 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of vanadium(V) hydrazido complexes with tris(2-hydroxyphenyl)amine ligands†
Toshiyuki
Moriuchi
*a,
Kousuke
Ikeuchi
a and
Toshikazu
Hirao
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: moriuchi@chem.eng.osaka-u.ac.jp; hirao@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7415; Tel: +81-6-6879-7413
bJST, ACT-C, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 12th April 2013
Abstract
The reaction of the oxidovanadium(V) complexes with N,N-dimethylhydrazine was demonstrated to afford the corresponding vanadium(V) dimethylhydrazido complexes. The substituent at the 3-position of the tris(2-hydroxyphenyl)amine ligand was found to influence the electronic environment of the vanadium center. The crystal structure of the non-substituted vanadium(V) dimethylhydrazido complex exhibited a distorted trigonal bipyramidal geometry with phenolate oxygen atoms in equatorial positions and the near-linear V(1)–N(2)–N(3) angle. The vanadium(V) diphenylhydrazido complexes could be obtained by the reaction of the oxidovanadium(V) complexes with N,N-diphenylhydrazine. A distorted trigonal bipyramidal geometry with phenolate oxygen atoms in equatorial positions was also observed in the crystal structure of the non-substituted vanadium(V) diphenylhydrazido complex.
Introduction
Much attention has been devoted to metal hydrazido compounds due to their intermediacy in dinitrogen activation.1 In particular, most of the studies have focused on molybdenum hydrazido compounds because molybdenum is present in an active site of nitrogenases.2 On the other hand, the biochemical roles of vanadium have gained growing interest which is related to the insulinomimetic ability of vanadium compounds3 and vanadium-dependent haloperoxidases.4 Although vanadium is known to be an essential constituent in certain nitrogenases,1a–d,5 vanadium hydrazido complexes have attracted less attention.6 We have already demonstrated that the substituents in the para position on the benzene rings of (arylimido)vanadium(V) complexes affect the nature of the imido bonds through π conjugation.7 The coordination from the apical nitrogen in the (arylimido)vanadium(V) triethanolaminates was also performed to influence the electron donation from the imido nitrogen to vanadium metal.8 The substituent effect of the basal ligand in the vanadium(V) hydrazido complex has not been investigated so far. From these points of view, we herein report the synthesis of the vanadium(V) hydrazido complexes with tris(2-hydroxyphenyl)amine ligands to reveal the influence of the apical nitrogen and the substituent of the tris(2-hydroxyphenyl)amine ligand on the vanadium center.Results and discussion
The substituted tris(2-hydroxyphenyl)amines 2 were prepared by the deprotection of methoxy groups of the ligand precursors 1, which were synthesized by Ullmann-type reaction of the substituted 2-methoxyaniline with 1-iodo-2-methoxybenzene. The reaction of VO(OiPr)3 with the substituted tris(2-hydroxyphenyl)amines 2 in dichloromethane at room temperature afforded the corresponding oxidovanadium(V) complexes 3 as shown in Scheme 1.9 The vanadium(V) dimethylhydrazido complexes 4 with the tris(2-hydroxyphenyl)amine ligand could be obtained by the treatment of the corresponding oxidovanadium(V) complexes 3 with N,N-dimethylhydrazine in dichloromethane at room temperature. 51V NMR measurements were performed in order to clarify the substituent effect on the electronic environment of the vanadium species. The 51V chemical shift of the non-substituted vanadium(V) dimethylhydrazido complex 4b was detected at 335 ppm. In the 51V NMR spectrum of 4a with the electron-donating substituent, the 51V chemical shift was observed at 328 ppm. In contrast, the electron-withdrawing substituent caused the 51V chemical shift to higher frequency (4c: 381 ppm; 4d: 378 ppm). The substituent at the 3-position of the tris(2-hydroxyphenyl)amine ligand was found to influence the electron density of the vanadium center directly. A similar substituent effect was observed in the vanadium(V) diphenylhydrazido complexes 5 (5a: 164 ppm; 5b: 178 ppm; 5c: 206 ppm; 5d: 209 ppm), which were prepared by the reaction of the corresponding oxidovanadium(V) complexes 3 with N,N-diphenylhydrazine. 