoIII(O2)(bpbp)(CH3COO)]2+, formed through acetonitrile substitution by O2 at one Co site (that in which the acetonitrile is trans to the amine in Fig. 4). The distal O atom of the resultant superoxide then attacks the second Co site and the cispyridine on that Co moves around, thereby expelling the remaining acetonitrile.
![Optimised structure of deoxy [Co2(bpbp)(CH3COO)(CH3CN)2]2+.](/image/article/2013/DT/c3dt50617g/c3dt50617g-f4.gif) |
| | Fig. 4 Optimised structure of deoxy [Co2(bpbp)(CH3COO)(CH3CN)2]2+. | |
Conclusions
We have demonstrated that dicobalt(II) complexes of the bpbp−ligand can be easily tuned with respect to oxygen affinity and reversible chemisorption by changing the electronic properties of the bridging carboxylato ligand. The oxy forms of these complexes have been structurally characterised. DFT calculations suggest that the deoxygenated complexes have a pseudo octahedral coordination environment completed by the coordination of two solvent molecules.
Experimental section
Physical measurements
IR spectra were measured as KBr discs using a Hitachi 270-30 IR spectrometer. UV-visible absorption spectra were recorded on a Shimadzu UV-3100 spectrophotometer with an inbuilt Shimadzu CPS-240A thermo-electrical temperature controlled cell holder. Spectrophotometric measurements of 1–5 in acetonitrile, acetone, ethanol and methanol were carried out at 40 °C under various O2 partial pressures p(O2). Argon and dioxygen gas were mixed using two Hastings HFC-202 mass flow controllers and passed into ∼0.1 mM solutions (gas flow 30 mL min−1) of the complex in a quartz cell (1 cm path length). The gas mixture was bubbled through the solution for 5 min before the measurement to ensure that equilibrium had been reached; longer bubbling times gave identical results. Absorption shoulders, absent in the deoxy form, that grow on oxygenation at 380 and 480 nm were monitored. Up to 5% of the solvent evaporated during the collection of a data series and the weight of the cuvette plus solution was measured between each data collection so that the measured absorptions could be corrected for the change in concentration. The binding affinity is given as p(O2)50% = K−1 and represents the partial pressure of O2 needed to oxygenate 50% of the complex. The strong binding affinity resulted in saturation effects at high p(O2) and measurements at these partial pressures could not be used in determination of K. Gas mixtures with p(O2) lower than ∼1.5% could not be obtained without exceeding the calibration range of the flow controllers. Elemental analyses were performed at the Chemistry Department II, Copenhagen University, Denmark and Atlantic Microlab, Inc., Norcross, Georgia, USA. Cyclic Voltammetry (CV) was recorded in acetonitrile solution under dry anaerobic conditions using an Autolab system (Eco Chemie, The Netherlands), controlled by the GPES software. The working electrode was a platinum disk, the auxiliary electrode a platinum wire and the reference electrode a Ag/Ag+ (0.01 M AgNO3). TBAClO4 (TBA = t-butylammonium) 0.1 M was used as electrolyte and all potentials are given versus the ferrocene/ferrocenium (Fc0/+) redox couple (E1/2 = 88 mV vs. Ag/Ag+, ΔE = 75–80 mV). Resonance Raman spectra were measured with a commercially available confocal scanning Raman microscope (Alpha300R) using linearly polarized illumination at the wavelength of 532 nm, 600 lines per mm diffraction grating, and ×100 objective (N.A. = 0.90).
CAUTION! Although we encountered no problems during preparation of the perchlorate salts, perchlorate salts of metal complexes are potentially explosive and should be handled with caution in small quantities.
Synthesis
2,6-Bis[bis(2-pyridylmethyl)aminomethyl]-4-tert-butylphenol (Hbpbp) was prepared as described previously.31
[Co2(bpbp)(O2)(CH3COO)](BF4)2·H2O (1-oxy(BF4)2·H2O).
Hbpbp (708.3 mg, 1.24 mmol) dissolved in 24 mL acetone and Co(BF4)2·6H2O (843.7 mg, 2.48 mmol) dissolved in 6 mL H2O were mixed giving a light brown solution. NaO2CCH3 (101.7 mg, 1.24 mmol) dissolved in 3 mL H2O was added, whereby the solution changed to a dark brown color. Slow evaporation in air at room temperature yielded black crystals overnight. These were isolated by filtration, washed with small amounts of H2O and dried in vacuo overnight. Yield 1057.1 mg (88%). Anal. Calcd for C38H42B2Co2F8N6O5·H2O: C, 46.94; H, 4.56; N, 8.64. Found: C, 46.99; H, 4.39; N, 8.55. ESI-MS (CH3CN, mild source conditions, Finnigan): m/z 374.3 (100%, [Co2(bpbp)(CH3COO)]2+), 390.2 (50%, [Co2(bpbp)(O2)(CH3COO)]2+), 394.8 (30%, [Co2(bpbp)(CH3COO)(NCCH3)]2+), 835.5 (40%, [Co2(bpbp)(CH3COO)(BF4)]+). UV-Vis (CH3CN) λmax/nm (ε/M−1 cm−1): 307 (8700, sh); 350 (7700); 475 (4450, sh); 700 (400, sh).
