 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of vanadium haloperoxidases in the formation of volatile brominated compounds and their impact on the environment
Ron Wever* and Michael A. van der Horst
University of Amsterdam, Van't Hoff Institute for Molecular Sciences, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: r.wever@uva.nl
First published on 12th April 2013
Abstract
Vanadium haloperoxidases differ strongly from heme peroxidases in substrate specificity and stability and in contrast to a heme group they contain the bare metal oxide vanadate as a prosthetic group. These enzymes specifically oxidize halides in the presence of hydrogen peroxide into hypohalous acids. These reactive halogen intermediates will react rapidly and aspecifically with many organic molecules. Marine algae and diatoms containing these iodo- and bromoperoxidases produce short-lived brominated methanes (bromoform, CHBr3 and dibromomethane CH2Br2) or iodinated compounds. Some seas and oceans are supersaturated with these compounds and they form an important source of bromine to the troposphere and lower stratosphere and contribute significantly to the global budget of halogenated hydrocarbons. This perspective focuses, in particular, on the biosynthesis of these volatile compounds and the direct or indirect involvement of vanadium haloperoxidases in the production of huge amounts of bromoform and dibromomethane. Some of the global sources are discussed and from the literature a picture emerges in which oxidized brominated species generated by phytoplankton, seaweeds and cyanobacteria react with dissolved organic matter in seawater, resulting in the formation of intermediate brominated compounds. These compounds are unstable and decay via a haloform reaction to form an array of volatile brominated compounds of which bromoform is the major component followed by dibromomethane.
 Ron Wever and Michael A. van der Horst | Ron Wever (right) (Amsterdam, The Netherlands, 1947) received his PhD in 1975 at the University of Amsterdam. In 2001 he was nominated as a professor in Biocatalysis at the same university. His research was focussed on metalloproteins and their reactions with oxygen or oxygen derivatives. His group discovered the new class of enzymes in which vanadium is the prosthetic group, the vanadium bromo- and chloroperoxidases. These enzymes are still being studied in his group. More recently he became interested in enzymes that are able to phosphorylate or sulfate compounds with the aim to replace chemical synthesis methods by biocatalytic procedures. Michael Alois van der Horst (left) (Vienna, Austria, 1975) finished his chemistry studies at the University of Amsterdam in 1999. He received his PhD in Microbiology in 2004 working on bacterial signal transduction with Prof. K. J. Hellingwerf at the University of Amsterdam. During his PhD and post-doctoral (KU Leuven, Belgium) positions, his research focused on the interface of microbiology, biochemistry and biophysics. During his most recent positions in the life-science consulting company InnoTact Consulting and in the Biocatalysis research group of Prof. R. Wever, Michael's interest shifted towards green chemistry and bio-based products. |
Introduction
Vanadium haloperoxidases occur widely in nature and are found in a large number of brown, red and green seaweeds, in diatoms, in bacteria and in a group of fungi, the dematiaceous hyphomycetes. The prosthetic group in these enzymes consists of metal oxide vanadate,1 and unlike the heme peroxidases, UV-VIS spectra of these vanadium enzymes show only a modest absorption in the optical spectra around 315 nm. The bromo and chloroperoxidases are remarkably stable,2–6 are highly resistant towards elevated temperatures and denature only at temperatures above 70 °C.7,8 Detergents such as SDS or water-miscible organic solvents e.g. ethanol or dioxane hardly denature these enzymes. Also oxidative agents such as H2O2 in high concentrations, HOCl and singlet oxygen have little effect on the activity of the chloroperoxidase.9 The enzymes are also resistant towards proteolytic degradation. This makes the vanadium haloperoxidases unique enzymes and also raises the question of their persistence in the environment after death and decay of the host organism. Haloperoxidases catalyze the two-electron oxidation of halides (Cl−, Br−, I−) by H2O2 to hypohalous acids:| H2O2 + H+ + X− → HOX + H2O |
Most enzymes during turnover release these hypohalous acids or related halogenating intermediates, such as OX−, X3−, and X+ from the active site in the medium, although there are exceptions in which vanadium haloperoxidases regiospecifically halogenate substrates.10–12 In most cases these halogenating intermediates react nonspecifically with a variety of organic compounds (RH) that are susceptible for electrophilic attack. This will result in the production of a wide array of halogenated compounds (RX).
| HOX + R − H → RX + H2O |
When a nucleophilic acceptor is absent a reaction may also occur between HOX and H2O2 resulting in the formation of singlet oxygen:
| HOX + H2O2 → 1O2 + H2O + HX |
The historical nomenclature convention of the vanadium haloperoxidases is based on the most electronegative halide which these enzymes are able to oxidize. Chloroperoxidases catalyze the oxidation of Cl−, Br− and I−, bromoperoxidases catalyze only the oxidation of Br− and I− and iodoperoxidases13 are specific for iodide oxidation. It should be noted however that vanadium chloroperoxidase has a much higher rate in the oxidation of bromide (kcat about 250 s−1) than in the oxidation of chloride (kcat about 20 s−1).1
Many red, brown and green macro-algae and ice microalgae from the Arctic, Antarctic, shores of temperate zones and shores of the subtropics and tropics produce CH2Br2 and CHClBr2 at very high rates.14–22 Also diatoms found in oceans produce these halogenated compounds.23–26 The short-lived brominated and volatile methanes bromoform (CHBr3) and dibromomethane (CH2Br2) are a very important source of bromine to the troposphere and lower stratosphere.27,28 These compounds represent the largest natural contributions to atmospheric organic bromine and 20–30% of stratospheric and tropospheric O3 depletion is due to these compounds.29 A recent review30 highlights the role of reactive halogens species in the chemistry and oxidizing capacity of the troposphere. The depletion of ozone through efficient catalytic cycles is well known but these compounds also oxidize dimethyl sulphide, affect the oxidation of organic compounds and reduce the lifetime of the greenhouse gas methane.