51V chemical shifts of 5 were observed at the lower frequency compared to those of 4. The electron-withdrawing diphenyl substituent, in which the nitrogen atom of the imido bond becomes more electronegative to increase 51V nuclear shielding, is considered to cause the 51V chemical shift to the lower frequency. These results are consistent with those of the (arylimido)vanadium(V) trichlorides reported.7,8,10 Structural characterization of the vanadium(V) dimethylhydrazido complex 4b was demonstrated by single-crystal X-ray structure determination to elucidate the influence of the coordination from the apical nitrogen on the vanadium center in the hydrazido structure (Table 1). The important bond lengths and angles are listed in Table 2. The crystal structure of 4b, in which two independent molecules exist in the asymmetric unit, exhibited the near-linear V(1)–N(2)–N(3) angles of 176.60(17) and 173.11(16)° with the V(1)–N(2) distances of 1.6891(19) and 1.6837(19) Å as shown in Fig. 1, suggesting the high participation of an sp-hybridized character in the nitrogen of the imido bond. The geometry at the metal center is a distorted trigonal bipyramidal geometry with the phenolate oxygen atoms in equatorial positions. The vanadium atom is pulled out of the plane formed by the phenolate oxygen atoms in the direction of the hydrazido nitrogen. Three equatorial vanadium–oxygen bond distances are statistically identical. The V(1)–N(2) distances of 4b with the coordination from the apical nitrogen is longer than that of the vanadium(V) dimethylhydrazido complex, [V(NNMe2)(OC6H3iPr2-2,6)3], with tetrahedral geometry at the vanadium center (V(1)–N(2), 1.653(3) Å),6d probably due to the coordination of the apical nitrogen. The electron donation from N(2) to vanadium metal might be weakened to some extent.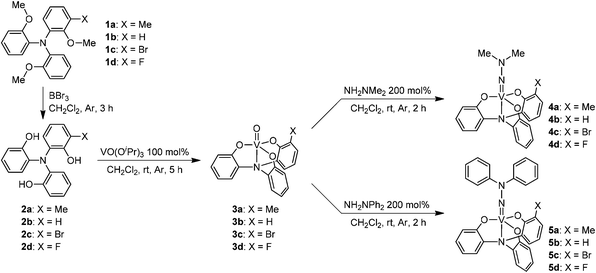 | ||
| Scheme 1 Synthesis of the vanadium(V) hydrazido complexes 4 and 5. | ||
 | ||
| Fig. 1 Molecular structure of 4b. | ||
| 4b | 4d-DMH-4d | 5b | |
|---|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|. b wR2 = [Σw(Fo2| − |Fc2|)2/Σw(Fo2)2]1/2. | |||
| Formula | C20H18N3O3V | C42H42N8O6F2V2 | C30H22N3O3V |
| Formula weight | 399.31 | 894.72 | 523.45 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n (no. 14) | P21 (no.4) | P21/n (no. 14) |
| a (Å) | 16.2235(5) | 9.3304(11) | 20.7200(15) |
| b (Å) | 11.8240(3) | 18.403(2) | 9.1557(6) |
| c (Å) | 18.6632(5) | 12.1985(14) | 25.6467(18) |
| β (°) | 96.4640(10) | 102.631(3) | 99.487(2) |
| V (Å3) | 3557.32(18) | 2043.9(4) | 4798.8(6) |
| Z | 8 | 2 | 8 |
| D calcd (g cm−3) | 1.491 | 1.454 | 1.449 |
| T (°C) | −170 | −150 | −150 |
| λ(Mo Kα) (Å) | 0.71075 | 0.71075 | 0.71075 |
| R 1 | 0.046 | 0.074 | 0.066 |
| wR2b | 0.117 | 0.180 | 0.177 |
| 4b | 5b | |||
|---|---|---|---|---|
| a Two independent molecules exist in the asymmetric unit. | ||||
| Bond lengths | ||||
| V1–N1 | 2.2751(18) | 2.2782(18) | 2.255(4) | 2.268(4) |
| V1–N2 | 1.6891(19) | 1.6837(19) | 1.683(4) | 1.692(4) |
| V1–O1 | 1.8685(16) | 1.8572(16) | 1.841 (3) | 1.830(3) |
| V1–O2 | 1.8536(16) | 1.8548(16) | 1.845(3) | 1.852(3) |
| V1–O3 | 1.8538(15) | 1.8574(16) | 1.848(3) | 1.836(3) |
| N2–N3 | 1.304(3) | 1.312(3) | 1.315(5) | 1.316(5) |
| Bond angles | ||||
| V1–N2–N3 | 176.60(17) | 173.11(16) | 173.7(4) | 161.8(3) |
| N1–V1–N2 | 178.88(8) | 177.02(8) | 175.63(18) | 174.57(17) |
| N1–V1–O1 | 81.03(7) | 81.18(7) | 81.08(14) | 80.12(14) |
| N1–V1–O2 | 80.92(7) | 80.40(7) | 80.18(14) | 80.95(14) |
| N1–V1–O3 | 81.02(6) | 81.35(6) | 81.52(14) | 81.81(14) |
| N2–V1–O1 | 99.00(8) | 96.69(8) | 102.33(18) | 95.39(17) |
| N2–V1–O2 | 98.09(8) | 99.04(8) | 95.59(17) | 98.56(17) |
| N2–V1–O3 | 99.95(8) | 101.42(8) | 99.42(17) | 103.30(17) |
| O1–V1–O2 | 117.35(7) | 121.21(7) | 123.79(16) | 117.08(15) |
| O1–V1–O3 | 116.25(7) | 111.48(8) | 113.74(15) | 123.42(16) |
| O2–V1–O3 | 119.17(7) | 120.00(7) | 115.06(16) | 112.16(16) |
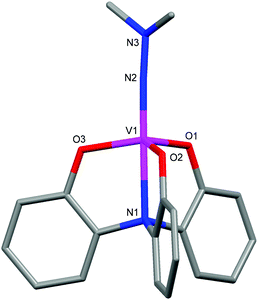
The N,N-dimethylhydrazine-bridged dinuclear vanadium(V) hydrazido complex 4d-DMH-4d could be obtained by recrystallization of 4d in the presence of N,N-dimethylhydrazine. The structure of 4d-DMH-4d was confirmed by single-crystal X-ray structure determination. Table 3 shows the selected bond lengths and angles. The solid-state molecular structure reveals that two 4d units are bridged by N,N-dimethylhydrazine to form the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex in anti-configuration as depicted in Fig. 2. The 3-fluoro group on the benzene ring might decrease the electron density of the vanadium center, resulting in the coordination of N,N-dimethylhydrazine to the vanadium center to form a hexacoordinate geometry. This hydrazido complex 4d-DMH-4d showed the near-linear V(1)–N(2)–N(3) angles of 171.8(7) and 174.0(7)° with the V(1)–N(2) distances of 1.663(7) and 1.690(7) Å, respectively. The V(1)–N(7) distance (2.349(7) Å) is longer than that of the V(2)–N(8) bond (2.245(7) Å) probably due to the steric hindrance of the dimethyl moieties.