[Co2(bpbp)(O2)(CH2ClCOO)](BF4)2·2.25H2O (2-oxy(BF4)2·2.25H2O).
The same procedure was followed as described for 1(BF4)2·H2O, using Hbpbp (693.9 mg, 1.21 mmol), Co(BF4)2·6H2O (826.1 mg, 2.43 mmol), and NaO2CH2Cl (141.5 mg, 1.21 mmol). Yield 1154.2 mg (93%). Anal. Calcd for C38H41B2ClCo2F8N6O5·2H2O: C, 44.54; H, 4.43; N, 8.20. Found: C, 44.87; H, 4.29; N, 8.14. ESI-MS (CH3CN, mild source conditions, Finnigan): m/z 391.3 (90%, [Co2(bpbp)(CH2ClCOO)]2+), 407.4 (50%, [Co2(bpbp)(O2)(CH2ClCOO)]2+), 411.8 (30%, [Co2(bpbp)(CH2ClCOO)(NCCH3)]2+), 875.7 (40%, [Co2(bpbp)(CH2ClCOO)2]+). UV-Vis (CH3CN) λmax/nm (ε/M−1 cm−1): 305 (8000, sh); 355 (7000); 470 (4350, sh); 720 (360, sh).
[Co2(bpbp)(O2)(CHCl2COO)](BF4)2·1.25H2O (3-oxy(BF4)2·1.25H2O).
Hbpbp (101.3 mg, 0.18 mmol) dissolved in 5 mL acetone and Co(BF4)2·6H2O (118.9 mg, 0.35 mmol) dissolved in 1 mL H2O were mixed giving a light brown solution. A mixture of Cl2CHCOOH (14.5 μL, 0.18 mmol) in ½ mL water adjusted to pH 7 using 4 M NaOH, was added. The solution turned dark brown. Slow evaporation of the solvent under 100% O2 atmosphere gave dark brown crystals overnight. The product was isolated by filtration, washed with small amounts of H2O and dried in vacuo. Yield 160.6 mg (86%). Anal. Calcd for C38H40B2Cl2Co2F8N6O5·H2O: C, 43.84; H, 4.07; N, 8.07. Found: C, 44.33; H, 4.00; N, 7.81. ESI-MS (CH3CN, mild source conditions, Finnigan): m/z 408.3 (100%, [Co2(bpbp)(CHCl2COO)]2+), 424.4 (50%, [Co2(bpbp)(O2)(CHCl2COO)]2+), 428.8 (100%, [Co2(bpbp)(CHCl2COO)(NCCH3)]2+), 903.5 (100%, [Co2(bpbp)(CHCl2COO)(BF4)]+), 945.7 (40%%, [Co2(bpbp)(CHCl2COO)2]+). UV-Vis (CH3CN) λmax/nm (ε/M−1 cm−1): 365 (5100); 470 (3300, sh); 720 (300, sh).
[Co2(bpbp)(O2)(CCl3COO)](BF4)2·2H2O (4-oxy(BF4)2·2H2O).
Hbpbp (100.4 mg, 0.18 mmol) dissolved in 3 mL acetone and Co(BF4)2·6H2O (119.3 mg, 0.36 mmol) dissolved in ½ mL H2O were mixed giving a light brown solution. Cl3CCO2H (28.9 mg, 0.18 mmol) dissolved in 0.25 mL H2O was added 4 M NaOH until pH 7 before being added to the solution giving a dark brown solution. Slow solvent evaporation at −30 °C under an O2 atmosphere yielded dark brown crystals over 4–6 days. The product was isolated by filtration, washed with small amounts of H2O and dried in a desiccator (CaCl2) for 2 days at ambient pressure. Yield 137.4 mg (74%). Anal. Calcd for C38H39B2Cl3Co2F8N6O5·2H2O: C, 41.73; H, 3.96; N, 7.68. Found: C, 41.30; H, 3.70; N, 7.30. ESI-MS (CH3CN, mild source conditions, Finnigan): m/z 362.3 (60%, [Co2(bpbp)Cl]2+), 426.0 (100%, [Co2(bpbp)(CCl3COO)]2+), 951.5 (50%, [Co2(bpbp)(CCl3COO)(BF4)]+), 1013.6 (20%, [Co2(bpbp)(CCl3COO)2]+). UV-Vis (CH3CN) λmax/nm (ε/M−1 cm−1): 370 (9400); 475 (62 000, sh); 720 (500, sh). The crystal structure of 4 was obtained from the perchlorate salt 4·2ClO4 which was obtained by a similar procedure except for the use of Co(ClO4)2·6H2O as starting material.