Most of these seaweeds have been shown to contain vanadium bromo- and iodoperoxidases31–37 and the majority of these halogenated products are formed directly or indirectly by the enzymatic activity of the vanadium enzymes. However, some seaweeds also contain heme peroxidases and for diatoms both the presence of heme and vanadium peroxidase has been reported.23,26 The major source of atmospheric CHBr3 in the marine environment21,38,39 is the sea-to-air flux originating from macroalgal and planktonic sources, and is therefore of importance as a source of reactive halogens to the troposphere and lower stratosphere. However, this flux of CHBr3 alone cannot account40 for the high atmospheric mixing ratios observed in the tropical Atlantic Ocean.38–42 Possibly terrestrial contributions for CHBr3 are involved and it is conceivable that soils in the tropical area produce brominated compounds. A global three-dimensional chemical model used to simulate atmospheric bromoform also indicates that emissions are likely to be significantly larger than previously assumed.44
The vanadium iodoperoxidases in seaweeds, their role in the formation of I2 and iodinated compounds and the impact on tropospheric photochemistry, via the intermediate formation of iodine radicals, will not be discussed here. For this the reader is referred to recent reviews.45–47 Similarly the vanadium chloroperoxidases and their involvement in the formation of chlorinated1,48 and brominated compounds in the terrestrial environment will be discussed elsewhere.
Formation of HOBr by seaweeds
It has been shown that in some brown33 and a green macro-alga22 the bromoperoxidases are located at or near the surface of the seaweed. This was demonstrated for the brown seaweed Ascophyllum nodosum (knotted wrack) by exposing the intact plant in seawater to hydrogen peroxide. Under these conditions the plants produce HOBr by oxidation of bromide (1 mM) present in seawater resulting in the bromination of phenol red to bromophenol blue. This phenomenon was also observed for the brown seaweeds Laminaria digitata,49L. saccharina, Pelvetia canaliculata, Fucus vesiculosis33 and more recently the marine red seaweed Delisea pulchra.11 However, seaweeds are very different in their morphology and physiology and are difficult to compare, e.g. in some red seaweeds the peroxidases are present in glands.The possible role of the extracellular enzymatic system may be to control the colonization of the surfaces of the seaweed by generating HOBr which is directly bactericidal.50,51 In addition, HOBr in very low concentrations reacts with bacterial homoserine lactones, resulting in inactivated brominated lactones.49 Many bacteria secrete these lactones (Fig. 1) and they play an important role in bacterial signalling systems and interference with these systems inhibits quorum sensing and bacterial biofilm formation, a first step in the fouling of surfaces.49
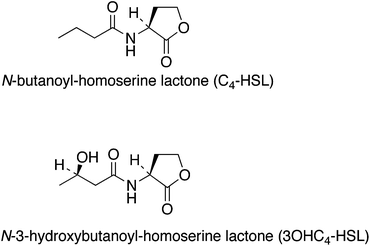 | ||
| Fig. 1 Chemical structures of some of the homoserine lactones produced by bacteria. | ||
Also these lactones are coregulatory ligands required for the control of the expression of genes encoding virulence traits in many Gram-negative bacterial species. Thus, HOBr is not only directly bactericidal but the oxidized halogen species also inactivate homoserine lactones and disturb communication signals between bacteria. Some red macro-algae produced halogenated furanones11,52–54 (Fig. 2) and the halogenated furanones inhibit N-acyl-L-homoserine lactones-dependent gene expression by bacteria.
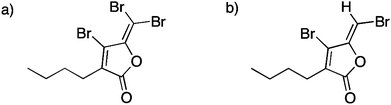 | ||
| Fig. 2 Structures of halogenated furanones produced by Delisea pulchra. (a) 4-Bromo-3-butyl-5-(dibromomethylene)-2(5H)-furanone. (b) 4-Bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone. | ||
The furanones are encapsulated in gland cells in the seaweed and this provides a mechanism for the delivery of the metabolites to the surface of the algae. In the macroalga Delisea pulchra a vanadium bromoperoxidase11 halogenates these furanones.
When the intact seaweed A. nodosum in seawater was exposed to direct sunlight the observation was made that this seaweed generated HOBr.33 The amounts formed under these conditions were 68 nmol g−1 h−1. Although the biomass of this seaweed is not known exactly, it grows in large quantities in the North Atlantic and western Russian polar seas55 and a biomass of 1011 is certainly an accepted value. From this biomass an input of 2 × 109 g of HOBr in the biosphere, produced annually by this particular seaweed, can be estimated. From the rate of release of halometabolites by several algae and a global biomass56 of 1013 g, Gschwend et al.14 estimated an annual global input in the biosphere of 104 tons per year and concluded that the macroalgae are an important source of bromine-containing material released in the atmosphere. More recent data show that these quantities for coastal seawater are even higher. Carpenter and Liss21 arrived at values of 1.6 Gmol Br per year from macroalgae and Carpenter et al.42 estimated on the basis of air–sea fluxes and bulk seawater and atmospheric concentrations of CHBr3 and CH2Br2 a coastal flux of 2.5 Gmol Br per year corresponding to annual fluxes of 1.3–2 × 105 tons per year of bromine in brominated compounds, respectively.