:1 complex in anti-configuration as depicted in Fig. 2. The 3-fluoro group on the benzene ring might decrease the electron density of the vanadium center, resulting in the coordination of N,N-dimethylhydrazine to the vanadium center to form a hexacoordinate geometry. This hydrazido complex 4d-DMH-4d showed the near-linear V(1)–N(2)–N(3) angles of 171.8(7) and 174.0(7)° with the V(1)–N(2) distances of 1.663(7) and 1.690(7) Å, respectively. The V(1)–N(7) distance (2.349(7) Å) is longer than that of the V(2)–N(8) bond (2.245(7) Å) probably due to the steric hindrance of the dimethyl moieties.
 | ||
| Fig. 2 Molecular structure of 4d-DMH-4d. | ||
| Bond lengths | |||
| V1–N1 | 2.295(7) | V2–N4 | 2.270(7) |
| V1–N2 | 1.663(7) | V2–N5 | 1.690(7) |
| V1–N7 | 2.349(7) | V2–N8 | 2.245(7) |
| V1–O1 | 1.903(6) | V2–O4 | 1.913(6) |
| V1–O2 | 1.902(6) | V2–O5 | 1.895(6) |
| V1–O3 | 1.862(6) | V2–O6 | 1.910(6) |
| N2–N3 | 1.315(10) | N5–N6 | 1.300(10) |
| N7–N8 | 1.512(9) | ||
| Bond angles | |||
| V1–N2–N3 | 171.8(7) | V2–N5–N6 | 174.0(7) |
| V1–N7–N8 | 106.9(4) | V2–N8–N7 | 133.4(5) |
| N1–V1–N2 | 172.0(3) | N4–V2–N5 | 173.6(3) |
| N1–V1–N7 | 101.9(2) | N4–V2–N8 | 90.1(2) |
| N1–V1–O1 | 79.8(3) | N4–V2–O4 | 79.5(2) |
| N1–V1–O2 | 77.7(2) | N4–V2–O5 | 77.6(2) |
| N1–V1–O3 | 79.5(2) | N4–V2–O6 | 80.1(2) |
| N2–V1–N7 | 85.6(3) | N5–V2–N8 | 96.3(3) |
| N2–V1–O1 | 92.6(3) | N5–V2–O4 | 102.3(3) |
| N2–V1–O2 | 101.0(3) | N5–V2–O5 | 102.4(3) |
| N2–V1–O3 | 104.6(3) | N5–V2–O6 | 93.6(3) |
| N7–V1–O1 | 178.2(3) | N8–V2–O4 | 76.9(3) |
| N7–V1–O2 | 81.0(2) | N8–V2–Q5 | 85.3(3) |
| N7–V1–O3 | 82.3(3) | N8–V2–O6 | 168.6(3) |
| O1–V1–O2 | 98.9(3) | 04–V2–O5 | 150.9(2) |
| O1–V1–O3 | 98.6(3) | 04–V2–O6 | 95.4(3) |
| O2–V1–O3 | 148.2(2) | 05–V2–O6 | 98.2(2) |
The structure of the vanadium(V) diphenylhydrazido complex 5b was also confirmed by single-crystal X-ray structure determination. The important bond lengths and angles are shown in Table 2. As 5b crystallized in the space group P21/n with Z = 8, two independent molecules exist in the asymmetric unit. The metal centers are distorted trigonal bipyramidal geometries with phenolate oxygen atoms in equatorial positions as shown in Fig. 3. One molecule of 5b shows the V(1)–N(2)–N(3) angle of 173.7(4)° with the V(1)–N(2) distance of 1.683(4) Å, indicating the high participation of an sp-hybridized character in the nitrogen of the imido bond. This hydrazido structure is almost similar to that of 4b. The V(1)–N(2)–N(3) angle of 161.8(3)° with the V(1)–N(2) distance of 1.692(4) Å based on some contribution of an sp2-hybridized character in the nitrogen of the imido bond was observed in the crystal structure of another molecule as depicted in Fig. 3.
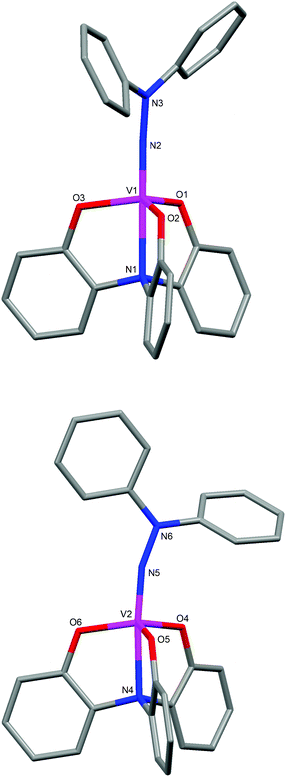 | ||
| Fig. 3 Molecular structures of two independent molecules in the asymmetric unit of 5b. | ||
Conclusions
In conclusion, the vanadium(V) hydrazido complexes with the tris(2-hydroxyphenyl)amine ligand were demonstrated to reveal the influence of the coordination from the apical nitrogen and the substituent of the tris(2-hydroxyphenyl)amine ligand on the vanadium center in solid and/or solution states. The coordination of the apical nitrogen to the vanadium center was performed to weaken the V–N multiple bond through the coordination from the apical nitrogen. The substituent at the 3-position of the tris(2-hydroxyphenyl)amine ligand was found to influence the electronic environment of the vanadium center directly. Further characterization of the hydrazido structures and the application of the vanadium(V) hydrazido complexes for catalysis are now in progress.Experimental
General methods
All reagents and solvents were purchased from commercial sources and were further purified by the standard methods, if necessary. All manipulations were carried out under nitrogen. Melting points were determined on a Yanagimoto Micromelting Point Apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded on a JEOL JNM-ECP 400 (400 and 100 MHz, respectively) spectrometer with tetramethylsilane as an internal standard. 51V NMR spectra were obtained with a JEOL JNM-ECP 400 (105 MHz) spectrometer with VOCl3 as an external standard. Mass spectra were run on a JEOL JMS DX-303 spectrometer.Synthesis of 2-methoxy-3-methyl-N,N-bis(2-methoxyphenyl)aniline (1a)
A mixture of 2-methoxy-3-methylaniline (549 mg, 4.0 mmol), 1-iodo-2-methoxybenzene (1.56 mL, 12.0 mmol), Cu (890 mg, 14.0 mmol), K2CO3 (2.76 g, 20.0 mmol) and 18-crown-6 (137 mg, 0.52 mmol) in 1,2-dichlorobenzene (50 mL) was refluxed under Ar for 24 h. After cooling, the insoluble inorganic material was filtered off. The solvent of the resulting filtrate was removed under reduced pressure and the residue was chromatographed on a silica-gel column (eluent, hexane–dichloromethane 1:1) to give the desired product 1a (572 mg, 1.64 mmol) as a white solid.