[Co2(bpbp)(O2)(O2C-napht)](ClO4)2 (5-oxy(ClO4)2).
The same procedure was followed as described for 1(BF4)2·H2O, using Hbpbp (101.7 mg, 0.18 mmol), Co(ClO4)2·6H2O (130.5 mg, 0.36 mmol), and naphthalene-1-carboxylic acid, HO2C-napht, (30.5 mg, 0.18 mmol) which was converted into the sodium salt by adding 1.8 mL 0.1 M NaOH to the acid dissolved in 2 mL acetone. To induce crystallisation ∼100 mg NaClO4 was added to the solution and slow evaporation yielded black hexagonal crystals. Yield 156.0 mg (71%). Anal. Calcd for C47H46Cl2Co2N6O13·C3H6O·½H2O: C, 51.83; H, 4.61; N, 7.25. Found: C, 51.73; H, 4.42; N, 7.29. ESI-MS (CH3CN): m/z 430.1 (100%, [Co2(bpbp)(O2C-napht)]2+), 515.6 (40%, [Co2(bpbp)(O2C-napht)2]2+), 759.1 (60%, [Co2(bpbp)Cl2]+), 895.1 (60%, [Co2(bpbp)(O2C-napht)Cl]+), 1031.2 (60%, [Co2(bpbp)(O2C-napht)2]+).
Single-crystal X-ray diffraction
Selected crystallographic data are presented in Table 4. Diffraction data were collected using a Bruker-Nonius X8 APEX-II instrument (Mo-Kα radiation). Structure solution and refinement was carried out using SHELXTL.33 H atoms on C atoms were placed at calculated positions and allowed to ride during subsequent refinement. H atoms of water molecules were either located in difference Fourier maps and their positions refined with restrained O–H distances, or placed to form reasonable H-bonds and subsequently allowed to ride on the O atom. Complexes 2-oxy and 3-oxy are isostructural. In both cases, one of the two distinct BF4− anions exhibits rotational disorder, which was modelled as two anion orientations with restrained geometry and relatively large displacement ellipsoids for F. The structure of 2-oxy contains three water molecule sites, two of which are fully occupied, and one of which is modelled with site occupancy 0.25 in order to provide a reasonable displacement ellipsoid. The structure of 3-oxy appears to contain water molecules in only two of these three sites, but including the site with 0.25 site occupancy. Thus, the water content in 2-oxy and 3-oxy appears to be less than or equal to 3 H2O. Both 4-oxy and 5-oxy contain large voids with diffuse solvent that could not be modelled effectively using discrete atom sites. Therefore, the structures of 4-oxy and 5-oxy are based on data processed by the SQUEEZE routine.32 For 4-oxy, SQUEEZE identifies one void of ca. 1575 Å3 per unit cell and suggests that it is occupied by ca. 700 electrons. For 5-oxy, there are four equivalent voids of ca. 270 Å3 per unit cell, each of which is occupied by ca. 130 electrons. In both structures, the solvent content could comprise water and/or acetone. The application of SQUEEZE did not change the values of the peroxide O–O bonds significantly. The perchlorate anions are rotationally disordered and the anion geometry was restrained to be a regular tetrahedron. In 4-oxy, relatively large residual electron density remains in the vicinity of one perchlorate anion, indicative of residual rotational disorder not accounted for by the model. In 5-oxy, there are two complexes in the crystallographic asymmetric unit, related by local inversion pseudosymmetry.
| |
1-oxy
|
2-oxy
|
3-oxy
|
4-oxy
|
5-oxy
|
|
Structure at 298 K: CCDC 913564.