Mechanisms of the formation of bromoform and dibromomethane by algae and diatoms
Many seaweeds that contain bromoperoxidases produce volatile brominated compounds like CHBr3 or dibromomethane in very large amounts. These seaweeds produce other volatile and non-volatile halogenated compounds as well but in general to a lesser extent and they will not be considered in this review. For a detailed discussion on the occurrence of other halogenated metabolites see the review by Paul and Pohnert.57 For a long time the biosynthetic pathways by which these compounds are produced were puzzling but that bromoperoxidases are involved is obvious. Two biosynthetic pathways are conceivable. The first possibility is based upon observations on the red seaweed Bonnemaisonia hamifera that ketones present in seaweeds are brominated by the peroxidase. These ketones are unstable and decay via the haloform reaction to form volatile brominated compounds.58 This requires however that ketones are present near or at the thallus surface and this has not been studied.A more likely possibility as originally proposed33 is that HOBr in the case of A. nodosum and other brown seaweeds is just released in seawater and reacts with dissolved organic matter (DOM) to form unstable brominated compounds which decay to form CHBr3 and other brominated compounds. This process is similar to the production of trihalomethanes (which include CHCl3, CHBr3, CHCl2Br, and CHClBr2) as by-products of drinking water disinfection using chlorination.59 The reaction of HOBr and DOM is very rapid, with a half-life in the millisecond range in high-DOM-containing surface waters.60 Not only polybromomethanes will be formed, but also as known from water disinfection procedures the released HOBr will react with reactive components of DOM to form non-volatile brominated organics.61
Substantial evidence that bromination of DOM by bromoperoxidase leads to polybrominated compounds comes from studies by Manley and coworkers. It was shown22 for example that the addition of H2O2 to the green seaweed Ulva lactuca that contains a bromoperoxidase located outside of the cell membrane resulted in an enhanced production of CHBr3 and that removal of DOM decreased the CHBr3 production. CHBr3 production was nearly tripled in the light compared to the dark. The results also show that H2O2 produced as a result of photosynthetic and respiratory electron transport is available to the bromoperoxidase for bromination leading to the production of volatile polybromomethanes. Similarly a study26 on eleven polar, temperate and warm water marine diatoms showed that 6 species have an extracellular bromoperoxidase which is located in the apoplast, the space between the cell membrane and silicified frustule. These diatoms showed a significant release of reactive bromine and iodine (primarily HOBr and HOI) in the presence of hydrogen peroxide. This is an important phenomenon that has not been reported before for diatoms. The species Porosira glacaialis which is known to produce CHBr323 exhibits the highest bromoperoxidase activity.26 Based on its resistance towards high concentrations of H2O2 the bromoperoxidase in this diatom is probably a vanadium enzyme. Most of the HOBr and HOI released by these diatoms into the sea will react with DOM to form polybromo- and poly-iodomethanes. Importantly the rate of HOBr release was much greater than the CHBr3 emissions measured by others from laboratory cultures and sea ice algae in the field indicating that most of the HOBr released may react with dissolved organic matter to form non-volatile brominated compounds.26 The release of the reactive brominating species may present a defence mechanism against pathogenic marine bacteria and viruses. The subsequent reaction of HOBr or HOI with DOM may be responsible for oceanic CHBr3 production, and CHBr3, CH2Br2 CHI3, and CH2I2 production in polar regions. Direct evidence that reactive bromine and iodine generated by a vanadium bromoperoxidase reacts with DOM, resulting in CHBr3 and dibromomethane, was published recently.62 It was shown that coastal waters contain organic compounds that are susceptible to both bromination by HOBr formed enzymatically by a bromoperoxidase or HOBr from the bottle (bromine water). In both cases the same polybromomethanes were produced with CHBr3 the dominant species. Thus it is very likely that the release of HOBr and HOI by phytoplankton is responsible for oceanic production of CHBr3, CH2Br2, CH3 and CH2I2.29,41
The equations below summarize the sequence of events leading to the formation of CHBr3 and other polybromomethanes:
| H2O2 + H + Br− → HOBr + H2O |
| HOBr + DOM (humic material) → DOM-Br |
| DOM(Br)n → CHBr3 + DOM(Br)n − 3 |
It should be noted that not all the brominated DOM will decay to CHBr3 but other brominated compounds will be formed as well with unknown stability and fate. Some of the brominated material may actually end up in marine sediments. Indeed, it has been shown that marine sediments also contain organic brominated species associated with humic material.63
If these other compounds are formed as well this raises an important question: is the variability and amount of the polybromomethanes produced by either phytoplankton or seaweeds not a function of the origin, nature and concentration of DOM in seawater? Indeed DOM in coastal waters may differ from ocean DOM in terms of chemical composition and it may react and decay differently when exposed to HOBr. Near coastal waters contain humic material that originates mostly from degradation products of leaves and this differs from DOM of marine ecosystems.64 This suggests strongly that the source of the seawater used, the site at which water samples were taken, and in which phytoplankton and seaweeds were incubated determine to a large extent the nature and the amounts of volatile halogenated compounds formed. This may for example explain the differences in distribution of CH2Br2 and CHBr3 in warm and nitrogen depleted surface waters and cold nitrogen enriched deeper waters in the Mauritanian upwelling observed in the study by Quack et al.65 Similarly Carpenter et al.42 suggested that the surface seawater concentration ratio of CH2Br2 and CHBr3 is dependent upon location, that is, open ocean and coastal regions.
Effect of light, H2O2 and stress on bromoform production by algae and diatoms
It is now clear why the addition of H2O2 to seaweeds resulted in the formation of brominated compounds. The vanadium bromoperoxidases that in general are extracellular are accessible to added hydrogen peroxide, start producing HOBr that will in turn react with DOM in seawater or in some cases with ketoacids from the seaweed. Consequently volatile halogenated compounds will be formed as shown by several authors.66–72It is well known that plant cells during photosynthesis and photorespiration generate H2O2.73 Also intra- and intercellular levels of H2O2 increase during environmental stresses experienced by plant cells. Several groups showed that illumination of seaweeds indeed results in the formation of volatile halogenated compounds.66,67,71,74,75 When six algal species (a diatom, a brown algae and four filamentous green algae) were illuminated a strong correlation was observed72 between the release of hydrogen peroxide and the release of brominated compounds to the medium. Surprisingly these species also generated substantial amounts of chloroform with the diatom Pleurosira laevis as one of the major producers. Nightingale et al.16 reported that several seaweed species also release chloroform, although substantially less than CHBr3. This suggests strongly that some of the seaweed species contain chloroperoxidase activity. This requires however that the reaction of enzymatically generated HOCl with DOM be faster than the reaction of HOCl with Br− present in seawater; otherwise only CHBr3 would be detected.