1a: yield 41%; mp 80–82 °C; 1H NMR (400 MHz, CDCl3) δ 7.04 (td, 2H, J = 7.3, 1.8 Hz), 6.89–6.78 (m, 8H), 6.65 (dd, 1H, J = 7.3, 2.2 Hz), 3.63 (s, 3H), 3.56 (s, 6H), 2.22 (s, 3H); 13C NMR (100 MHz, CDCl3) 153.9, 151.7, 141.7, 137.8, 132.1, 125.5, 125.2, 124.2, 123.4, 122.3, 121.0, 113.1, 58.8, 56.0, 16.4 ppm; HRMS (EI) m/z Calcd for C22H23N1O3 (M+), 349.1678; Found, 349.1677.
Synthesis of 3-bromo-2-methoxy-N,N-bis(2-methoxyphenyl)aniline (1c)
A mixture of 3-bromo-2-methoxyaniline (1.62 g, 8.0 mmol), 1-iodo-2-methoxybenzene (3.12 mL, 24.0 mmol), Cu (1.78 g, 28.0 mmol), K2CO3 (5.53 g, 40.0 mmol) and 18-crown-6 (275 mg, 1.04 mmol) in 1,2-dichlorobenzene (80 mL) was refluxed under Ar for 24 h. After cooling, the insoluble inorganic material was filtered off. The solvent of the resulting filtrate was removed under reduced pressure and the residue was chromatographed on a silica-gel column (eluent, hexane–ethyl acetate 5:1) to give the desired product 1c (1.39 g, 3.36 mmol) as a white solid.
1c: yield 42%; mp 94–95 °C; 1H NMR (400 MHz, CDCl3) δ 7.15 (dd, 1H, J = 7.6, 1.8 Hz), 7.04 (td, 2H, J = 7.8, 2.0 Hz), 6.89–6.81 (m, 6H), 6.74 (t, 1H, J = 7.6 Hz), 6.71 (dd, 1H, J = 8.0, 2.0 Hz), 3.65 (s, 3H), 3.57 (s, 6H); 13C NMR (100 MHz, CDCl3) 154.0, 150.1, 143.4, 137.1, 126.6, 126.0, 124.8, 124.6, 123.3, 121.1, 118.2, 113.0, 59.3, 55.9 ppm; HRMS (FAB) m/z Calcd for C21H20Br1N1O3 (M+), 413.0627; Found, 413.0631.
Synthesis of 3-fluoro-2-methoxy-N,N-bis(2-methoxyphenyl)aniline (1d)
A mixture of 3-fluoro-2-methoxyaniline (602 μL, 5.0 mmol), 1-iodo-2-methoxybenzene (1.95 mL, 15.0 mmol), Cu (1.05 g, 16.5 mmol), K2CO3 (3.46 g, 25.0 mmol) and 18-crown-6 (172 mg, 0.65 mmol) in 1,2-dichlorobenzene (60 mL) was refluxed under Ar for 24 h. After cooling, the insoluble inorganic material was filtered off. The solvent of the resulting filtrate was removed under reduced pressure and the residue was chromatographed on a silica-gel column (eluent, hexane–dichloromethane 1:1) to give the desired product 1d (1.3 g, 3.7 mmol) as a white solid.
1d: yield 74%; mp 107–108 °C; 1H NMR (400 MHz, CDCl3) δ 7.06 (td, 2H, J = 7.8, 1.8 Hz), 6.9–6.7 (m, 8H), 6.56 (dt, 1H, J = 8.2, 1.8 Hz), 3.59 (s, 6H), 3.52 (s, 3H); 13C NMR (100 MHz, CDCl3) 156.8 (d, 1JF–C = 244.4 Hz), 154.0, 143.5 (d, 3JF–C = 3.8 Hz), 141.1 (d, 2JF–C = 12.5 Hz) 137.4, 125.8, 124.7, 122.7 (d, 3JF–C = 9.6 Hz), 121.1, 119.3, 113.1, 110.5 (2JF–C = 19.2 Hz), 60.1 (d, 4JF–C = 3.8 Hz), 56.0 ppm; HRMS (EI) m/z Calcd for C21H20F1N1O3 (M+), 353.1427; Found, 353.1429.