Refinement values based on data after application of SQUEEZE.32Solvent content is unknown.
|
| Formula |
[C38H42Co2N6O5]2+·2(BF4)·H2O |
[C38H41ClCo2N6O5]2+·2(BF4)·2.25H2O |
[C38H40Cl2Co2N6O5]2+·2(BF4)·1.25H2O |
[C38H39Cl3Co2N6O5]2+·2(ClO4) |
[C47H46Co2N6O5]2+·2(ClO4) |
|
M/g mol−1 |
972.27 |
1029.23 |
1045.66 |
1082.86 |
1091.66 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
|
a/Å |
12.0799(2) |
10.6177(4) |
10.6153(12) |
12.340(2) |
26.3433(11) |
|
b/Å |
21.2024(4) |
11.6326(4) |
11.3123(15) |
16.437(3) |
18.4942(7) |
|
c/Å |
15.9892(3) |
18.3035(6) |
18.840(2) |
18.524(3) |
21.7739(10) |
|
α/° |
90 |
82.523(1) |
81.346(5) |
77.664(6) |
90 |
|
β/° |
91.633(1) |
84.474(1) |
83.427(5) |
88.953(6) |
109.509(2) |
|
γ/° |
90 |
74.401(1) |
75.393(4) |
73.611(6) |
90 |
|
V/Å3 |
4093.54(13) |
2154.46(13) |
2157.4(4) |
3517.8(10) |
9999.2(7) |
|
Z
|
4 |
2 |
2 |
2 |
8 |
|
D
c/g cm−3 |
1.578 |
1.587 |
1.610 |
1.022 |
1.450 |
|
μ(Mo Kα)/mm−1 |
0.901 |
0.923 |
0.981 |
0.706 |
0.839 |
| Crystal size/mm |
0.25 × 0.20 × 0.06 |
0.25 × 0.25 × 0.20 |
0.16 × 0.08 × 0.03 |
0.25 × 0.25 × 0.20 |
0.30 × 0.24 × 0.10 |
| Crystal colour |
Brown |
Brown |
Brown |
Brown |
Brown |
|
θ Range/° |
3.56–25.67 |
3.66–28.40 |
3.58–25.29 |
3.52–25.03 |
3.52–25.74 |
| Refl. collected |
23478 |
48901 |
19324 |
59425 |
192465 |
| Unique refl. |
8586 |
10577 |
7445 |
12127 |
18985 |
|
R
int
|
0.035 |
0.031 |
0.072 |
0.106 |
0.083 |
| Observed refl. [I > 2σ(I)] |
6563 |
8437 |
4197 |
6429 |
12708 |
| No. of parameters |
567 |
623 |
623 |
581 |
1289 |
| No. of restraints |
3 |
102 |
102 |
20 |
49 |
|
R
1 [I > 2σ(I)] |
0.039 |
0.044 |
0.054 |
0.074 |
0.056 |
| wR2 (all data) |
0.102 |
0.124 |
0.125 |
0.203 |
0.149 |
| GOF on F2 |
1.03 |
1.04 |
1.01 |
0.95 |
1.05 |
| CCDC |
913563
|
913565
|
913566
|
913567
|
913568
|
Computational details
Density functional theory (DFT) calculations were performed with the Gaussian09 software package and the functional TPSSh by Tao, Perdew, Staroverov and Scuseria.34,35 The SDD basis set36 (Stuttgart/Dresden ECP) was used for Co and the TZVP37 basis set on the peroxido ligand and for the calculations on molecular dioxygen. For all other atoms, the split-valence basis set of Ahlrich and co-workers38 was applied with polarisation functions taken from the turbomole basis set library.39 All structures were optimised in the gas phase and were confirmed to be minima on their potential energy surfaces by calculation of their vibrational frequencies. Solvent effects were estimated through single point calculations on the optimised gas phase structures with a continues polarisation model40 as implemented in Gaussian09.
Acknowledgements
We thank the Danish Councils for Independent Research (FTP and FNU) for financial support.
Notes and references
- B. M. Hoffman and D. H. Petering, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 637–643 CrossRef CAS.
- C. Floriani and F. Calderaz, J. Chem. Soc. A, 1969, 946–953 RSC.
- B. Ramdhanie, J. Telser, A. Caneschi, L. N. Zakharov, A. L. Rheingold and D. P. Goldberg, J. Am. Chem. Soc., 2004, 126, 2515–2525 CrossRef CAS.
- R. Guilard, F. Jerome, C. P. Gros, J. M. Barbe, Z. P. Ou, J. G. Shao and K. M. Kadish, C. R. Acad. Sci., Ser. IIc: Chim., 2001, 4, 245–254 CrossRef CAS.
- J. P. Collman, Acc. Chem. Res., 1977, 10, 265–272 CrossRef CAS.
- N. Herron, Inorg. Chem., 1986, 25, 4714–4717 CrossRef CAS.
