 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenylvinylcobalamin: an alkenylcobalamin featuring a ligand with a large trans influence†
Christopher B.
Perry
*,
Naree
Shin
,
Manuel A.
Fernandes
and
Helder M.
Marques
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, P.O. Wits, Johannesburg, 2050, South Africa. E-mail: Christopher.Perry@matthey.com; Helder.Marques@wits.ac.za
First published on 21st March 2013
Abstract
Cob(I)alamin reacts with phenylacetylene to produce two diastereomers in which the organic ligand is coordinated to the upper (β) and lower (α) face of the corrin ring, respectively. The isomers were separated chromatographically and characterised by ESI-MS and, in the case of the β isomer, by 1H and 13C NMR. Only the β isomer crystallised and its molecular structure, determined by X-ray diffraction, shows that the organic ligand coordinates Co(III) through the β carbon of the phenylvinyl ligand. The Co–C bond length is 2.004(8) Å while the Co–N bond length to the trans 5,6-dimethylbenzimidazole (dmbzm) base is 2.217(8) Å, one of the longest Co–Ndmbzm bond lengths known in an organocobalamin. Unlike benzylcobalamin (BzCbl), phenylvinylcobalamin (PhVnCbl) is stable towards homolysis. DFT calculations (BP86/TZVP) on model compounds of BzCbl and PhVnCbl show that the Co–C bond dissociation energy for homolysis to Co(II) and an organic radical in the former is 8 kcal mol−1 lower than in the latter. An analysis of the electron density at the Co–C bond critical point using Bader's QTAIM approach shows that the Co–C bond in PhVnCbl is shorter, stronger and somewhat more covalent than that in BzCbl, and has some multiple bond character. Together with calculations that show that the benzyl radical is more stable than the phenylvinyl radical, this rationalises the stability of PhVnCbl compared to BzCbl. The phenylvinyl ligand has a large trans influence. The pKa for deprotonation of dmbzm and its coordination by the metal in β-PhVnCbl is 4.60 ± 0.01, one of the highest values reported to date in cobalamin chemistry. The displacement of dmbzm ligand by CN− in β-PhVnCbl occurs with log K = 0.7 ± 0.1; the trans influence order of C-donor ligands is therefore CN− < CCH < CHCH2 = PhVn < Me < Et.
Introduction
The initial step in the coenzyme B12-dependent enzymatic reactions involves homolysis of the Co–C bond between Co(III) and 5′-deoxyadenosine to produce Co(II) and the 5′-deoxyadenosyl radical.1–3 In an attempt to observe the formation of the free radical in the solid state, we prepared and crystallized the compound 1-phenylvinylcob(III)alamin (PhVnCbl) anticipating that its photolysis in the solid state would lead to a stable radical that could be observed spectroscopically. Although we were unable to find any evidence for the formation of such a stable radical species, PhVnCbl nevertheless has some interesting properties that we report in this paper. In particular we show that the phenylvinyl ligand has a particularly large trans influence, manifest in a long bond between Co and the 5,6-dimethylbenzimidazole (dmbzm) base in the solid state (Fig. 1), in the position of the base-on–base-off equilibrium, and in the stability constant for the reactions of PhVnCbl with CN−. | ||
| Fig. 1 Standard view and numbering of the organocobalamins in which the upper or β ligand, R, of the corrin in an organic group. We report in this paper the molecular structure and properties of R = phenylvinyl. | ||
Results and discussion
PhVnCbl was synthesised by the reaction of cob(I)alamin and phenylacetylene. HPLC showed the formation of two products, the ratio of which depended on the temperature of the reaction. It was anticipated4 that reaction of cob(I)alamin with phenylacetylene using this procedure would lead to the formation of diastereomeric organocobalamins with the organic ligand on either the upper (β) face of the corrin or on the lower (α) face; in the latter case, the dmbzm ligand is displaced from the coordination sphere of the metal.At room temperature, two peaks of approximately equal intensity with retention times of 8.3 and 8.8 min on reverse phase HPLC emerge while a peak due to H2OCbl+ (from oxidation of unreacted cob(I)alamin, 6.7 min), and one due to unreacted CNCbl (7.3 min) decrease. If the reaction proceeds with the reaction vessel in a stream of warm air, the ratio of the area of the earlier to the later eluting peak is approximately 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55. When the reaction is carried out with the reaction vessel emerged in an ice bath the ratio becomes 65:35.
:55. When the reaction is carried out with the reaction vessel emerged in an ice bath the ratio becomes 65:35.