Synthesis of 2-hydroxy-3-methyl-N,N-bis(2-hydroxyphenyl)aniline (2a)
To a dichloromethane (15 mL) solution of 2-methoxy-3-methyl-N,N-bis(2-methoxyphenyl)aniline (1a) (349 mg, 1.0 mmol) was added a 1 M BBr3 in dichloromethane (3.0 mL, 3.0 mmol) solution under Ar at −78 °C. The resulting mixture was stirred at −78 °C for a few minutes. The mixture was allowed to warm to room temperature and was stirred for 3 h. The resulting mixture was diluted with ethyl acetate and washed with water. After evaporation of the solution, the desired product 2a was isolated as a white solid (239 mg, 0.78 mmol) by reprecipitation from ethyl acetate and hexane.2a: yield 78%; mp 120–121 °C; 1H NMR (400 MHz, CDCl3) δ 7.11 (td, 2H, J = 8.0, 1.4 Hz), 7.01–6.77 (m, 9H), 5.33 (s, 1H), 5.15 (s, 2H), 2.24 (s, 3H); 13C NMR (100 MHz, CDCl3) 150.1, 148.6, 133.5, 132.8, 128.6, 126.4, 125.5, 123.3, 121.7, 120.8, 117.4, 16.1 ppm; HRMS (EI) m/z Calcd for C19H17N1O3 (M+), 307.1208; Found, 307.1210.
Synthesis of 3-bromo-2-hydroxy-N,N-bis(2-hydroxyphenyl)aniline (2c)
To a dichloromethane (15 mL) solution of 3-bromo-2-methoxy-N,N-bis(2-methoxyphenyl)aniline (1c) (207 mg, 0.5 mmol) was added a 1 M BBr3 in dichloromethane (2.0 mL, 2.0 mmol) solution under Ar at room temperature. The resulting mixture was stirred at room temperature for 3 h. The resulting mixture was diluted with ethyl acetate and washed with water. After evaporation of the solution, the desired product 2c was isolated as a white solid (80 mg, 0.21 mmol) by reprecipitation from ethyl acetate and hexane.2c: yield 43%; mp 138–139 °C; 1H NMR (400 MHz, CDCl3) δ 7.29 (dd, 1H, J = 8.2, 1.4 Hz), 7.12 (td, 2H, J = 8.0, 1.4 Hz), 6.98–6.85 (m, 7H), 6.72 (t, 1H J = 7.8 Hz) 5.52 (s, 1H), 5.47 (s, 2H); 13C NMR (100 MHz, CDCl3) 150.7, 147.0, 135.0, 133.5, 128.8, 127.1, 126.2, 124.7, 121.9, 121.3, 117.0 ppm; HRMS (FAB) m/z Calcd for C18H14Br1N1O3 (M+), 371.0157; Found, 371.0168.
Synthesis of 3-fluoro-2-hydroxy-N,N-bis(2-hydroxyphenyl)aniline (2d)
To a dichloromethane (15 mL) solution of 3-fluoro-2-methoxy-N,N-bis(2-methoxyphenyl)aniline (1d) (353 mg, 1.0 mmol) was added a 1 M BBr3 in dichloromethane (3.0 mL, 3.0 mmol) solution under Ar at −78 °C. The resulting mixture was stirred at −78 °C for a few minutes. The mixture was allowed to warm to room temperature and was stirred for 3 h. The resulting mixture was diluted with ethyl acetate and washed with water. After evaporation of the solution, the desired product 2d was isolated as a white solid (168 mg, 0.54 mmol) by reprecipitation from ethyl acetate and hexane.2d: yield 54%; mp 138–139 °C; 1H NMR (400 MHz, CDCl3) δ 7. 11 (td, 2H, J = 7.6, 1.6 Hz), 6.99–6.85 (m, 7H), 6.78 (td, 1H J = 8.2, 6.0 Hz), 6.72 (dt, 1H, J = 8.2, 1.4 Hz), 5.56 (s, 2H), 5.33 (s, 1H); 13C NMR (100 MHz, CDCl3) 152.2 (d, 1JF–C = 239.6 Hz), 150.6, 138.3 (d, 2JF–C = 15.3 Hz), 135.8 (d, 3JF–C = 2.9 Hz), 133.4, 127.1, 126.2, 121.4, 120.4 (d, 3JF–C = 8.6 Hz), 120.2, 117.1, 112.4 (2JF–C = 18.2 Hz) ppm; HRMS (EI) m/z Calcd for C18H14F1N1O3 (M+), 311.0958; Found, 311.0954.
Synthesis of oxidovanadium(V) complex 3a
A mixture of the tris(2-hydroxyphenyl)amine ligand 2a (154 mg, 0.50 mmol) and VO(OiPr)3 (118 μL, 0.50 mmol) was stirred in dichloromethane (15 mL) under Ar at room temperature for 3 h. After evaporation of the solution, the oxidovanadium(V) complex 3a was isolated as a blue-purple solid (122 mg, 0.33 mmol) by reprecipitation from dichloromethane and hexane.3a: yield 66%; 1H NMR (400 MHz, CDCl3) δ 7.72 (dd, 2H, J = 8.0, 1.4 Hz), 7.56 (d, 1H, J = 8.2 Hz), 7.22 (t, 2H, J = 7.8 Hz), 7.08 (d, 1H, J = 7.3 Hz), 6.93 (t, 2H, J = 7.8 Hz), 6.85 (t, 1H, J = 7.8 Hz), 6.59 (d, 2H, J = 7.8 Hz), 2.18 (s, 3H); 13C NMR (100 MHz, CDCl3) 164.2, 162.7, 140.8, 140.7, 131.8, 130.4, 127.5, 126.1, 124.8, 123.9, 123.7, 115.7, 16.7 ppm; 51V NMR (105 MHz, CDCl3) −292 ppm; HRMS (FAB) m/z Calcd for C19H14N1O4V1 (M+), 371.0362; Found, 371.0362.