The highest production rates of halocarbons by macroalgae were observed when photosynthesis was maximal in a rock pool containing macroalgae76 under near natural conditions. The concentrations of halocarbons corresponded well with the concentration of H2O2. This again establishes a clear relationship between photosynthesis, H2O2 and halocarbon formation. In line with this, bromination of externally added phenol red occurs33,66 when the brown macroalga A. nodosum and the red alga Meristiella gelidium, which contain (vanadium) bromoperoxidases, were exposed to light. This demonstrates again the direct release of HOBr by these algae.
Stress experienced by the red seaweed Soliera chordalis also affects the release of volatile halogenated compounds, e.g. hypo-osmotic conditions strongly enhanced the production rate of volatile halogenated compounds.74
In line with this exposure of laboratory-grown and harvested wild individuals of L. digitata to oligoguluronates, carbohydrates which are released when the plant is wounded and stressed resulted in the release of H2O2.77 Further, concomitantly L. digitata produces a number of volatile brominated and iodinated compounds.
Thus, illumination and stressing of seaweeds result in the formation of hydrogen peroxide and as a consequence of the presence of the bromoperoxidases, halogenated compounds will be formed via the intermediate formation of HOBr, as has been demonstrated for several different algae under a variety of conditions.66–72,74–77 However, although Antarctic macro algae release CHBr3 and dibromomethane no clear light-dependent relationship was found.78 Similarly the exposure of three species of marine microalgae that were selected to represent three different algal groups that are common to the marine environment, to light stress did not result in the release of iodinated or brominated volatile compounds.79
Bromoform and dibromomethane in oceans and coastal areas
The Gulf Stream may also play a role in this by transporting CHBr3-rich waters from coastal areas to the Arctic Ocean. For some time the CHBr3 released from seaweed and sea-ice algae24,88–90 was taken as the major source of bromine responsible for the ozone loss in the arctic troposphere at polar sunrise.87 Recent studies91,92 showed that the photolysis of CHBr3 was too slow to account for the formation of reactive bromine species and subsequently the reactive gas phase bromine “Bromine explosion” has been explained by heterogeneous autocatalytic reactions taking place on salt-laden ice surfaces.93,94 Globally, however, the direct emission of brominated compounds and molecular iodine contributes significantly to halogen loading in coastal marine boundary layers95,96 and oceans and it may be a new link between climate change and the composition of the global atmosphere.28,29,38
Interestingly a study97 of the marine boundary layer at Roscoff, Britanny, where large macroalgal beds exposed to a large tidal range are present, showed in addition to significant I2 emission the formation of BrO. This pointed to extremely localized emissions of reactive bromine. It is tempting to speculate that the brown algae, mainly Laminariacea, are responsible for this since it is known that they produce HOBr directly. Overall these brominated compounds either abiotic or biotically produced play an important role in tropospheric and lower stratospheric ozone chemistry.
Values of 50 pmol L−1 have been reported by Moore et al.99 at Resolute Bay (Arctic waters) during spring ice algal bloom and by Fogelqvist and Tunhua100 of 54 pmol L−1 in close proximity to an ice flow in the Weddel Sea in Antarctica. Similarly near the King George Island values of up to 190 pmol L−1 (50 ng L−1) were found.101 Strong seasonality in CHBr3 and CH2Br2 concentrations was observed102 and the highest bromocarbon concentrations (up to 276.4 ± 13.0 pmol CHBr3 L−1 and 30.0 ± 0.4 pmol CH2Br2 L−1) were found to coincide with the annual microalgal bloom during the austral summer. The timing of the initial increase in bromocarbon concentrations was related to the sea-ice retreat and onset of the microalgal bloom. The high sea–air flux rate of bromocarbon emissions observed at the seasonal ice edge zone bloom, which occurs each year over large areas of the Southern Ocean during the austral summer, could have an important impact on the chemistry of the Antarctic atmosphere.102 Similarly seawater CHBr3 concentrations in samples collected in coastal waters of the western Antarctic Peninsula were found103 to be significantly higher (122 pmol L−1) in samples collected in diatom bloom compared to non-bloom waters (42.9 [12.0–126] pmol L−1). A comparison of sea-to-air flux rates suggests that a switch from diatom bloom to non-bloom conditions results in a factor of 3–4 decrease in average CHBr3 sea-to-air emission rates which will reduce the supply of biogenic bromine to the atmosphere. These concentrations of bromoform are still 1–2 orders of magnitude less than global near shore values influenced by macro-algae.38
General remarks