The products were separated by column chromatography on C18 and analysed by ESI-MS and 1H and 13C NMR. Crystallisation of the two isomers was attempted by vapour diffusion of acetone into a concentrated neutral aqueous solution of each isomer at 4 °C. After three weeks, deep red crystals of the isomer that eluted second from the column were obtained and analysed by X-ray diffraction methods.
The two compounds have somewhat different UV-visible (UV-vis) spectra (Fig. S1 of the ESI†). What we have shown unambiguously by XRD, confirming the ESI-MS result (see below), to be β-PhVnCbl has, at pH 7.0 and hence in the base-on form, the γ band (ε/104 M−1 cm−1) at 335 nm (1.90) with a shoulder (which is quite prominent) at 342 nm; a band at 440 nm (0.66); and the α and β bands at 480 (0.75) and 516 (0.80) nm, respectively. In the base-off form (pH 1.5), the γ band (ε/104 M−1 cm−1) is at 326 nm (2.17) with a shoulder at 347 nm; and the α and β bands at 420 (1.75) and 455 nm (1.01) with a shoulder at 505 nm. The other compound, which ESI-MS suggests is the α diastereomer, has the γ band at 328 nm (2.97) with a weak shoulder at about 350 nm, and the α and β bands at 440 (sh) and 470 nm (1.27), respectively.
Organocobalamins are light-sensitive, resulting in homolysis of the Co–C bond.5 When samples of the two products were dissolved in aqueous solution (pH 7.0, 0.1 M phosphate) and exposed to visible light for some 3 hours, the UV-vis spectrum of the resultant product was in each case identical to that of H2OCbl+.
Molecular structure
The molecular structure of β-PhVnCbl is illustrated in Fig. 2, which gives the standard numbering scheme used for cobalt corrinoids. The crystallographic numbering is given in Fig. S2 of the ESI.† | ||
| Fig. 2 The structure of β-PhVnCbl with non-H atoms drawn as 30% probability ellipsoids. H atoms are omitted for clarity. | ||
There are 4 molecules in the unit cell, hydrogen bonded to neighbouring molecules both directly and through the included water solvent molecules (Fig. S3 of the ESI†). The hydrogen bonding results in the formation of approximately helical voids in the crystal structure that are occupied by solvent water (72 molecules were located in the unit cell) and acetone (4 molecules in the unit cell), leading to a relatively open structure (Fig. S4 of the ESI†).
Contrary to our expectations, the organic ligand is coordinated to the metal through its β carbon rather than its α carbon. The Co–C bond length is 2.004(8) Å while the Co–N bond length to the dmbzm base is 2.217(8) Å. The fold angle of the corrin is 14.6°. (The corrin fold angle is defined as the angle between the mean planes through N21, C4, C5, C6, N22, C9, C10 and through N24, C16, C15, C14, N23, C11, C10.6) The structures of two other vinylcobalamins have been reported, vinylcobalamin itself (VnCbl, R = CH2CH2)7 and chlorovinylcobalamin (ClVnCbl, R = CH2CHCl).7 The Co–C bond lengths are shorter than in the present case (1.912 and 1.953 Å, respectively), presumably because of the greater steric bulk of the Ph substituent in the present compound.
Organocobalamins usually exhibit an inverse trans influence8,9 where the Co–N bond to dmbzm increases as the Co–C bond to the β organic ligand increases. It is thought that this effect is primarily steric in origin and this has been verified recently by DFT calculations.10 As the bulkiness of the β ligand increases, the steric pressure on the corrin increases (a steric cis influence) which in turn is transmitted through to the dmbzm ligand. A plot of Co–C bond length against Co–Ndmbzm gives a strong correlation (Fig. S5 of the ESI†). PhVnCbl has a relatively large trans influence with the Co–Ndmbzm bond length, at 2.217(7) Å only some 0.06 Å shorter than the longest Co–Ndmbzm bond length known in an organocobalamin, that trans to iso-amyl (2.277 Å).11 Surprisingly, its corrin fold angle, at 14.6°, is actually larger than that in VnCbl (12.5°) and in ClVnCbl (5.7°); it might have been anticipated that the steric bulk of the PhVn ligand would have led to considerable steric pressure on the corrin, and hence a small fold angle. Doyle and co-workers12 have shown, from an analysis of five high resolution cobalamin structures that are available, that crystal packing forces affect the orientation of the corrin side chains, and that this is likely to significantly affect the fold of the corrin. Molecular dynamics simulations have also demonstrated that the corrin ring is quite flexible.13–18 We conclude that the difference in the fold angle between PhVnCbl and VnCbl is probably insignificant, and the very flat corrin of ClVnCbl is likely a consequence of the unusual packing in that structure which does not fit into the standard packing schemes normally observed for the cobalamins.12,19,20
It is interesting to note that benzylcobalamin (BzCbl) itself is unstable towards homolysis in the dark at room temperature.21 We found no evidence for decomposition of PhVnCbl over several weeks which suggests that the presence of the vinyl group stabilises the complex. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length [1.333(11) Å] is not statistically significantly longer than that of free styrenes (from a search of the Cambridge Structural Database, 1.29(7) Å, n = 4) which argues against a significant displacement of electron density from this bond to the Co–C bond.