- H. Nishide, M. Kuwahara, M. Ohyanagi, Y. Funada, H. Kawakami and E. Tsuchida, Chem. Lett., 1986, 43–46 CrossRef CAS.
- H. Nishide, M. Ohyanagi, O. Okada and E. Tsuchida, Macromolecules, 1986, 19, 494–496 CrossRef CAS.
- E. Tsuchida, H. Nishide, M. Ohyanagi and H. Kawakami, Macromolecules, 1987, 20, 1907–1912 CrossRef CAS.
- M. Suzuki, H. Furutachi and H. Okawa, Coord. Chem. Rev., 2000, 200, 105–129 CrossRef.
- M. Suzuki, H. Kanatomi and I. Murase, Chem. Lett., 1981, 1745–1748 CrossRef CAS.
- M. Suzuki, I. Ueda, H. Kanatomi and I. Murase, Chem. Lett., 1983, 185–188 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS.
- A. C. Sharma and A. S. Borovik, J. Am. Chem. Soc., 2000, 122, 8946–8955 CrossRef CAS.
- N. Preethi, H. Shinohara and H. Nishide, React. Funct. Polym., 2006, 66, 851–855 CrossRef CAS.
- M. Shoji, K. Oyaizu and H. Nishide, Polymer, 2008, 49, 5659–5664 CrossRef CAS.
- R. K. Seidler-Egdal, F. B. Johansson, S. Veltze, E. M. Skou, A. D. Bond and C. J. McKenzie, Dalton Trans., 2011, 40, 3336–3345 RSC.
- M. Ghiladi, J. T. Gomez, A. Hazell, P. Kofod, J. Lumtscher and C. J. McKenzie, Dalton Trans., 2003, 1320–1325 RSC.
- T. B. Freedman, C. M. Yoshida and T. M. Loehr, J. Chem. Soc., Chem. Commun., 1974, 1016–1017 RSC.
- C. G. Barraclough, G. A. Lawrance and P. A. Lay, Inorg. Chem., 1978, 17, 3317–3322 CrossRef CAS.
- T. Kayatani, Y. Hayashi, M. Suzuki and A. Uehara, Bull. Chem. Soc. Jpn., 1994, 67, 2980–2989 CrossRef CAS.
- H. Sugimoto, T. Nagayama, S. Maruyama, S. Fujinami, Y. Yasuda, M. Suzuki and A. Uehara, Bull. Chem. Soc. Jpn., 1998, 71, 2267–2279 CrossRef CAS.
- M. Suzuki, H. Kanatomi and I. Murase, Bull. Chem. Soc. Jpn., 1984, 57, 36–42 CrossRef CAS.
- M. Suzuki, T. Sugisawa and A. Uehara, Bull. Chem. Soc. Jpn., 1990, 63, 1115–1120 CrossRef CAS.
- T. J. Beugelsdijk and R. S. Drago, J. Am. Chem. Soc., 1975, 97, 6466–6472 CrossRef CAS.
- N. J. Rose and R. S. Drago, J. Am. Chem. Soc., 1959, 81, 6138–6141 CrossRef CAS.
- T. Shibata, S. Nagao, M. Fukaya, H. Tai, S. Nagatomo, K. Morihashi, T. Matsuo, S. Hirota, A. Suzuki, K. Imai and Y. Yamamoto, J. Am. Chem. Soc., 2010, 132, 6091–6098 CrossRef CAS.
- R. K. Egdal, A. D. Bond and C. J. McKenzie, Daltons Trans., 2009, 3833–3839 RSC.
- F. B. Larsen, A. Boisen, K. J. Berry, B. Moubaraki, K. S. Murray, V. McKee, R. C. Scarrow and C. J. McKenzie, Eur. J. Inorg. Chem., 2006, 3841–3852 CrossRef CAS.
- M. Ghiladi, K. B. Jensen, J. Jiang, C. J. McKenzie, S. Morup, I. Sotofte and J. Ulstrup, J. Chem. Soc., Dalton Trans, 1999, 2675–2681 RSC.
- M. Ghiladi, C. J. McKenzie, A. Meier, A. K. Powell, J. Ulstrup and S. Wocadlo, J. Chem. Soc., Dalton Trans., 1997, 4011–4018 RSC.
- P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194–201 CrossRef.
-
G. M. Sheldrick, SHELXTL, version 6.10, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, J. Chem. Phys., 2004, 121, 11507–11507 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
-
http://www.rz.uni-karlsruhe.de/~cd177/basissets.php
.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- P. D. Southon, D. J. Price, P. K. Nielsen, C. J. McKenzie and C. J. Kepert, J. Am. Chem. Soc., 2011, 133, 10885–10891 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a