From the studies described it is clear that in oceans marine phytoplankton is the main source of bromoform and dibromomethane. Similarly sea ice algae present in the Arctic or Antarctic also produce these brominated compounds in substantial amounts. In this case their formation correlates strongly with microalgal bloom in spring. Some tropical regions are even supersaturated with bromoform and dibromomethane which by sea–air flux leads to halogen-mediated ozone destruction in the atmosphere. The concentrations in the pelagic zone are in general lower than in the coastal waters38,42,82,104 and in going from the open ocean via shelf to coastal areas, saturations and sea–air fluxes of bromoform and dibromomethane in general increase. Several studies have established that in these coastal waters marine macro algae are responsible for the high concentrations of the brominated compounds formed. Tidal cycles and day light seem to affect their formation as was shown for the Iberian upwelling off Portugal.105In the upwelling along the coast, an additional coastal source was present that further increased the bromocarbon concentration. This coastal source was correlated to the diurnal and tidal cycles, showing that intertidal macro-algae beds were involved in halocarbon production.105 Indeed ample evidence exists that photosynthesis plays an important role in the formation of these brominated compounds as has been shown for many seaweed species in laboratory experiments66,67,71,73,75 but also under near natural conditions.76 Phytoplankton and macro algae are not the only source of volatile brominated compounds. Data obtained from a cruise in the Baltic established106 that cyanobacteria form also significant amounts of CHBr3, CH3I, CH2Br2 and CHBr2Cl and in particular during summer, cyanobacteria become highly dominant in the Baltic Sea, forming extensive blooms. The calculated total production of bromoform, in the Baltic Sea, could be of the order of 110 Mg per year for a cyanobacterial bloom period of 1 month and a maximum bloom coverage of 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km2. For the other compounds a range of Mg per year is calculated. Interestingly the addition of hydrogen peroxide to a culture of the cyanobacterium Nodularia spumigena resulted in an increased production of halocarbons, indicating haloperoxidase activity.106 Indeed it has been shown that in several sequenced genomes of marine cyanobacteria, putative vanadium bromoperoxidases are present.107,108 Protein extracts of cultures of Synechococcus sp. CC9311 show bromoperoxidase activity establishing a functional vanadium bromoperoxidase in this strain. Homologous bromoperoxidase sequences are also present in the genomes of Acaryochloris marina, Crocosphaera watsonii and Nodularia spumigena.108 As has also been observed for many seaweed species16,74,76 the presence of halocarbons in surface waters of the Baltic Sea showed a diurnal variation with the highest concentrations around noon.106
000 km2. For the other compounds a range of Mg per year is calculated. Interestingly the addition of hydrogen peroxide to a culture of the cyanobacterium Nodularia spumigena resulted in an increased production of halocarbons, indicating haloperoxidase activity.106 Indeed it has been shown that in several sequenced genomes of marine cyanobacteria, putative vanadium bromoperoxidases are present.107,108 Protein extracts of cultures of Synechococcus sp. CC9311 show bromoperoxidase activity establishing a functional vanadium bromoperoxidase in this strain. Homologous bromoperoxidase sequences are also present in the genomes of Acaryochloris marina, Crocosphaera watsonii and Nodularia spumigena.108 As has also been observed for many seaweed species16,74,76 the presence of halocarbons in surface waters of the Baltic Sea showed a diurnal variation with the highest concentrations around noon.106
Conclusions
Fig. 3 summarizes and simplifies the events occurring in the formation of CHBr3 and its ventilation to the atmosphere. Seaweeds, phytoplankton and cyanobacteria triggered by light or stress start releasing HOBr in seawater. This will react with DOM, resulting in brominated DOM that partly decays via the haloform reaction to volatile brominated compounds, mainly CHBr3 and CH2Br2. The brominated DOM that does not decay may end up in the marine sediment.63 The volatile compounds are ventilated to the atmosphere and as a result of photolysis in the troposphere and lower stratosphere bromine radicals will be formed that cause depletion of ozone.30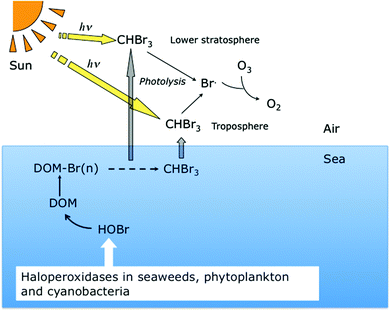 | ||
| Fig. 3 The formation of bromine radicals results from the photolysis of brominated methanes formed by the reaction of DOM with HOBr produced by seaweed, phytoplankton and cyanobacteria. By a catalytic cycle these radicals will cause depletion of ozone.30 | ||
References
- R. Wever and W. Hemrika, in Handbook of Metalloproteins, ed. A. Messerschmidt, R. Hubert, T. Poulos and K. Wieghardt, John Wiley & Sons, Ltd, Chichester, 2001, pp. 1417–1428 Search PubMed.
- M. Almeida, S. Filipe, M. Humanes, M. F. Maia, R. Melo, N. Severino, J. A. L. da Silva, J. J. R. F. da Silva and R. Wever, Phytochemistry, 2001, 57, 633–642 CrossRef CAS.
- D. J. Sheffield, T. Harry, A. J. Smith and L. J. Rogers, Phytochemistry, 1993, 32, 21–26 CrossRef.
- M. G. M. Tromp, G. Olafsson, B. E. Krenn and R. Wever, Biochim. Biophys. Acta, 1990, 1040, 192–198 CrossRef CAS.
- B. Zhang, X. Cao, X. Cheng, P. Wu, T. Xiao and W. Zhang, Biotechnol. Lett., 2011, 33, 545–548 CrossRef CAS.
- E. Garcia-Rodriguez, T. Ohshiro, T. Aibara, Y. Izumi and J. Littlechild, J. Biol. Inorg. Chem., 2005, 10, 275–282 CrossRef CAS.
- H. Plat, B. E. Krenn and R. Wever, Biochem. J., 1987, 248, 277–279 CAS.
- J. W. P. M. van Schijndel, P. Barnett, J. Roelse, E. G. M. Vollenbroek and R. Wever, Eur. J. Biochem., 1994, 22, 151–157 CrossRef.
- R. Renirie, C. Pierlot, J. M. Aubry, A. F. Hartog, H. E. Schoemaker, P. L. Alsters and R. Wever, Adv. Synth. Catal., 2003, 345, 849–858 CrossRef CAS.
- A. Butler and M. Sandy, Nature, 2009, 460, 848–854 CrossRef CAS.
- M. Sandy, J. N. Carter-Franklin, J. D. Martiny and A. Butler, Chem. Commun., 2011, 47, 12086–12088 RSC.
- J. M. Winter, M. C. Moffit, E. Zazopolos, J. B. McAlpine, P. C. Dorrestein and B. S. Moore, J. Biol. Chem., 2007, 282, 16362–16368 CrossRef CAS.
- C. Colin, C. Leblanc, E. Wagner, L. Delage, E. Leize-Wagner, A. van Dorsselaer, B. Kloareg and P. Potin, J. Biol. Chem., 2003, 278, 23545–23552 CrossRef CAS.