Synthesis of oxidovanadium(V) complex 3c
A mixture of the tris(2-hydroxyphenyl)amine ligand 2c (186 mg, 0.50 mmol) and VO(OiPr)3 (118 μL, 0.50 mmol) was stirred in dichloromethane (15 mL) under Ar at room temperature for 3 h. After evaporation of the solution, the oxidovanadium(V) complex 3c was isolated as a blue-green solid (218 mg, 0.50 mmol) by reprecipitation from dichloromethane and hexane.3c: yield 100%; 1H NMR (400 MHz, CDCl3) δ 7.66 (m, 3H), 7.44 (dd, 1H, J = 8.2, 1.4 Hz), 7.23 (td, 2H, J = 1.8, 8.2 Hz), 6.93 (td, 2H, J = 7.8, 1.4 Hz), 6.80 (t, 1H, J = 8.2 Hz), 6.58 (dd, 2H, J = 8.0, 1.2 Hz); 13C NMR (100 MHz, CDCl3) 164.2, 140.9, 140.4, 137.5, 133.9, 130.6, 127.3, 127.1, 124.4, 124.2, 115.7 ppm; 51V NMR (105 MHz, CDCl3) −299 ppm; HRMS (FAB) m/z Calcd for C18H11Br1N1O4V1 (M+), 434.9311; Found, 434.9314.
Synthesis of oxidovanadium(V) complex 3d
A mixture of the tris(2-hydroxyphenyl)amine ligand 2d (156 mg, 0.50 mmol) and VO(OiPr)3 (118 μL, 0.50 mmol) was stirred in dichloromethane (15 mL) under Ar at room temperature for 3 h. After evaporation of the solution, the oxidovanadium(V) complex 3d was isolated as a blue-purple solid (184 mg, 0.49 mmol) by reprecipitation from dichloromethane and hexane.3d: yield 98%; 1H NMR (400 MHz, CDCl3) δ 7.68 (dd, 2H, J = 8.0, 1.4 Hz), 7.50 (d, 1H, J = 7.8 Hz), 7.21 (t, 2H, J = 8.7 Hz), 6.99 (t, 1H J = 8.7 Hz), 6.92 (td, 2H, J = 8.7 Hz), 6.85 (td, 1H, J = 8.2, 5.5 Hz), 6.59 (d, 2H, J = 8.2 Hz); 13C NMR (100 MHz, CDCl3) 164.3, 152.5 (d, 2JF–C = 11.5 Hz), 146.5 (d, 1JF–C = 251.1 Hz), 141.2, 139.7, 130.2, 127.1, 124.2, 123.0 (d, 3JF–C = 7.7 Hz), 122.4 (d, 3JF–C = 3.8 Hz), 117.1 (d, 2JF–C = 17.3 Hz), 115.4 ppm; 51V NMR (105 MHz, CDCl3) −304 ppm; HRMS (FAB) m/z Calcd for C18H11F1N1O4V1 (M+), 375.0112; Found, 375.0112.
Synthesis of vanadium(V) dimethylhydrazido complexes 4a–d
A mixture of the corresponding oxidovanadium(V) complex 3 (0.10 mmol), N,N-dimethylhydrazine (15 μL, 0.20 mmol) and a small amount of MS4A (4 Å molecular sieves) was stirred in dichloromethane (10 mL) under an atmosphere of nitrogen in a drybox at room temperature for 2 h. The resulting solution was filtered off, and the filtrate was evaporated under reduced pressure. The resulting red-brown solid was washed with hexane to give the desired vanadium(V) dimethylhydrazido complex 4.4a: yield 56%; 1H NMR (400 MHz, CDCl3) δ 7.57 (dd, 2H, J = 8.0, 1.4 Hz), 7.46 (d, 1H, J = 6.9 Hz), 7.09 (td, 2H, J = 7.8, 1.4 Hz), 6.75–6.69 (m, 4H), 6.67 (t, 1H, J = 7.8 Hz), 3.57 (s, 6H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3) 164.1, 162.8, 137.5, 136.8, 130.7, 129.6, 126.0, 125.9, 123.4, 120.2, 119.9, 117.1, 43.7, 16.1 ppm; 51V NMR (105 MHz, CDCl3) 328 ppm; HRMS (FAB) m/z Calcd for C21H20N3O3V1 (M+), 413.0944; Found, 413.0955.
4b: yield 71%; 1H NMR (400 MHz, CDCl3) δ 7.60 (dd, 3H, J = 8.1, 1.4 Hz), 7.14 (td, 3H, J = 8.1, 1.4 Hz), 6.76 (m, 6H), 3.58 (s, 6H); 13C NMR (100 MHz, CDCl3) 164.3, 137.3, 129.6, 125.8, 120.3, 117.1, 43.7 ppm; 51V NMR (105 MHz, CDCl3) 335 ppm; HRMS (FAB) m/z Calcd for C20H18N3O3V1 (M+), 399.0788; Found, 399.0805.
4c: yield 62%; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, 1H, J = 7.8 Hz), 7.48 (d, 2H, J = 7.8 Hz), 7.33 (d, 1H, 7.8 Hz), 7.08 (t, 2H, J = 7.6 Hz), 6.75–6.68 (m, 4H), 6.59 (t, 1H, J = 7.8), 3.56 (s, 6H); 13C NMR (100 MHz, CDCl3) 164.3, 161.9, 137.5, 137.0, 132.3, 129.2, 125.3, 124.6, 119.9, 119.8, 117.0, 110.6, 43.8 ppm; 51V NMR (105 MHz, CDCl3) 382 ppm; HRMS (FAB) m/z Calcd for C20H17Br1N3O3V1 (M+), 476.9893; Found, 476.9911.