C bond length [1.333(11) Å] is not statistically significantly longer than that of free styrenes (from a search of the Cambridge Structural Database, 1.29(7) Å, n = 4) which argues against a significant displacement of electron density from this bond to the Co–C bond.
NMR assignments
The methodology developed by Brown4,22–25 for assigning the NMR spectrum of cobalamins was used to assign the spectrum of β-PhVnCbl. The full assignment is given in Table S2 of the ESI.† For reasons that are not obvious, the NMR data for the putative α-PhVnCbl diastereomer were unfortunately too poor for a reliable assignment.The aromatic region (5.9–7.8 ppm vs. TMS) of the 1H NMR spectrum showed signals assigned to the R1, C10, B4, B7 and B2 protons at 6.20, 6.50, 6.82, 7.21, and 7.61 ppm, respectively (Table S1†). Hence signals at 6.04, 6.87, and 7.05 ppm are due to the phenyl protons. From the COSY and ROESY spectra, the resonance at 6.87 shows strong coupling to the resonances at 6.04 and 7.05, whereas the latter two are only weakly coupled to each other. The resonance at 6.04 ppm is thus assigned to the phenyl meta H, ph5 (see Fig. S6 of the ESI† for nomenclature).
Strong nOe interactions between ph5 and C46 (0.94 ppm) and C54 (1.54 ppm) are also observed, confirming that the phenylvinyl ligand is indeed coordinated to cobalt at the upper-axial position (Table S3 of the ESI†). Of the remaining two aromatic signals, 6.05 and 7.05 ppm, the former shows many more nOe interactions with groups on the upper face of the corrin ring; the signal at 7.05 ppm only shows very weak nOe interactions between C46 (0.94 ppm) and C37 (1.72 ppm). If we assume that the preferred orientation of the phenylvinyl ligands relative to the corrin ring in solution is the same at that found in the solid state, then the signal at 6.05 ppm must be due to ph4, and the remaining one at 7.05 ppm corresponds to ph6.
These assignments were confirmed by observing that the multiplicities of the three signals at 6.05, 6.87, and 7.05 ppm are a doublet, and two triplets, respectively. The coupling constant for all three signals is between 7.5 and 8.0 Hz, indicating ortho-coupling (meta coupling is not observed). The only possibility for a doublet is due to ph4 coupling to ph5, as there is no proton on ph3. The doublet at 6.05 ppm is thus due to ph4. Both the ROESY and COSY spectra show that the signal at 6.05 couples much more strongly to the signal at 6.87 ppm than the one at 7.05 ppm. The former thus corresponds to ph5 and the latter to ph6. Assignment of the two non-equivalent ph1 protons (3.01 and 3.06 ppm, in the alkene region of the 1H spectrum) is based on the fact that one or both of them shows strong nOe interaction with ph4, C46, C54, C37, and C26.
13C signals for the ph1, ph4, ph5, and ph6 carbon nuclei were obtained directly from the HSQC spectrum and are assigned as the resonances at 118.07, 127.39, 128.31, and 127.39 ppm, respectively (the signals for ph4 and ph6 overlap).
The HMBC spectrum was used to assign the ph2 and ph3 resonances. Both the ph5 proton and the protons on ph1 (but oddly not the ph4 proton) show a correlation to a 13C signal at 148.93 ppm which we assign to ph3. Both ph1 protons, as well as the ph4 proton show a correlation with a signal at 143.24 ppm, which we assign to ph2.
In the solid state, the preferred orientation of the PhVn ligand with respect to the corrin ring has the phenyl group positioned in the “southern” quadrant of the corrin, over the C pyrrole ring when viewed from above. This is the same conformation adopted by the adenine moiety of the adenosyl group in coenzyme B12 (AdoCbl) in the solid state.26 In the ROESY spectrum one of the germinal ph1 protons shows nOe interactions with C54 and C46, which suggests that conformations where the phenyl group of the PhVn ligand occupies positions over the “northern” and “western” quadrants of the corrin ring are also accessible at room temperature. There is therefore some free rotation about the Co–C bond at room temperature. A “northern” conformation of the PhVn ligand would place the phenyl protons in close proximity to the C26 and C37 protons of the a and c side chains, respectively, and indeed, nOe interactions are observed between one of the diastereotopic C37 protons and ph4, as well as ph6. In addition, ph4 shows through-space interactions with one of the C26 protons. A “western” conformation of the PhVn ligand is confirmed since both ph4 and ph5 show nOe interactions with C54, and through-space interactions are also observed between ph4 and C26′, and ph5 and C26′′. Previous studies involving NMR-restrained molecular modelling of AdoCbl, and the related compound Co-β-5′-deoxyadenosylimidazolylcobamide (Ado(Im)Cbl, a coenzyme B12 analogue with an imidazole axial nucleoside) have shown that the “northern” and “western” conformations (as well as some “eastern” conformations) of the Ado ligand are accessible in solution at room temperature.27,28
Molecular modelling
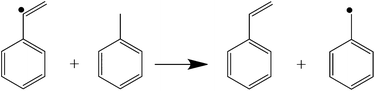
To investigate the nature of the bonding in PhVnCbl, and in particular why it is significantly more stable than BzCbl, we performed DFT calculations on models of the two compounds (see Experimental section). The Co–C bond in the PhVnCbl model was significantly shorter than in that of BzCbl (1.991 and 2.064 Å, respectively; the experimental value for the former is 2.004(8) Å, see above) which, in the absence of the substituents of the corrin, induced a normal (i.e., electronic) trans influence (Co–Ndmbzm = 2.139 and 2.128 Å, respectively, in the PhVnCbl and BzCbl models). The Co–C–C angle in the former (115.1°) is marginally larger than in the latter (113.7°). The ph2–ph3 bond lengths were similar (1.478 Å in the PhVnCbl model; 1.474 Å in the BzCbl model) and not significantly different to that in free styrene itself (1.470 Å). The Co–C bond dissociation energy (eqn (1)) for the PhVnCbl model (42.3 kcal mol−1) was significantly larger than that for the BzCbl model (34.3 kcal mol−1). The isodesmic reaction of eqn (2) was found to have a ΔE of −10.2 kcal mol−1; thus, the benzyl radical is more stable than the PhVn radical.| [Im–Co(III)(Corrin)–R]+ → [Im–Co(II)(Corrin)]+ + R˙ |
| BDE = (E[Im–Co(II)(Corrin)]+ + ER˙) − E[Im–Co(III)(Corrin)–R]+ | (1) |
 | (2) |
An examination of the properties of the Co–C bond using Bader's quantum theory of atoms in molecules (QTAIM)29,30 indicates that the electron density, ρ, at the bond critical point, which is a measure of the strength of a chemical bond,31–34 is larger for PhVnCbl (0.109 au) than for BzCbl (0.094 au; 1 au of ρ = 6.7483 e Å−3), consistent with the shorter bond length. The ratio of the kinetic and potential energy densities at a bond critical point, |Vb|/Gb can be used to characterise the nature of a bond.35 Interactions with |Vb|/Gb < 1 are characteristic of ionic interactions; those with |Vb|/Gb > 2 are typically covalent interactions; and 1 < |Vb|/Gb < 2 are diagnostic of interactions of intermediate character. The values of |Vb|/Gb for PhVnCbl and BzCbl are 1.649 and 1.639, respectively; the bonding in the former therefore has a somewhat more covalent character than the latter.
The ellipticity, εb, at a bond critical point is a function of the ratio of the rate of electron density decrease in the two directions perpendicular to the bond path at that point.29 Values of εb significantly different from zero may be diagnostic of multiple bonding character.36 For example, in the model of PhVbCbl the value of εb for the C–C bond in the imidazole ligand is 0.321; values for C–H bonds are typically ∼0.01. In the case of PhVnCbl, εb for Co–C = 0.0375, but 0.0132 in BzCbl. This suggests there is a measure of multiple bond character between the metal and the alkenyl ligand in PhVnCbl.
We conclude that the greater stability of PhVnCbl compared to BzCbl is due to stronger bonding between the metal and the β organic ligand, which results in a shorter, stronger, somewhat more covalent bond with some multiple bond character. The greater stability of the benzyl radical compared to the phenylvinyl radical may further explain the significantly larger BDE of PhVnCbl than BzCbl.
An assessment of the trans influence of the phenylvinyl ligand
The pKa for the deprotonation of dmbzm and its coordination by the metal (to covert from the base-off to the base-on form) is a measure of the trans influence of the organic ligand. Titration of β-PhVnCbl from pH 8.5 to pH 1.5 causes a significant red shift in the bands of the UV-vis spectrum (Fig. S7A of the ESI†) with pKa = 4.60 ± 0.01. This is one of the highest values reported to date in cobalamin chemistry (to our knowledge only NOCbl, with a pKa of 5.1,37 has a higher pKa – see Table S4 of the ESI,† and Fig. 3). | ||
| Fig. 3 Dependence of the pKa for the deprotonation and coordination of dmbzm by Co(III) on the crystallographically-measured Co–C bond length. The red dot is for PhVnCbl. Data in Table S5 of the ESI.† | ||
The reaction of β-PhVnCbl with CN− displaces the dmbzm ligand by CN−. A titration at pH 11.2 (0.5 M CAPS, 25 °C) gave log K = 0.7 ± 0.1. This can be compared to values of 4.0 when the β ligand is CN−,38 2.7 (CCH),39 0.7 (CHCH2),39 0.1 (Me)39 and <0 (Et).40 These values indicate that the trans influence of ligands where the donor atom is C is CN− < CCH < CHCH2 = PhVn < Me < Et.
It is interesting to note that this order only approximately parallels the trans influence order observed crystallographically as measured by the Co–Ndmbzm bond length: CN− < Me < CCH2 ≪ PhVn < Et (Table S5 of the ESI†). This could well be a consequence of crystal packing forces in the solid state perturbing the Co–Ndmbzm bond length and probably cautions against an over-reliance on solid state data to infer the properties of ligands in the coordination chemistry of the cobalamins.
Experimental section
Reactants and methods
Deionised water was produced by a Millipore RO unit, and further purified by reverse osmosis using a Millipore MilliQ Ultra-Pure system (18 MΩ cm). pH measurements were performed using a Metrohm 6.05 pH meter and a 6.0234 combination glass electrode calibrated against the standard potassium hydrogen phthalate, KH2PO4/K2HPO4 and borax buffers. HPLC was performed on a Phenomenex 5μ micron (150 × 4.6 mm) C18 reverse phase analytical column, a Dionex Ultimate 3000 pump and photodiode array detector at a constant flow rate of 1.00 ml min−1. The mobile phase used consisted of 25 mM phosphate buffer, pH 3, and CH3CN with gradient elution from 98:2 to 75:25 buffer:CH3CN over 12 minutes. All UV-vis spectra were recorded on either a Cary 1E or a Cary 3E spectrophotometer, using 1.00 cm pathlength quartz cuvettes. The cell compartment temperature was kept constant with a water-circulating bath or a Peltier device set at 25.0 °C. ESI mass spectra were obtained using a Thermo LXQ ion trap mass spectrometer with ESI source. One-dimensional (1H, 13C{1H} and dept135), two-dimensional proton homonuclear (COSY, TOCSY, NOESY and ROESY) and two-dimensional heteronuclear (1H–13C) experiments (HSQC and HMBC) NMR spectra were recorded on 15 mg samples dissolved in 700 μL of MeOD at 300 K on a Bruker Avance III 500 spectrometer operating at 500.133 MHz (1H) and 125.770 MHz (13C) using a 5 mm PABBO broad band probe. One-dimensional (1H and 13C) NMR experiments were also recorded on an Avance III 300 spectrometer at 300 K operating at 3000.131 MHz (1H) and 75.475 MHz (13C) using a 5 mm BBI broad band probe. All NMR spectra were internally referenced to TMS. NMR spectra were processed using Bruker Topspin3.0 (Avance III 500) and Topspin2.1 (Avance III 300) software packages. All solvents (Merck) and reagents (Sigma Aldrich) were of the highest purity available and used as received. Cyanocobalamin and hydroxocobalamin were from Roussel.
Preparation of phenylvinylcobalamin
Freshly acid-washed Zn granules (1 g) were placed in a two-necked round bottom flask through which was passed a steady stream of Ar. Cyanocobalamin (CNCbl, 200 mg, 0.15 mmol) was dissolved in 50 mL of a 10% acetic acid/methanol solution in a separate flask; after deaeration (Ar), this was cannula-transferred to the flask containing the Zn metal at room temperature. The solution rapidly turned from red to brown to grey, consistent with the reduction to Co(I). After complete reduction (20 min) phenylacetylene (150 mmol) was added and left to react for three hours in the dark under Ar. The progress of the reaction was monitored by HPLC.All manipulations of the product were conducted under dim red light. The product mixture was cannula-transferred to a round bottom flask and rotatory evaporated to dryness (50 °C). The product was dissolved in a small amount of methanol, filtered through cotton wool, and chromatographed on a 230 × 15 mm C18 column using a Büchi Sepacore chromatographic system consisting of two C-605 pump modules, a C-615 pump manager, a C-660 fraction collector, and a C-635 UV photometer. Fractions (10 mL) were collected using a fraction collector. A gradient of phosphate buffer (25 mM, pH 3) and CH3CN from 0 to 40% CH3CN at a flow rate of 3 ml min−1 was used to elute two bands from the column as well as a small amount of unreacted CNCbl. Fractions were analysed by HPLC and those that contained >98% of the relevant compound were pooled and rotary evaporated to dryness. We isolated 84.5 mg (40.0% yield) of β-PhVnCbl and 32.3 mg of the other isomer (15.0% yield assuming this is indeed α-PhVnCbl).
Analyses
ESI-MS of the compound that crystallised showed a peak at m/z = 1433 (MH+; relative intensity 8%, calculated 1433.49), a base peak at 729 (MHNa2+, calculated 728.2) and a peak at 718 (M·2H2+, 43%, calculated 717.3), consistent with the molecular structure of the β-PhVnCbl diastereomer (Fig. S8†). ESI-MS of the first product eluted from the column gave a peak at m/z = 1434 (relative intensity 5%, Fig. S9†) which may be α-PhVnCbl+ which has lost the H2O ligand from the β coordination site (calculated, 1432.5); the loss of axial ligands from cobalt corrins even using mild ionisation methods is often observed is their mass spectra.41,42 The base peak occurs at 717 (M·H–H2O2+, calculated 716.7). A peak at 726 is assigned to M·H2+ (27%, calculated 725.8); and one at 736 to M·Na–H2O2+ (83%, calculated 736.8).Crystallography
Intensity data for PhVnCbl were collected at 173(2) K on a Bruker APEX II CCD area detector diffractometer with graphite monochromated Mo Kα radiation (50 kV, 30 mA) using the APEX 243 data collection software. The collection method involved ω-scans of width 0.5° and 512 × 512 bit data frames. Data reduction was carried out using the program SAINT+44 and multiscan absorption corrections applied using SADABS.44 The crystal structure was solved by direct methods using SHELXS-97.45 Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement (with the exception of the acetone molecule) by full matrix least-squares calculations based on F2 using SHELXL-97.45 Hydrogen atoms were first located in the difference map then positioned geometrically and allowed to ride on their respective parent atoms, with isotropic thermal parameters fixed at 1.2 (CH, CH2 and NH2) or 1.5 (CH3 and OH) times those of their corresponding parent atoms. Oxygen O15 on the ribose group was found to be disordered and refined over two positions with the aid of SADI, DELU and SIMU restraints. The final occupancies for O15A and O15B were 0.928(12) and 0.072(12), respectively. The acetone molecule was refined isotropically with FLAT, DFIX, DELU and SIMU being used in the final refinements to restrain the molecule to a reasonable geometry. The oxygen atom O12w of a solvent water molecule was refined over two positions (occupancy 0.50(2)). As suggested by a reviewer, hydrogen atoms were added to all the water molecules using geometrical criteria while ensuring that the hydrogen bonding pattern was self-consistent throughout the structure (water molecules do not donate to each other along the same hydrogen bond; the resulting pattern involves all H acceptors and donors; and H⋯H close contacts have been kept as far from each other as possible).Refinement converged as a final R1 = 0.0954 (wR2 = 0.2504 all data) for 9586 observed reflections [I > 2σ(I)]. Figures were prepared using ORTEP-3.46 Details of the crystallographic data are given in Table 1. The structure files have been deposited at the Cambridge Structural Database (deposition number 922431). A list of bond length and angles is given in Table S2 of the ESI.†
| CCDC Deposition Code | 922431 |
| Empirical formula | C73H131CoN13O30P |
| Formula weight | 1760.81 |
| Temperature | 173(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell dimensions | a = 15.7783(7) Å |
| b = 22.3400(9) Å | |
| c = 25.1607(14) Å | |
| Volume | 8868.8(7) Å3 |
| Z | 4 |
| Density (calculated) | 1.319 Mg m−3 |
| Absorption coefficient | 0.296 mm−1 |
| F(000) | 3768 |
| Crystal size | 0.48 × 0.17 × 0.15 mm3 |
| Theta range for data collection | 1.52 to 26.00° |
| Index ranges | −18 ≤ h ≤ 19, −27 ≤ k ≤ 27, −31 ≤ l ≤ 13 |
| Reflections collected | 47932 |
| Independent reflections | 17377 [R(int) = 0.0884] |
| Completeness to θ = 26.00° | 99.9% |
| Absorption correction | None |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 17377/59/1052 |
| Goodness-of-fit on F2 | 0.992 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0954, wR2 = 0.2504 |
| R indices (all data) | R 1 = 0.1631, wR2 = 0.2898 |
| Absolute structure parameter | 0.07(3) |
| Largest diff. peak and hole | 0.681 and −0.467 e Å−3 |
Molecular modelling
All calculations were conducted on a model system based on the crystal structure of PhVnCbl after suitable editing. All corrin side chains were replaced by hydrogen atoms, and the α-dmbzm ligand was truncated to imidazole. Since no geometrical parameters are available for BzCbl, the model system was based on the PhVnCbl crystal structure, constructed by removing the vinyl group from the β-phenylvinyl ligand, and replacing it with hydrogen atoms.DFT geometry optimizations, employing the RI-J approximation,47–53 were performed using the ORCA electronic structure package.54 Calculations were performed with the BP86 functional,55,56 the TZVP basis set57 and corresponding auxiliary basis set, and the empirical van der Waals correction of Grimme et al.58 Frequency calculations were performed to obtain the zero-point correction to the electronic energy, as well as to confirm that all structures were at stable energy minima.
Reported bond dissociation energies (BDE) include a correction for the zero-point vibrational energy, the dispersion energy, as well as a correction for the basis set superposition error (BSSE). The BSSE correction was obtained by performing Counterpoise59,60 calculations in Gaussian 09 using the optimized geometries obtained from ORCA at the same level of theory. Similarly, the wavefunction files required for the analysis of the topological properties of the electron charge density using the atoms in molecules (AIM) framework of Bader29,30 were generated in Gaussian 09 using ORCA-optimized geometries by performing a single point calculation at the same level of theory. The topological properties of the electron density (ρ) were evaluated at all the bond critical points (bcp's) using AIMALL.61
Acknowledgements
Funding for this work was provided by the Department of Science and Technology/National Research Foundation South African Research Chairs Initiative, and the University of the Witwatersrand. Dr Richard Mampa is thanked for recording the NMR spectra.References
- T. Toraya, Chem. Rev., 2003, 103, 2095–2127 CrossRef CAS.
- W. Buckel and B. T. Golding, Annu. Rev. Microbiol., 2006, 60, 27–49 CrossRef CAS.
- E. N. Marsh, D. P. Patterson and L. Li, ChemBioChem, 2010, 11, 604–621 CrossRef CAS.
- K. L. Brown and X. Zou, J. Am. Chem. Soc., 1992, 114, 9643–9651 CrossRef CAS.
- J. M. Pratt, The Inorganic Chemistry of Vitamin B12, Academic Press, London, 1972 Search PubMed.
- J. P. Glusker, in B12, ed. D. Dolphin, Wiley-Interscience, New York, 1982, vol. 1, pp. 23–107 Search PubMed.
- K. M. McCauley, D. A. Pratt, S. R. Wilson, J. Shey, T. J. Burkey and W. A. van der Donk, J. Am. Chem. Soc., 2005, 127, 1126–1136 CrossRef CAS.
- N. Bresciani-Pahor, M. Porcolin, L. G. Marzilli, L. Randaccio, M. F. Summers and P. J. Toscano, Coord. Chem. Rev., 1985, 63, 1–125 CrossRef CAS.
- L. Randaccio, N. Bresciani-Pahor and E. Zangrando, Chem. Soc. Rev., 1989, 18, 225–250 RSC.
- J. Kuta, J. Wuerges, L. Randaccio and P. M. Kozlowski, J. Phys. Chem. A, 2009, 113, 11604–11612 CrossRef CAS.
- C. B. Perry, M. A. Fernandes and H. M. Marques, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, m165–m167 CAS.
- N. Marino, A. E. Rabideau and R. P. Doyle, Inorg. Chem., 2011, 50, 220–230 CrossRef CAS.
- K. L. Brown, X. Zou and H. M. Marques, J. Mol. Struct. (THEOCHEM), 1998, 453, 209–224 CrossRef CAS.
- H. M. Marques and K. L. Brown, Coord. Chem. Rev., 1999, 190–192, 127–153 CrossRef CAS.
- H. M. Marques and K. L. Brown, J. Mol. Struct. (THEOCHEM), 2000, 520, 75–95 CrossRef CAS.
- K. L. Brown and H. M. Marques, J. Inorg. Biochem., 2001, 83, 121–132 CrossRef CAS.
- J. M. Sirovatka, A. K. Rappé and R. G. Finke, Inorg. Chim. Acta, 2000, 300–302, 545–555 CrossRef CAS.
- C. B. Perry, K. L. Brown, X. Zou and H. M. Marques, J. Mol. Struct. (THEOCHEM), 2005, 737, 245–258 CrossRef CAS.
- L. Randaccio, S. Geremia, G. Nardin and J. Wuerges, Coord. Chem. Rev., 2006, 250, 1332–1350 CrossRef CAS.
- L. Randaccio, S. Geremia, G. Nardin, M. Šlouf and I. Srnova, Inorg. Chem., 1999, 38, 4087–4092 CrossRef CAS.
- G. N. Schrauzer and J. H. Grate, J. Am. Chem. Soc., 1981, 103, 541–546 CrossRef CAS.
- K. L. Brown, in Chemistry and Biochemistry of B12, ed. R. Banerjee, John Wiley & Sons, Inc., New York, 1999, pp. 197–237 Search PubMed.
- K. L. Brown, H. B. Brooks, B. D. Gupta, M. Victor, H. M. Marques, D. C. Scooby, W. J. Goux and R. Timkovich, Inorg. Chem., 1991, 30, 3430–3438 CrossRef CAS.
- K. L. Brown, X. Zou, G. Z. Wu, J. D. Zubkowski and E. J. Valente, Polyhedron, 1995, 14, 1621–1639 CrossRef CAS.
- K. L. Brown, X. Zou, S. R. Savon and D. W. Jacobsen, Biochemistry, 1993, 32, 8421–8428 CrossRef CAS.
- L. Z. Ouyang, P. Rulis, W. Y. Ching, G. Nardin and L. Randaccio, Inorg. Chem., 2004, 43, 1235–1241 CrossRef CAS.
- H. M. Marques, X. Zou and K. L. Brown, J. Mol. Struct. (THEOCHEM), 2000, 520, 75–95 CrossRef CAS.
- K. L. Brown, X. Zou, R. R. Banka, C. B. Perry and H. M. Marques, Inorg. Chem., 2004, 43, 8130–8142 CrossRef CAS.
- R. F. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- S. T. Howard and T. M. Krygowski, Can. J. Chem., 1997, 75, 1174–1181 CrossRef CAS.
- S. E. O'Brien and P. L. Popelier, Can. J. Chem., 1999, 77, 28–36 CrossRef CAS.
- R. F. W. Bader, C. F. Matta and F. Cortés-Guzmán, Organometallics, 2004, 23 Search PubMed.
- I. Vidal, S. Melchor, I. Alkorta, J. Elguero, M. R. Sundberg and J. A. Dobado, Organometallics, 2006, 25, 5638–5647 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- P. R. Varadwaj, A. Varadwaj and H. M. Marques, J. Phys. Chem. A, 2011, 115, 5592–5601 CrossRef CAS.
- H. A. Hassanin, M. F. El-Shahat, S. DeBeer, C. A. Smith and N. E. Brasch, Dalton Trans., 2010, 39, 10626–10630 RSC.
- P. George, D. H. Irvine and S. C. Glauser, Ann. N. Y. Acad. Sci., 1960, 88, 393–415 CrossRef CAS.
- G. C. Hayward, H. A. O. Hill, J. M. Pratt, N. J. Vanston and R. J. P. Williams, J. Chem. Soc. A, 1965, 6485–6493 CAS.
- R. A. Firth, H. A. O. Hill, J. M. Pratt and R. G. Thorp, Anal. Biochem., 1968, 23, 429–432 CrossRef CAS.
- H. M. Schiebel and H.-R. Schulten, Mass Spectrom. Rev., 1986, 5, 249–311 CrossRef CAS.
- S. M. Chemaly, K. L. Brown, M. A. Fernandes, O. Q. Munro, C. Grimmer and H. M. Marques, Inorg. Chem., 2011, 50, 8700–8718 CrossRef CAS.
- APEX2, 2009.1-0, Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed.
- SAINT+, 7.60A (includes XPREP and SADABS), Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–566 CrossRef CAS.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- B. I. Dunlap, J. W. D. Connolly and J. R. Sabin, J. Chem. Phys., 1979, 71, 3396–3402 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- R. A. Kendall and H. A. Früchtl, Theor. Chem. Acc., 1997, 97, 158–163 CrossRef CAS.
- C. Van Alsenoy, J. Comput. Chem., 1988, 9, 620–626 CrossRef CAS.
- J. L. Whitten, J. Chem. Phys., 1973, 58, 4496–4501 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 73–78 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- AIMAll (Version 11.12.19), 08.05.04, http://aim.tkgristmill.com, Overland Park, KS, 2011.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 922431. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt50336d |
| This journal is © The Royal Society of Chemistry 2013 |