- P. Gschwend, J. MacFarlane and K. Newman, Science, 1985, 227, 1033–1035 CAS.
- S. L. Manley, K. Goodwin and W. J. North, Limnol. Oceanogr., 1992, 37, 1652–1659 CrossRef CAS.
- P. D. Nightingale, G. Malin and P. S. Liss, Limnol. Oceanogr., 1995, 40, 680–689 CrossRef CAS.
- F. Laturnus, Mar. Chem., 1996, 55, 359–366 CrossRef CAS.
- N. Itoh, M. Tsujita, T. Ando, G. Hisatomi and T. Higashi, Phytochemistry, 1996, 45, 67–73 CrossRef.
- K. D. Goodwin, W. J. North and M. E. Lidstrom, Limnol. Oceanogr., 1997, 42, 1725–1734 CrossRef CAS.
- F. Laturnus, C. Wiencke and H. Klöser, Mar. Environ. Res., 1996, 41, 169–181 CrossRef CAS.
- L. Carpenter and P. S. Liss, J. Geophys. Res., [Atmos.], 2000, 105, 539–548 CrossRef.
- S. L. Manley and P. E. Barbero, Limnol. Oceanogr., 2001, 46, 1392–1399 CrossRef CAS.
- R. M. Moore, M. Webb, R. Tokarczyk and R. Wever, J. Geophys. Res., [Atmos.], 1996, 101, 20899–20908 CrossRef CAS.
- W. T. Sturges, G. F. Cota and P. T. Buckley, Nature, 1992, 358, 660–662 CrossRef CAS.
- W. T. Sturges, C. W. Sullivan, R. C. Schnell, L. E. Heidt and W. H. Pollock, Tellus Ser. B, 1993, 45, 120–126 CrossRef.
- V. L. Hill and S. L. Manley, Limnol. Oceanogr., 2009, 54, 812–822 CrossRef CAS.
- World Meteorological Organization, Scientific assessment of ozone depletion: 2006, Global Ozone Res. Monit. Proj. 50, Geneva, Switzerland, 2007, p. 498 Search PubMed.
- X. Yang, R. A. Cox, N. J. Warwick, J. A. Pyle, G. D. Carver and N. H. Savage, J. Geophys. Res., [Atmos.], 2005, 110, 1984–2012 Search PubMed.
- R. Salawitch, Nature, 2006, 439, 275–277 CrossRef CAS.
- A. Saiz-Lopez and R. von Glasow, Chem. Soc. Rev., 2012, 41, 6448–6472 RSC.
- E. de Boer, M. G. M. Tromp, H. Plat, G. E. Krenn and R. Wever, Biochim. Biophys. Acta, 1986, 872, 104–115 CrossRef CAS.
- B. E. Krenn, Y. Izumi, H. Yamada and R. Wever, Biochim. Biophys. Acta, 1989, 998, 63–68 CrossRef CAS.
- R. Wever, M. G. M. Tromp, B. E. Krenn, A. Marjani and M. van Tol, Environ. Sci. Technol., 1991, 25, 446–449 CrossRef CAS.
- R. Wever, M. G. M. Tromp, J. W. P. M. van Schijndel and E. Vollenbroek, in Biogeochemistry of Global Change, ed. R. D. Oremland, Chapman and Hall, New York, 1993, pp. 811–824 Search PubMed.
- T. Ohshiro, S. Nakano, Y. Takahashi, M. Suzuki and Y. Izumi, Phytochemistry, 1999, 52, 1211–1215 CrossRef CAS.
- J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060–15066 CrossRef CAS.
- T. Suthiphongchai, P. Boonsiri and B. Panijpan, J. Appl. Phycol., 2008, 20, 271–278 CrossRef CAS.
- B. Quack and D. W. R. Wallace, Global Biogeochem. Cycles, 2003, 17, 1023 CrossRef.
- L. J. Carpenter, D. J. Wevill, J. R. Hopkins, R. M. Dunk, C. E. Jones, K. E. Hornsby and J. B. McQuaid, Geophys. Res. Lett., 2007, 34, L11810, DOI:10.1029/2007gl029893.
- B. Quack, I. Peeken, G. Petrick and K. Nachtigall, J. Geophys. Res., 2007, 112, C10006, DOI:10.1029/2006jc003803.
- B. Quack, E. Atlas, G. Petrick, V. Stroud, S. Schauffler and D. W. R. Wallace, Geophys. Res. Lett., 2004, 31, L23S05, DOI:10.1029/2004gl020597.
- L. J. Carpenter, C. E. Jones, R. M. Dunk, K. E. Hornsby and J. Woeltjen, Atmos. Chem. Phys., 2009, 9, 1805–1816 CrossRef CAS.
- K. A. Read, A. S. Mahajan, L. J. Carpenter, M. J. Evans, B. V. E. Faria, D. E. Heard, J. R. Hopkins, J. D. Lee, S. J. Moller, A. C. Lewis, L. Mendes, J. B. McQuaid, H. Oetjen, A. Saiz-Lopez, M. J. Pilling and J. M. C. Plane, Nature, 2008, 453, 1232–1235 CrossRef CAS.
- N. J. Warwick, J. A. Pyle, G. D. Carver, X. Yang, N. H. Savage, F. M. O'Connor and R. A. Cox, J. Geophys. Res., 2006, 111, D24305, DOI:10.1029/2006jd007264.
- R. Wever, in Vanadium Biochemical and Molecular Biological Approaches, ed. H. Michibata, Springer, Dordrecht, Heidelberg, London, New York, 2012, pp. 95–125 Search PubMed.