4d: yield 53%; 1H NMR (400 MHz, CDCl3) δ 7.55 (d, 2H, J = 7.8 Hz), 7.35 (d, 1H, J = 8.2 Hz), 7.12 (td, 2H, J = 7.8, 1.4 Hz), 6.92 (td, 1H, J = 9.0, 1.4 Hz), 6.76–6.71 (m, 4H), 6.64 (td, 1H, J = 8.2, 5.5 Hz); 13C NMR (100 MHz, CDCl3) 164.3, 153.1 (d, 2JF–C = 13.4 Hz), 149.5 (d, 1JF–C = 247.2 Hz), 139.1 (d, 3JF–C = 3.8 Hz), 136.9, 129.6, 125.8, 121.0, 120.3, 118.8 (d, 3JF–C = 7.7 Hz), 117.1, 116.0 (d, 2JF–C = 18.2 Hz), 43.7 ppm; 51V NMR (105 MHz, CDCl3) 378 ppm; HRMS (FAB) m/z Calcd for C20H17F1N3O3V1 (M+), 417.0694; Found, 417.0700.
Synthesis of vanadium(V) diphenylhydrazido complexes 5a–d
A mixture of the corresponding oxidovanadium(V) complex 3 (0.10 mmol), N,N-diphenylhydrazine (33 μL, 0.20 mmol) and a small amount of MS4A (4 Å molecular sieves) was stirred in dichloromethane (10 mL) under an atmosphere of nitrogen in a drybox at room temperature for 2 h. The resulting solution was filtered off, and the filtrate was evaporated under reduced pressure. The resulting red-purple solid was washed with hexane to give the desired vanadium(V) diphenylhydrazido complex 5.5a: yield 68%; 1H NMR (400 MHz, CDCl3) δ 7.75 (dd, 4H, J = 8.0, 0.8 Hz), 7.66 (dd, 2H, J = 7.6, 1.2 Hz), 7.52 (d, 1H, J = 6.8 Hz), 7.50 (t, 4H, J = 7.6 Hz), 7.18 (td, 2H, J = 8.4, 2.0 Hz), 7.08 (d, 1H, J = 7.2 Hz), 6.82 (td, 2H, J = 8.0, 1.2 Hz), 6.76 (t, 1H, J = 7.6 Hz), 6.72 (dd, 2H, J = 8.4, 1.6 Hz); 13C NMR (100 MHz, CDCl3) 164.1, 149.4, 141.3, 137.8, 131.1, 130.1, 129.5, 129.4, 129.2, 126.8, 126.2, 123.6, 122.1, 120.9, 120.7, 120.1, 119.6, 118.0, 117.1, 16.2 ppm; 51V NMR (105 MHz, CDCl3) 164 ppm; HRMS (FAB) m/z Calcd for C31H24N3O3V1 (M+), 537.1257; Found, 537.1256.
5b: yield 68%; 1H NMR (400 MHz, CDCl3) δ 7.71 (dd, 4H, J = 8.7, 0.9 Hz), 7.66 (dd, 3H, J = 8.0, 1.4 Hz), 7.50 (t, 4H, J = 8.7 Hz), 7.31–7.15 (m, 5H), 7.17 (td, 3H, J = 8.2, 1.4 Hz), 6.83 (td, 3H, J = 7.8, 1.4 Hz), 6.73 (dd, 3H, J = 8.2, 1.4 Hz); 13C NMR (100 MHz, CDCl3) 117.2, 119.6, 120.1, 121.1, 126.2, 126.9, 129.2, 129.4, 130.2, 137.8, 141.2, 164.2 ppm; 51V NMR (105 MHz, CDCl3) 178 ppm; HRMS (FAB) m/z Calcd for C30H22N3O3V1 (M+), 523.1101; Found, 523.1099.
5c: yield 71%; 1H NMR (400 MHz, CDCl3) δ 7.77–7.75 (m, 3H), 7.62 (dd, 1H, J = 6.0, 1.6 Hz), 7.60 (dd, 1H, J = 6.4, 1.2 Hz), 7.52–7.47 (m, 3H), 7.31–7.16 (m, 8H), 6.98 (td, 1H, J = 8.0, 1.2 Hz), 6.81 (td, 2H, J = 8.0, 1.2 Hz), 6.73–6.71 (m, 2H); 13C NMR (100 MHz, CDCl3) 164.0, 149.3, 141.1, 138.5, 137.3, 133.3, 130.4, 129.6, 129.2, 127.1, 126.0, 125.4, 122.1, 121.4, 121.2, 120.2, 119.6, 117.2 ppm; 51V NMR (105 MHz, CDCl3) 206 ppm; HRMS (FAB) m/z Calcd for C30H21Br1N3O3V1 (M+), 601.0206; Found, 601.0223.
5d: yield 61%; 1H NMR (400 MHz, CDCl3) δ 7.71 (dd, 4H, J = 8.7, 0.9 Hz), 7.64 (dd, 2H, J = 8.2, 1.4 Hz), 7.50 (t, 4H, J = 7.8 Hz), 7.45 (d, 1H, J = 8.4 Hz), 7.39–7.17 (m, 7H), 6.84 (td, 2H, J = 7.8, 1.4 Hz), 6.78–6.71 (m, 3H); 13C NMR (100 MHz, CDCl3) 164.1, 152.7 (d, 2JF–C = 12.5 Hz), 149.3, 148.9 (d, 1JF–C = 249.2 Hz), 141.0, 139.4 (d, 3JF–C = 3.8 Hz), 137.2, 130.3, 129.2, 127.0, 126.0, 122.1, 121.6 (d, 3JF–C = 2.9 Hz), 119.6, 117.2, 116.6 (d, 2JF–C = 18.2 Hz) ppm; 51V NMR (105 MHz, CDCl3) 209 ppm; HRMS (FAB) m/z Calcd for C30H21F1N3O3V1 (M+), 541.1007; Found, 541.1004.