- G. McFiggans, C. S. E. Bale, S. M. Ball, J. M. Beames, W. J. Bloss, L. J. Carpenter, J. Dorsey, R. Dunk, M. J. Flynn, K. L. Furneaux, M. W. Gallagher, D. E. Heard, A. M. Hollingsworth, K. Hornsby, T. Ingham, C. E. Jones, R. L. Jones, L. J. Kramer, J. M. Langridge, C. Leblanc, J.-P. LeCrane, J. D. Lee, R. J. Leigh, I. Longley, A. S. Mahajan, P. S. Monks, H. Oetjen, A. J. Orr-Ewing, J. M. C. Plane, P. Potin, A. J. L. Shillings, F. Thomas, R. von Glasow, R. Wada, L. K. Whalley and J. D. Whitehead, Atmos. Chem. Phys., 2010, 10, 2975–2999 CrossRef CAS.
- S. La Barre, P. Potin, C. Leblanc and L. Delage, Mar. Drugs, 2010, 8, 988–1010 CrossRef CAS.
- P. Ortiz-Bermúdez, K. C. Hirth, E. Srebotnik and K. E. Hammel, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3895–3900 CrossRef.
- S. A. Borchardt, E. J. Allain, J. J. Michels, G. W. Stearns, R. F. Kelly and W. F. Mccoy, Appl. Environ. Microbiol., 2001, 67, 3174–3179 CrossRef CAS.
- R. Renirie, A. Dewilde, C. Pierlot, R. Wever, D. Hober and J.-M. Aubry, J. Appl. Microbiol., 2008, 105, 264–270 CrossRef CAS.
- E. H. Hansen, L. Albertsen, T. Schafer, C. Johansen, J. C. Frisvad, S. Molin and L. Gram, Appl. Environ. Microbiol., 2003, 69, 4611–4617 CrossRef CAS.
- T. B. Rasmussen, M. Manefield, J. B. Andersen, L. Eberl, U. Anthoni, C. Christophersen, P. Steinberg, S. Kjelleberg and M. Givskov, Microbiology, 2000, 146, 3237–3244 CAS.
- S. Kjelleberg and P. Steinberg, Microbiology Today, 2001, 28, 134–135 Search PubMed.
- M. Manefield, T. B. Rasmussen, M. Henzter, J. B. Andersen, P. Steinberg, S. Kjelleberg and M. Givskov, Microbiology, 2002, 148, 1119–1127 CAS.
- L. Zenkevitch, Biology of the Seas of the USSR, Interscience Publishers, New York, 1963, 955 Search PubMed.
- R. J. Waaland, in The Biology of Seaweeds, ed. C. S. Lobban and M. J. Wynne, University of California Press, Berkeley and Los Angeles, 1981, pp. 726–741 Search PubMed.
- C. Paul and G. Pohnert, Nat. Prod. Rep., 2011, 28, 186–195 RSC.
- R. Theiler, J. C. Cook, L. P. Hager and J. F. Siuda, Science, 1978, 202, 1094–1096 CAS.
- G. R. Helz and R. Y. Hsu, Limnol. Oceanogr., 1978, 23, 858–869 CrossRef CAS.
- D. A. Jaworske and G. R. Helz, Environ. Sci. Technol., 1985, 19, 1188–1191 CrossRef CAS.
- S. D. Boyce and J. F. Hornig, Environ. Sci. Technol., 1983, 17, 202–211 CrossRef CAS.
- C. Y. Lin and S. L. Manley, Limnol. Oceanogr., 2012, 56, 1857–1866 CrossRef.
- A. C. Leri, J. A. Hakala, M. A. Marcus, A. Lanzirotti, C. M. Reddy and S. C. B. Myneni, Global Biogeochem. Cycles, 2010, 24, GB4017, DOI:10.1029/2010gb003794.
- S. Opsahl and R. Benner, Nature, 1997, 6, 480–482 CrossRef.
- B. Quack, E. Atlas, G. Petrick and D. Wallace, J. Geophys. Res., 2007, 112, D09312, DOI:10.1029/2006jd007614.
- M. Pedersen, J. Collen, K. Abrahamsson and A. Ekdahl, Sci. Mar., 1996, 60, 257–263 CAS.
- J. Collen, A. Ekdahl, K. Abrahamsson and M. Pedersen, Phytochemistry, 1994, 36, 1197–1203 CrossRef CAS.
- J. Sundström, J. Collin, K. Abrahamsson and M. Pedersén, Phytochemistry, 1996, 42, 1527–1530 CrossRef.
- N. Ohsawa, Y. Ogata, N. Okada and N. Itoh, Phytochemistry, 2001, 58, 683–692 CrossRef CAS.
- R. A. Marshall, D. B. Harper, W. McRoberts and M. J. Dring, Limnol. Oceanogr., 1999, 44, 1348–1352 CrossRef CAS.
- L. Mata, H. Gaspar, F. Justini and R. Santos, J. Appl. Phycol., 2011, 23, 827–832 CrossRef CAS.
- K. Abrahamsson, K.-S. Choo, M. Pedersen, G. Johansson and P. Snoeijs, Phytochemistry, 2003, 64, 725–734 CrossRef CAS.
- I. Ślesak, M. Libik, B. Karpinska, S. Karpinski and Z. Miszalski, Acta Biochim. Pol., 2007, 54, 39–50 Search PubMed.
- S. Bondu, B. Cocquempot, E. Deslandes and P. Morin, Bot. Mar., 2008, 51, 485–492 CrossRef CAS.
- M. S. P. Mtolera, J. Collen, M. Pedersen, A. Ekdahl, K. Abrahamsson and A. K. Semesi, Eur. J. Phycol., 1996, 31, 89–95 CrossRef.
- A. Ekdahl, M. Pedersen and K. Abrahamsson, Mar. Chem., 1998, 63, 1–8 CrossRef CAS.
- F. Thomas, A. Cosse, S. Goulitquer, S. Raimund, P. Morin, M. Valero, C. Leblanc and P. Potin, PLoS One, 2011, 6, e21475 CAS.
- F. Laturnus, C. Wiencke and F. C. Adam, Mar. Environ. Res., 1998, 45, 285–294 CrossRef CAS.