X-ray structure analysis
All measurements for 4d-DMH-4d and 5b were made on a Rigaku R-AXIS RAPID diffractometer using graphite monochromated Mo Kα radiation. All measurements for 4b were made on a Rigaku R-AXIS RAPID diffractometer using filtered Mo Kα radiation. The structures of 4b, 4d-DMH-4d and 5b were solved by direct methods and expanded using Fourier techniques. All calculations were performed using the Crystal Structure crystallographic software package except for the refinement, which was performed using SHELXL-97. The non-hydrogen atoms were refined anisotropically. The H atoms were placed in idealized positions and allowed to ride with the C atoms to which each was bonded. Crystallographic details are given in Table 1. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-925607 for 4b, CCDC-925608 for 4d-DMH-4d and CCDC-925609 for 5b.Acknowledgements
This work was supported partly by a Grant-in-Aid for Scientific Research ACT-C from JST, Japan. We thank Dr Hideki Sugimoto and Prof. Shinobu Itoh for the single-crystal X-ray structure determination, and Dr Masato Ohashi and Prof. Sensuke Ogoshi for its discussion. Thanks are due to the Analytical Center, Graduate School of Engineering, Osaka University.Notes and references
- (a) D. Sutton, Chem. Rev., 1993, 93, 995 CrossRef CAS; (b) M. Hidai and Y. Mizobe, Chem. Rev., 1995, 95, 1115 CrossRef CAS; (c) R. L. Richards, Coord. Chem. Rev., 1996, 154, 83 CrossRef CAS; (d) B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385 CrossRef CAS; (e) R. R. Schrock, Acc. Chem. Res., 2005, 38, 955 CrossRef CAS.
- (a) J. Kim and D. C. Rees, Science, 1992, 257, 1677 CAS; (b) B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983 CrossRef CAS.
- K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561 CrossRef CAS.
- (a) D. Rehder, Angew. Chem., Int. Ed. Engl., 1991, 30, 148 CrossRef; (b) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937 CrossRef CAS; (c) A. Butler, Coord. Chem. Rev., 1999, 187, 17 CrossRef CAS; (d) A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89 CrossRef CAS; (e) D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849 CrossRef CAS; (f) J. Hartung, Y. Dumont, M. Greb, D. Hach, F. Köhler, H. Schulz, M. Časný, D. Rehder and H. Vilter, Pure Appl. Chem., 2009, 81, 1251 CrossRef CAS; (g) K. Kikushima, T. Moriuchi and T. Hirao, Tetrahedron, 2010, 66, 6906 CrossRef CAS; (h) T. Moriuchi and T. Hirao, Bioinspired catalytic bromination systems for bromoperoxidase, in Vanadium: Biochemical and Molecular Biological Approaches, ed. H. Michibata, Springer-Verlag, Berlin, 2012, pp. 127–142 Search PubMed.
- (a) T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69 CrossRef CAS; (b) R. R. Eady, Chem. Rev., 1996, 96, 3013 CrossRef CAS; (c) D. Rehder, Coord. Chem. Rev., 1999, 182, 297 CrossRef.
- (a) N. Wiberg, H.-W. Häring, G. Huttner and P. Friedrich, Chem. Ber., 1978, 111, 2708 CrossRef CAS; (b) J. Bultitude, L. F. Larkworthy, D. C. Povey, G. W. Smith, J. R. Dilworth and G. J. Leigh, J. Chem. Soc., Chem. Commun., 1986, 1748 RSC; (c) S. C. Davies, D. L. Hughes, Z. Janas, L. Jerzykiewicz, R. L. Richards, J. R. Sanders and P. Sobota, Chem. Commun., 1997, 1261 RSC; (d) R. A. Henderson, Z. Janas, L. B. Jerzykiewicz, R. L. Richards and P. Sobota, Inorg. Chim. Acta, 1999, 285, 178 CrossRef CAS; (e) S. C. Davies, D. L. Hughes, Z. Janas, L. B. Jerzykiewicz, R. L. Richards, J. R. Sanders, J. E. Silverston and P. Sobota, Inorg. Chem., 2000, 39, 3485 CrossRef CAS; (f) S. C. Davies, D. L. Hughes, M. Konkol, R. L. Richards, J. R. Sanders and P. Sobota, J. Chem. Soc., Dalton Trans., 2002, 2811 RSC; (g) S. Banerjee and A. L. Odom, Dalton Trans., 2008, 2005 RSC.
- (a) T. Moriuchi, K. Ishino, T. Beppu, M. Nishina and T. Hirao, Inorg. Chem., 2008, 47, 7638 CrossRef CAS; (b) T. Moriuchi, M. Nishina and T. Hirao, Angew. Chem., Int. Ed., 2010, 49, 83 CrossRef CAS; (c) M. Nishina, T. Moriuchi and T. Hirao, Dalton Trans., 2010, 39, 9936 RSC; (d) T. Moriuchi and T. Hirao, Coord. Chem. Rev., 2011, 255, 2371 CrossRef CAS.
- (a) T. Moriuchi, K. Ishino and T. Hirao, Chem. Lett., 2007, 1486 CrossRef CAS; (b) T. Moriuchi, T. Beppu, K. Ishino, M. Nishina and T. Hirao, Eur. J. Inorg. Chem., 2008, 12, 1969 CrossRef; (c) M. Nishina, T. Moriuchi and T. Hirao, Bull. Chem. Soc. Jpn., 2012, 85, 606 CrossRef CAS.
- D. C. Crans, H. Chen, O. P. Anderson and M. M. Miller, J. Am. Chem. Soc., 1993, 115, 6769 CrossRef CAS.
- D. D. Devore, J. D. Lichtenhan, F. Takusagawa and E. A. Maatta, J. Am. Chem. Soc., 1987, 109, 7408 CrossRef CAS.
Footnote |
| † CCDC 925607–925609. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt50533b |
| This journal is © The Royal Society of Chemistry 2013 |