- C. Hughes, G. Malin, P. D. Nightingale and P. S. Liss, Limnol. Oceanogr., 2006, 51, 2849–2854 CrossRef CAS.
- A. S. Mahajan, J. M. C. Plane, H. Oetjen, L. Mendes, R. W. Saunders, A. Saiz-Lopez, C. E. Jones, L. J. Carpenter and G. B. McFiggans, Atmos. Chem. Phys., 2010, 10, 4611–4624 CrossRef CAS.
- S. Tegtmeier, K. Krüger, B. Quack, E. L. Atlas, I. Pisso, A. Stohl and X. Yang, Atmos. Chem. Phys., 2012, 12, 10633–10648 CrossRef CAS.
- Y. Liu, S. A. Yvon-Lewis, L. Hu, J. E. Salisbury and J. E. O'Hern, J. Geophys. Res., 2011, 116, C10004, DOI:10.1029/2010jc006729.
- D. Dyrssen and E. Fogelqvist, Ocean. Acta, 1981, 43, 313–317 Search PubMed.
- E. Fogelqvist, J. Geophys. Res., [Atmos.], 1985, 90, 9181–9193 CrossRef CAS.
- E. Fogelqvist, B. Josefsson and C. Roos, Environ. Sci. Technol., 1982, 16, 479–482 CrossRef CAS.
- M. Krysell, Mar. Chem., 1991, 33, 187–197 CrossRef CAS.
- L. A. Barrie, J. W. Bottenheim, R. C. Schnell, P. J. Crutzen and R. A. Rasmussen, Nature, 1988, 334, 138–141 CrossRef CAS.
- R. Wever, Nat. Correspondence, 1988, 335, 501 CrossRef.
- R. Wever, in Microbial growth on C1 Compounds, ed. J. C. Murell and D. P. Kelly, Intercept Ltd, Andover, 1993, pp. 35–45 Search PubMed.
- N. Itoh and M. Shinya, Mar. Chem., 1994, 45, 95–103 CrossRef CAS.
- M. Martinez, T. Arnold and D. Perner, Ann. Geophys., 1999, 17, 941–956 CrossRef CAS.
- W. R. Simpson, R. von Glasow, K. Riedel, P. Anderson, P. Ariya, J. Bottenheim, J. Burrows, L. J. Carpenter, U. Friess, M. E. Goodsite, D. Heard, M. Hutterli, H. W. Jacobi, L. Kaleschke, B. Neff, J. Plane, U. Platt, A. Richter, H. Roscoe, R. Sander, P. Shepson, J. Sodeau, A. Steffen, T. Wagner and E. Wolff, Atmos. Chem. Phys., 2007, 7, 4375–4418 CrossRef CAS.
- K. L. Foster, R. A. Plastridge, J. W. Bottenheim, P. B. Shepson, B. J. Finlayson-Pitts and C. W. Spicer, Science, 2001, 291, 471–474 CrossRef CAS.
- L. Kaleschke, A. Richter, J. Burrows, O. Afe, G. Heygster, J. Notholt, A. M. Rankin, H. K. Roscoe, J. Hollwedel, T. Wagner and H. W. Jacobi, Geophys. Res. Lett., 2004, 31, L16114, DOI:10.1029/2004gl020655.
- L. J. Carpenter, W. T. Sturges, S. A. Penkett, P. S. Liss, B. Alicke, K. Heibestreit and U. Platt, J. Geophys. Res., [Atmos.], 1999, 104, 1679–1689 CrossRef CAS.
- C. J. Palmer, T. L. Anders, L. J. Carpenter, F. C. Kupper and G. McFiggans, Environ. Chem., 2005, 2, 282–290 CrossRef CAS.
- D. Lowe, J. Ryder, R. Leigh, J. R. Dorsey and G. McFiggans, Atmos. Chem. Phys., 2011, 11, 979–994 CrossRef CAS.
- L. J. Carpenter, D. J. Wevill, C. J. Palmer and J. Michels, Mar. Chem., 2007, 103, 227–236 CrossRef CAS.
- R. M. Moore and R. Tokarczyk, Geophys. Res. Lett., 1992, 19, 1779–1782 CrossRef CAS.
- E. Fogelqvist and T. Tanhua, in Naturally-Produced Organohalogens, ed. A. Grimvall and E. W. B. de Leer, Kluwer Academic Publishers, Dordrecht, the Netherlands, 1995, pp. 295–306 Search PubMed.
- W. Reifenhäuser and K. G. Heumann, Chemosphere, 1992, 24, 1293–1300 CrossRef.
- C. Hughes, A. Chuck, H. Rossetti, P. J. Mann, S. M. Turner, A. Clarke, R. Chance and P. S. Liss, Global Biogeochem. Cycles, 2009, 23, GB2024, DOI:10.1029/2008gb003268.
- C. Hughes, M. Johnson, R. von Glasow, R. Chance, H. Atkinson, T. Souster, G. A. Lee, A. Clarke, M. Meredith, H. J. Venables, S. M. Turner, G. Malin and P. S. Liss, Global Biogeochem. Cycles, 2012, 26, GB3019, DOI:10.1029/2012gb004295.
- R. M. Moore and R. Tokarczyk, Global Biogeochem. Cycles, 1993, 7, 195–210 CrossRef CAS.
- S. Raimund, B. Quack, Y. Bozec, M. Vernet, V. Rossi, V. Garcon, Y. Morel and P. Morin, Biogeosciences, 2011, 8, 1551–1564 CrossRef CAS.
- A. Karlsson, N. Auer, D. Schulz-Bull and K. Abrahamsson, Mar. Chem., 2008, 110, 129–139 CrossRef CAS.
- B. Palenik, Q. Ren, C. Dupont, G. Myers, J. Heidelberg, J. Badger and R. Madupu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13555–13559 CrossRef CAS.
- T. L. Johnson, B. Palenik and B. Brahamsha, J. Phycol., 2011, 47, 792–801 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |